α-突触核蛋白聚集的基于结构的肽抑制剂的制作方法
本申请根据第119(e)要求2016年6月29日提交的题为“structure-basedpeptideinhibitorsofalpha-synucleinaggregation”的美国临时申请序列号62/356,410的优先权,该临时申请的内容通过引用并入本文。
政府支持声明
本发明根据国立卫生研究院授予的批准号ag029430在政府支持下完成。政府拥有本发明的某些权利。
序列表
本申请包含序列表,该序列表已经以ascii格式电子提交,并且据此通过引用整体并入。于___创建的所述ascii副本命名为___且大小为___字节。
背景技术:
帕金森病(pd)、路易体痴呆(dlb)和多系统萎缩(msa)一起归类为突触核蛋白病(synucleinopathies),这是一类特征在于α-突触核蛋白(α-syn)这种蛋白在神经元细胞中病理性积聚的神经退行性疾病。这些共同构成了第二大常见形式的神经退行性疾病。在脑的可溶性和膜相关部分中发现的突触前蛋白α-突触核蛋白(α-syn),在例如帕金森病(pd)中聚集。这些聚集体是路易体(这种神经退行性疾病的限定组织学特征)的主要组分,并且已被证明伴随神经元损伤7。不希望受任何特定机理的束缚,有人提出这一观察结果和两个其他观察结果指出聚集的α-syn是帕金森病的分子原因8。首先是具有遗传型pd的家族携带α-syn突变(诸如a53t)和丰富的路易体9,10,11。其次是具有编码α-syn的重复基因或三重基因的家族发展早发性pd,这可能是因为在高局部浓度下α-syn被迫进入淀粉样蛋白12,13。
基于结构研究,已经提出了许多不同的α-syn原纤维模型。有限的蛋白水解和nmr研究表明原纤维核由残基30-100组成(miakeh(2002)jbiolchem277(21):19213–19219)。晶体结构和nmr研究暗示了不同的α-syn原纤维模型。在一个基于短区段的晶体结构的模型中,每个折叠结构的两个单体形成延伸的立体拉链(rodriguezja等人(2015)nature525(7570):486-490)。在第二个基于ssnmr的模型中,已经显示了每个淀粉样蛋白层具有一个单体的希腊回纹拓扑结构(tuttlemd等人(2016)natstructmolbiol23(5):409-415)。总之,这些研究表明α-syn可以形成多态性纤维结构。α-syn的称为nacore的片段68-78可以形成α-syn原纤维的核。nacore位于淀粉样蛋白沉积物中发现的35个残基的nac(非淀粉样蛋白β组分)结构域内(rodriguezja等人(2015)nature525(7570):486-490)。nacore容易聚集,并且聚集体显示出类似于全长α-syn的性质,诸如衍射图和细胞毒性。此外,β-突触核蛋白(一种同源物)不含残基74-84且未在淀粉样蛋白沉积物中发现,并且之前已证实除去残基71-82可在体外和果蝇模型中减少聚集和毒性(giasson等人(2001)jbiolchem276(4):2380-2386和periquet等人(2007)jneurosci27(12):3338-3346)。此外,thr72处的修饰防止其聚集倾向。
本发明人最近表明,利用被设计成特异性“封端”正在生长的聚集体的短氨基酸抑制剂,可能有效地阻止阿尔茨海默病相关蛋白tau和精液源性hiv病毒感染增强因子(sevi)的聚集(sievers等人36;美国专利号8,754,03457)。他们还表明,相同的设计策略可以导致抑制p53淀粉样蛋白的形成、拯救癌细胞系中的突变体p53功能并减少肿瘤增殖的经设计的细胞穿透肽(美国专利申请2014/03738758)。因此,存在一个有吸引力的治疗窗,它靶向α-突触核蛋白淀粉样蛋白聚集,α-突触核蛋白是帕金森病和相关神经退行性疾病中的路易体(lb)形式的细胞内沉积物的主要组分。
需要鉴定防止和/或抑制α-突触核蛋白聚集和/或细胞毒性的剂。
技术实现要素:
尽管已经广泛表征了α-syn淀粉样蛋白形成,但是在开发能够抑制α-syn聚集或减少α-syn聚集体(“种子”)在细胞间的朊病毒样扩散的治疗剂方面取得的进展很小。有希望的方法包括螯合α-syn聚集体的抗体以及结合α-syn单体的小分子稳定剂(参见例如mandlerm等人(2015)molneurodegener10(1).doi:10.1186/s13024-015-0008-9;wrasidlow等人(2016)brain:aww238)。使用nacore的原子结构作为模板,我们开发了一类新的抑制剂,即结合α-syn种子并防止它们的生长和伸长的肽剂。如下所示,这些抑制剂抑制许多模型系统中α-突触核蛋白原纤维的形成和接种,以及α-突触核蛋白聚集体的扩散。
本文公开的发明具有许多实施方案。本发明的一个实施方案是包含至少一种抑制肽的物质的组合物,该抑制肽通过与α-突触核蛋白(seqidno:1)的残基68-78结合来抑制α-突触核蛋白聚集。在本发明的典型实施方案中,该抑制肽包含序列gavvwgvtavkk(seqidno:3)或ravvtgvtavae(seqidno:4)。任选地,该抑制肽包含序列gavvwgvtavkkkkk(seqidno:5)、gavvwgvtavkkgrkkrrqrrrpq(seqidno:6)或ygrkkrrqrrravvtgvtavae(seqidno:7)。在本发明的某些实施方案中,该组合物包含多种抑制肽。通常,抑制肽的长度为6至30个氨基酸。
在本发明的抑制肽组合物中,该抑制肽中的至少一个氨基酸可以包括非天然存在的氨基酸(例如d-氨基酸或包含n-甲基部分的氨基酸);和/或该抑制肽与异源肽标签偶联。此类异源肽标签包括增加肽溶解度的氨基酸序列;或有助于监测或操纵肽的氨基酸序列;或促进肽进入哺乳动物细胞的氨基酸序列。任选地,这些肽组合物包括药学上可接受的载体和肽稳定赋形剂。
本发明的另一个实施方案是编码抑制肽的表达载体,该抑制肽通过与α-突触核蛋白的残基68-78结合来抑制α-突触核蛋白聚集。相关实施方案是包含肽或编码这种肽的表达载体的试剂盒,该肽通过与α-突触核蛋白(seqidno:1)的残基68-78结合来抑制α-突触核蛋白聚集。本发明的实施方案还包括通过化学合成或重组产生的方式制备本文公开的肽的方法。本发明的另一个实施方案是包含α-突触核蛋白和通过与α-突触核蛋白的残基68-78结合来抑制α-突触核蛋白聚集的肽的复合物。
本发明的又一个实施方案是用于减少或抑制α-突触核蛋白(seqidno:1)聚集的方法,其包括使α-突触核蛋白淀粉样蛋白原纤维与本文公开的抑制肽以足以减少或抑制α-突触核蛋白聚集的量接触。任选地,在该方法中,α-突触核蛋白淀粉样蛋白原纤维处于体内环境中。替代性地,在该方法中,α-突触核蛋白淀粉样蛋白原纤维处于体外环境中。本发明的相关实施方案是调节α-突触核蛋白淀粉样蛋白原纤维的尺寸或生长速率的方法,其包括使该原纤维与一定量的至少一种抑制肽在某一环境中接触,该抑制肽通过与α-突触核蛋白(seqidno:1)的残基68-78结合而抑制α-突触核蛋白聚集,在该环境中该抑制肽接触α-突触核蛋白的残基68-78,使得所接触的α-突触核蛋白淀粉样蛋白原纤维表现出经调节的尺寸或生长速率。
本发明的又一个实施方案是观察生物样品中α-突触核蛋白淀粉样蛋白原纤维存在或不存在的方法,其包括将生物样品与结合α-突触核蛋白的残基68-78的肽组合,使肽结合可能存在于生物样品中的α-突触核蛋白淀粉样蛋白原纤维,然后监测该组合在α-突触核蛋白淀粉样蛋白原纤维与肽之间形成的复合物的存在;其中所述复合物的存在表明该生物样品中存在α-突触核蛋白淀粉样蛋白原纤维。在该实施方案中,我们的抑制剂之一可以与成像剂诸如放射性标记、不透射线标记、荧光染料、荧光蛋白、比色标记等结合(例如,从而有利于成像方法,诸如mri或pet),并且我们的抑制剂将与患者脑中的α-突触核蛋白原纤维结合,从而允许对原纤维成像以用于诊断接下来的疾病进展。
通过以下详细描述,本发明的其他目的、特征和优点对于本领域的技术人员而言将变得显而易见。然而,应该理解,详细描述和具体实例虽然指明了本发明的一些实施方案,但是它们是以说明而非限制的方式给出的。在不脱离本发明的精神的情况下,可以在本发明的范围内进行许多改变和修改,并且本发明包括所有这些修改。
附图说明
该专利或申请文件包含至少一幅彩色附图。
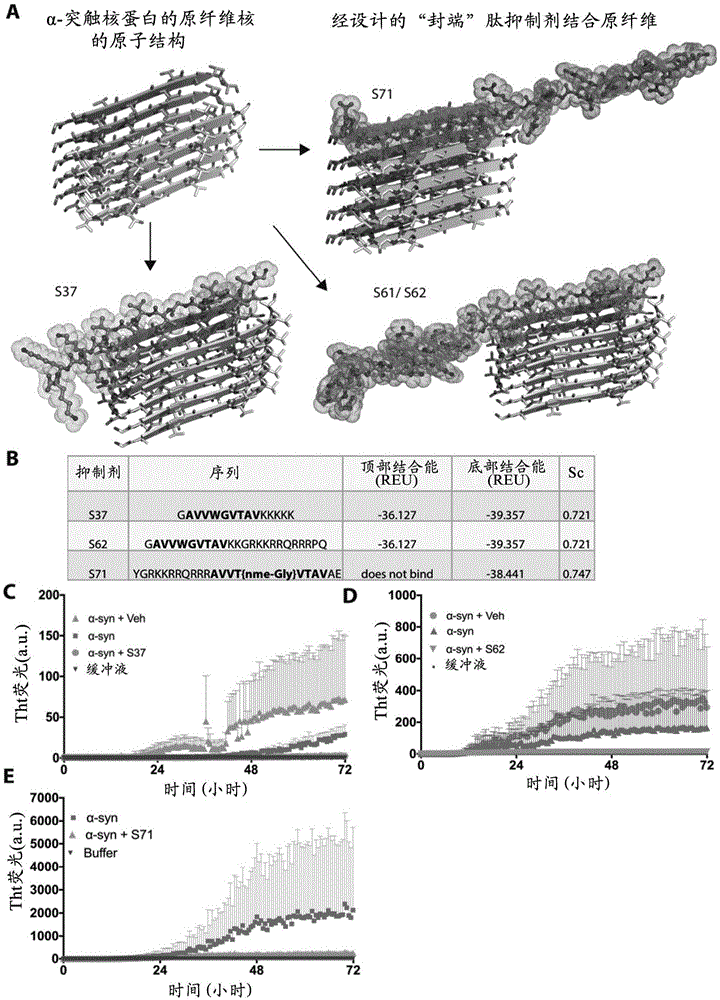
图1:α-syn接种抑制剂的设计。(a)α-syn聚集抑制剂的基于结构的设计。nacore的结构由两个形成立体拉链的自我互补β-折叠组成。鉴定了三种类型的结合一端或两端的抑制剂(褐红色、橙色和青色)。(b)由rossetta计算的不同抑制剂的结合能显示s37和s62结合两个界面,而预期s71仅结合一个界面。所有三种抑制剂的形状互补性都很高。(c)测量α-syn聚集和抑制剂作用的硫磺素t测定。添加50μm的α-syn和5倍摩尔过量的抑制剂。
图2:在抑制剂存在下形成的α-syn聚集体不具有接种能力。(a)细胞培养接种测定的实验设计。α-syn在抑制剂存在下聚集,将混合物在表达yfp标记的wtα-syn或a53tα-syn(绿色)的hek293细胞中转染。a53t是α-突触核蛋白的家族性变体,其导致早发性帕金森病。转染后,内源性α-syn形成荧光斑点(红色)(b)nacore不影响α-syn原纤维的接种能力。(c、d、e)如通过对表达wt和a53t的细胞中每个孔形成的颗粒总数进行计数所测量,在10、5、2和1倍过量的s37、s61和s62中聚集的50μmα-syn不具有接种能力。(f)如通过对表达wt和a53t的细胞中每个孔形成的颗粒总数进行计数所测量,在过量s71中聚集的50μmα-syn不具有接种能力。结果显示为平均值±sd(n=3)。通过双因素anova来分析统计学显著性。
图3:抑制剂防止细胞培养中的接种。(a)细胞培养接种测定的实验设计。用不同量的抑制剂转染125nm重组α-syn原纤维,并随时间监测聚集。(b)nacore不影响接种能力(c、d、e、f)s37、s61、s62和s71降低α-syn的接种能力。所有数据报告为每个孔的颗粒计数,并归一化为在缓冲液处理的孔中的颗粒计数。结果显示为平均值±sd(n=3)。通过双因素anova来分析统计学显著性。
图4:提取自pd脑组织的细丝在体外和细胞培养中接种α-syn聚集。(a)用于从pd脑组织中提取肌氨酰不溶性蛋白丝的方案。(b、c、d)来自3名不同受试者的2%种子诱导快速α-syn聚集,tht荧光增加4-10倍。(e、f、g)来自pd受试者的细丝接种的α-syn比单独的α-syn诱导更多的颗粒。所有数据报告为每个孔的颗粒计数,并归一化为在缓冲液处理的孔中的颗粒计数。结果显示为平均值±sd(n=3)。通过双因素anova来分析统计学显著性。
图5:抑制剂防止pd组织提取的细丝接种。测试了来自4名不同受试者的细丝接种α-syn聚集并通过tht测定进行监测。s61和s62对所有测试的种子都有效。s71对种子b和d有效。
图6:在pd细丝种子和抑制剂存在下形成的α-syn原纤维不具有接种能力。(a)细胞培养接种测定的实验设计(b、c、d)测试了抑制剂对三种不同的pd脑提取组织的接种的抑制作用。s71对所有三种种子均有效,而s61对种子b有效。所有数据报告为每个孔的颗粒计数,归一化为在缓冲液处理的孔中的颗粒计数。结果显示为平均值±sd(n=3)。通过双因素anova来分析统计学显著性。
图7:抑制剂在细胞培养中减少pd细丝接种的α-syn原纤维的接种。(a)细胞培养接种测定的实验设计。(b、c、d)将在两种不同的pd相关细丝存在下形成的α-syn原纤维在yfp-α-synhek细胞中转染,此后测量荧光持续6天。s37在12.5μm至1.25μm的浓度下防止接种。s61防止接种,在第2天效果明显。s71在6.25μm的浓度下减少接种。所有数据报告为每个孔的颗粒计数,归一化为在转染前第0天的颗粒计数。结果显示为平均值±sd(n=3)。通过双因素anova来分析统计学显著性。
具体实施方式
除非另外定义,否则本文使用的所有专门术语、符号和其他科学术语或用辞旨在具有本发明所属领域的技术人员通常所理解的含义。在一些情况下,为了清楚起见和/或为了方便参考,本文定义了具有通常理解的含义的术语,并且本文中包括这些定义不应被解释为表示与本领域通常理解的那些具有实质性差异。本文描述或引用的许多技术和程序均能由本领域的技术人员很好地理解,并且通常可利用常规方法实施。本文引用的所有出版物、专利和专利申请均据此通过引用整体并入以用于所有目的。在优选实施方案的描述中,可以参考形成其一部分的附图,并且其中通过说明的方式示出了可以实践本发明的具体实施方案。应当理解,在不脱离本发明的范围的情况下,可以利用其他实施方案并且可以进行结构变化。
α-突触核蛋白(一种140个氨基酸的蛋白质)发现于疾病状态下神经元细胞中的淀粉样蛋白沉积物中。α-syn淀粉样蛋白形成与疾病进展之间的因果联系得到以下发现的支持:基因重复和增加淀粉样蛋白负荷的家族性突变也引起早发性pd,并且超过90%的散发性pd患者对于α-syn沉积物染色呈阳性。本文公开的发明提供了一种基于结构的方法来停止α-syn聚集。我们假设nacore和prenac的原子结构保留在α-syn淀粉样蛋白种子中,并且使内源蛋白质募集到拉链构象中。使用nacore的原子结构,我们开发了阻碍原纤维形成的抑制剂,并测试了它们在体外和细胞培养中的功效。可以使用相同的程序来开发基于prenac的抑制剂。优化抑制剂以封端原纤维的末端,防止进一步添加单体。我们使用rosetta软件设计了有利地与nacore片段相互作用的肽序列。rosetta使用的能量函数考虑了静电相互作用、氢键、范德华力(vanderwaalsforce)等,以评估结合能。一旦证实特定残基在设计的肽的某个位置产生有利的结合,就可以将其固定并防止进行进一步设计,同时进一步细化该序列的其余部分。我们反复进行这个固定和再设计的过程,直到确定了一组最佳序列。这个合理的设计过程允许计算采样的抑制剂序列数量级比实验上测试可行的更多。
我们对nac结构域和相邻区域内的肽区段晶体进行了筛选,寻求关于α-syn聚集和毒性的分子基础的结构信息。使用zipperdb算法55,鉴定了nac结构域中可结晶的淀粉样蛋白形成区段。具体而言,我们鉴定并集中于α-syn的中心区段,即残基68-78,在本文中称为nacore,这是因为其在α-syn的聚集和细胞毒性中起重要作用,nacore是完整的140残基α-突触核蛋白中35个残基的nac(非淀粉样蛋白-β组分)结构域(残基61-95)的原纤维形成核。在一些研究中,我们利用nacore的亚片段,其由残基69-77组成。在其他研究中,我们结晶并确定了称为prenac的α-突触核蛋白的残基47-56的结构,其形成另一个立体拉链。鉴定的区段经化学合成和结晶,并通过微观结晶和微电子衍射(microed)确定其三维结构。
为了找到最有效的抑制剂,根据经验测试了近100种不同的设计,并对抑制剂设计进行了连续多轮优化。例如,我们观察到n或c末端上标签的位置会影响其功效。另外,添加的修饰类型也会影响其功效。在我们的例子中,只有thr72处的trp取代有效,而arg取代无效。虽然计算方法不足以鉴定一个成功的设计,但它可以缩小我们对候选抑制剂的搜索范围,然后可以通过合理的设计进行优化。
使用基于细胞的测定法测试封端抑制剂在防止接种方面的功效。在该系统中,纳摩尔量的α-syn种子的转染引起内源性蛋白质聚集。聚集体显示淀粉样蛋白形成特性-与淀粉样蛋白特异性小分子结合,细胞分裂时精确地转移和显著特异性(sandersdw等人(2014)neuron82(6):1271–1288)。例如,α-syn原纤维只能将α-syn蛋白接种到聚集体中。值得注意的是,在该系统中,在聚集体形成时我们没有观察到急性细胞死亡,仅细胞增殖轻度减缓。我们的抑制剂防止该系统中斑点形成,单次施用抑制剂,持续2-3天有效。
在淀粉样蛋白疾病的情况下,接种是沿着连接的组织顺序转移病理性蛋白质聚集体。该过程促成神经退行性疾病的进展和严重性。迄今为止,部分由于缺乏病理种子的结构特性信息,因此没有专门针对接种的治疗剂。本申请涉及例如,特异性结合α-突触核蛋白(α-syn)聚集体并阻断、抑制和/或减少α-syn聚集和/或α-syn细胞毒性的肽的设计、合成和功能表征。肽抑制剂特异性“封端”α-突触核蛋白的生长聚集体。在一些实施方案中,肽与细胞穿透肽融合,所述细胞穿透肽增强它们向细胞中的递送。
除了细胞内α-syn自发聚集成淀粉样蛋白原纤维外,促成疾病进展的第二种现象是α-syn聚集体的朊病毒样扩散(goedertm(2015).science349(6248):1255555-1–1255555-9)。braak分期已经显示病理随时间通过连接的脑区逐渐扩散,并且细胞培养和动物模型显示少量的α-syn聚集体可以充当种子并诱导天然蛋白质聚集(参见,例如braakh等人(2003)neurobiolaging24(2):197–211;braak等人(2009)advanatembryolcellbiol201:1–119;masuda-suzukakem等人(2013)brain136(4):1128–1138;desplatsp等人(2009)procnatlacadsciusa106(31):13010–13015;lukkc等人(2009)procnatlacadsci106(47):20051–20056)。虽然不同于可以在人与人之间传播的典型朊病毒,但这种‘接种’现象似乎是疾病进展的驱动因素。
观察到患者提取的原纤维的接种能力不同并且显示出类似菌株的特征。体外pd患者提取的原纤维引起α-syn聚集显著增加,并且在细胞培养模型中,接种的样品增加了斑点形成。值得注意的是,与先前的患者在细胞培养中得到α-syn细丝种子的报告不同,在我们的测定中,我们在细胞培养中没有观察到接种(prusinersb等人(2015)procnatlacadsci112(38):e5308–e5317;和woermanal等人(2015)procnatlacadsci112(35):e4949–e4958)。先前的报告利用黑质组织,而我们使用接种效力可能不同的额叶和颞叶组织。事实上在先前的报告中,已经证实从不同脑区提取的原纤维的接种能力与不同的菌株相似(prusinersb等人(2015)procnatlacadsci112(38):e5308–e5317)。此外,不同的抑制剂对不同种子的功效不同。例如,s61仅对种子a和b有效,而s71对种子b和d有效。最近,报道了全长α-syn原纤维的nmr结构,其中在延伸的拉链构象中未发现nacore区段,但是在原纤维的核中发现了区段68-78(tuttlemd等人(2016)natstructmolbiol23(5):409–415)。或者可能是其中描述的抑制剂可以与nmr结构中的nacore构象结合。还可以想像,nmr结构和立体拉链结构是不同的多晶型物。在缺乏鉴定人受试者中不同多晶型物的诊断方法的情况下,理论上可以使用靶向不同多晶型物的不同抑制剂的混合物。
我们使用计算方法和合理设计的组合来开发一系列靶向防止α-syn聚集体扩散的抑制剂。只能通过确定α-syn淀粉样蛋白原纤维核的原子结构来使我们的方法成为可能,这种方法可以用于其中接种在疾病进展中起一定作用的其他疾病。抑制剂防止体外和细胞培养模型中的α-突触核蛋白聚集。抑制剂还在体外和细胞培养模型中显示出防止患者来源的α-突触核蛋白原纤维接种的功效。我们的结果提供证据表明,α-syn的病理种子含有立体拉链,并提出了一种靶向扩散和进展的可适用于pd和相关突触核蛋白病的治疗方法。类似地,我们的抑制剂可适用于pd和相关突触核蛋白病的诊断方法。
我们假设突变、过表达或其他细胞因子可以使天然α-突触核蛋白结构不稳定,暴露出粘性、立体拉链区段,该区段被提议作为淀粉样蛋白聚集体的基本构件18,19。因此我们生成了α-突触核蛋白聚集体的淀粉样蛋白棘的高分辨率视图。然后我们应用基于rosetta的方法36,使用α-突触核蛋白69-77结构或α-突触核蛋白68-78结构作为模板设计特异性“封端”α-突触核蛋白的生长聚集体,从而破坏和抑制α-突触核蛋白进一步聚集的抑制剂。
我们利用nacore[68-gavvtgvtava-78](seqidno:46)的原子结构作为模板,并使用计算方法和基于结构的方法设计肽抑制剂。nacore的原子结构揭示了一对形成立体拉链的自我互补β-折叠(sawayamr等人(2007)nature447(7143):453–457)。通过基于rossetta的计算模型预测抑制剂,以结合立体拉链界面并‘封端’原纤维。我们鉴定了3种候选抑制剂;有利地结合拉链一端或两端的s37、s61和s71(图1)。三种抑制剂的结合能和形状互补性也是有利的(图1b)。所有抑制剂均保留nacore天然序列的大多数残基,但含有一个或多个经修饰的残基。rodriquez等人表明nacore[69-avvtgvtav-77](seqidno:48)中较小的9残基区段聚集比nacore慢,并且结构与nacore相似。rodriguez等人(2015)还描述了具有序列gvvhgvttv残基47-56的a-syn的第二区段,称为prenac。设计的该序列的抑制剂也可能抑制a-syn的原纤维形成。
为了防止我们设计的抑制剂自聚集,我们使用较短的区段连同一个或多个修饰。s37在thr72处具有w突变并且在c端具有另外的聚赖氨酸标签以诱导电荷-电荷排斥。预期它会结合立体拉链原纤维的两个尖端。s61和s62保留与s37相同的抑制剂序列,但是代替聚赖氨酸标签,添加tat标签以帮助溶解并防止自聚集。s71在gly73处具有弱化沿β-折叠的氢键的甲基化甘氨酸和溶解和细胞穿透的附加tat标签。
我们在体外聚集测定中测试了抑制剂的功效。使重组纯化的α-syn在抑制剂存在下聚集,并通过测量淀粉样蛋白结合染料硫磺素t的荧光来监测。三种抑制剂全部都能防止聚集,tht荧光显著降低(图1c、1d、1e)。
我们测试了抑制剂在细胞培养模型中防止聚集的功效。对于这些测定,我们利用了两种稳定表达yfp标记的全长wtα-syn和a53tα-syn的hek293t细胞(sandersdw等人(2014)neuron82(6):1271–1288)。在该模型中,lipofectamine介导的重组原纤维转染导致内源yfp标记的蛋白质聚集,其被视为荧光斑点。另外,这些斑点的尺寸和数量随着时间的推移而增加。这种聚集体随着时间推移的增殖指示‘接种’现象,借此少量淀粉样蛋白原纤维诱导内源蛋白质的聚集。首先,我们测试了亲本肽区段nacore,以检查其对接种的影响。α-syn在摩尔过量的nacore存在下聚集(图2b)。将混合物转染到细胞中并使斑点形成可视化,并对斑点数量计数为每孔的颗粒数。如预期的那样,nacore并未导致任一细胞系中斑点形成的显著减少。接下来,我们使50μmα-syn在对应于10、5、2和1倍过量的500μm、250μm、100μm和50μm抑制剂存在下聚集。然后将混合物转染到细胞中,并通过荧光成像随时间推移而监测聚集长达3天(图2a)。在wt和a53t表达细胞系中,s37导致接种显著减少长达2天。与s37类似,s61(图2d)也导致斑点形成减少,在两个细胞系中在2、5和10倍过量时功效最大。在s62存在下形成的聚集体(图2e)也无接种能力,在所有抑制剂浓度下形成的颗粒明显更少。s71以等摩尔和亚化学计量比进行测试,发现会降低聚集体的接种效力(图2f)。这些结果表明抑制剂防止形成有接种能力的聚集体。
我们测试了抑制剂在细胞培养模型中防止接种的功效(图3a)。我们将α-syn原纤维连同不同的抑制剂一起转染。nacore,亲本聚集肽不影响α-syn原纤维的接种效力(图3b)。在12.5μm和6.25μm浓度下s37导致两个细胞系中的接种显著减少,持续长达2天(图3c)。浓度为2.5μm和1.25μm的s61导致接种长时间减少,在wtα-syn细胞中持续长达6天和在a53tα-synhek细胞中持续2天(图3d)。在两个细胞系中,s62在12.5μm和6.25μm的浓度下均有效(图3e)。s71在1.25μm的低浓度下有效。有趣的是,我们观察到添加更高浓度的这些抑制剂并不能防止接种,这表明临界浓度范围具有最大功效。这些结果一起表明抑制剂可以封端原纤维种子并防止其伸长。
我们从冷冻尸检pd脑组织中提取不溶性蛋白质聚集体。我们从4个不同的受试者获得组织,包括一个受试者的黑质和额区以及其他受试者的颞区和额区。使用先前描述的方案(goedert等人(1992)neuron8(1):159–168),包括用离子型去垢剂肌氨酰进行沉淀,我们提取了不溶性蛋白质聚集体(图4a)。所有样品均在体外和我们的细胞培养模型中稳健地接种α-syn聚集。体外添加2%种子使tht荧光增加4至10倍(图4b、4c、4d)以及滞后时间小幅减少。然后将接种的样品转染到hek细胞中。与tht测定一致,所有接种的样品诱导斑点快速形成(图4e、4f、4g)。因此,从pd脑组织提取的原纤维接种重组蛋白质,并且在接种时形成的聚集体诱导细胞培养物中的斑点快速形成。
我们在pd提取的种子存在下测试了不同抑制剂防止α-syn聚集的作用。如通过tht荧光测定法所测量的,s71和s62最有效地显示出针对所有种子的功效(图5)。s61还减少了两种不同种子的聚集(图5b、5d),而s37显示出tht荧光的边际性减少(图5d)。接下来,我们测试了在pd种子和不同抑制剂存在下形成的α-syn聚集体的接种效力(图6a)。在s71存在下形成的聚集体(图6b、6c和6d)在表达wt和a53t的hek细胞中均不诱导斑点形成。s61也显示出功效(图6c)。与体外测定一致,s37在降低聚集体的接种效力方面无效。这些结果一起表明s62和s71可以防止形成有接种能力的原纤维。
我们测试了抑制剂在防止细胞培养中pd原纤维接种方面的功效。将在pd原纤维存在下形成的α-syn聚集体连同不同的抑制剂一起转染到wthek细胞中(图7a)。s37在12.5μm-1.25μm的浓度下防止两种不同pd细丝形成斑点,持续长达2天(图7b)。与s37类似,s61在12.5μm-1.25μm的浓度下也显示出长达2天的功效,并且s71也在相似浓度下防止接种。这些结果表明,抑制剂稳健地防止细胞培养中的接种。
上面的公开内容描述了与本发明的许多工作实施方案相关的工作。本文公开的发明提供了此类抑制肽;包含本发明抑制肽和药学上可接受的载体的药物组合物;使用抑制肽阻断、抑制和/或防止α-突触核蛋白聚集和/或α-突触核蛋白细胞毒性的方法,该方法包括使α-突触核蛋白分子(例如单体、小聚集体、寡聚体或原纤维)与有效量的本发明的肽抑制剂接触,或向受试者施用有效量的本发明的肽抑制剂;及计算机相关的实施方案,诸如基于本文所述结构设计和获得抑制肽或小分子的方法。
本发明抑制肽的优点包括:(1)合成肽并不昂贵。(2)由于其组成和小尺寸,细胞穿透和蛋白质稳定性不具挑战性。另外,所述肽可以与细胞穿透肽融合,所述细胞穿透肽增强它们向细胞中的递送。(3)它们出乎意料地稳定:它们不蛋白水解并且表现出足够长的半衰期以在体内(例如身体内)起作用。(4)肽抑制剂对其靶标具有特异性,因此提供与例如可能与许多靶标结合的小分子相比更少机会的副作用。
本文公开的发明具有许多实施方案。本发明的一个实施方案是包含至少一种抑制肽的物质的组合物,所述抑制肽通过与α-突触核蛋白的残基68-78结合来抑制α-突触核蛋白(seqidno:1)聚集。如下面实施例中所公开的,这些肽的工作实施方案包括s37、s62和s71,如下表1中所示:
表中,n-me-gly代表具有甲基化氨基的甘氨酸。粗体给出了抑制剂序列。非粗体显示标签,溶解标签或接头。
在本发明的典型实施方案中,该抑制肽包含序列gavvwgvtavkk(seqidno:3)或ravvtgvtavae(seqidno:4)。任选地,该抑制肽包含序列gavvwgvtavkkkkk(seqidno:5)、gavvwgvtavkkgrkkrrqrrrpq(seqidno:6)或ygrkkrrqrrravvtgvtavae(seqidno:7)。在本发明的某些实施方案中,该组合物包含多种抑制肽。通常,抑制肽的长度为6至30个氨基酸。还包括这些抑制肽中任一种的活性变体。具有前述序列的抑制肽,包括活性变体,在本文中有时称为“本发明的抑制肽”。
在本发明的抑制肽组合物中,抑制肽中的至少一个氨基酸包含非天然存在的氨基酸(例如d-氨基酸或包含n-甲基部分的氨基酸);和/或抑制肽与异源肽标签偶联。此类异源肽标签包括在体内或体外增加肽溶解性的氨基酸序列(例如多个精氨酸残基);或利于在体内或体外对肽进行监测或操纵的氨基酸序列(多个赖氨酸或组氨酸氨基酸);或利于肽进入哺乳动物细胞的氨基酸序列(例如细胞穿透肽序列)。本发明的另一方面是包含本发明的抑制肽和药学上可接受的载体的药物组合物。此类药物组合物有时在本文中称为“本发明的药物组合物”。任选地,本文公开的肽组合物包括药学上可接受的载体和肽稳定赋形剂。
本发明的另一个实施方案是编码抑制肽的表达载体,所述抑制肽通过与α-突触核蛋白的残基68-78结合来抑制α-突触核蛋白聚集。任选地,表达载体是用于将多肽递送至哺乳动物细胞的载体(诸如慢病毒)。关于这点,本发明的另一个实施方案是在哺乳动物细胞(例如离体或体内细胞)中递送编码抑制α-突触核蛋白聚集的抑制肽的dna的方法,该方法是通过使哺乳动物细胞与转导该细胞的载体接触,使得所述dna在该细胞内表达。
另一个实施方案是试剂盒,其包含通过结合α-突触核蛋白的残基68-78来抑制α-突触核蛋白(seqidno:1)聚集的肽或编码此类肽的表达载体。本发明的实施方案还包括通过化学合成或重组产生的方式制备本文公开的肽的方法。本发明的另一个实施方案是包含α-突触核蛋白和通过与α-突触核蛋白的残基68-78结合来抑制α-突触核蛋白聚集的肽的复合物。
另一个实施方案是在prenac结构(α-突触核蛋白的残基47-56:gvvhgvttva)上设计的用于抑制α-突触核蛋白的原纤维形成的肽。
本发明的又一个实施方案是用于减少或抑制α-突触核蛋白(seqidno:1)聚集的方法,其包括使α-突触核蛋白淀粉样蛋白原纤维与本文公开的抑制肽以足以减少或抑制α-突触核蛋白聚集的量接触。任选地,在该方法中,α-突触核蛋白淀粉样蛋白原纤维处于体内环境中。替代性地,在该方法中,α-突触核蛋白淀粉样蛋白原纤维处于体外环境中。本发明的相关实施方案是调节α-突触核蛋白淀粉样蛋白原纤维的尺寸或生长速率的方法,其包括使该原纤维与一定量的至少一种抑制肽在某一环境中接触,该抑制肽通过与α-突触核蛋白(seqidno:1)的残基68-78结合而抑制α-突触核蛋白聚集,在该环境中该抑制肽接触α-突触核蛋白的残基68-78,使得所接触的α-突触核蛋白淀粉样蛋白原纤维表现出经调节的尺寸或生长速率。
本文公开的肽抑制剂可用作中止帕金森病在脑内扩散的疗法。替代性地,本文公开的肽抑制剂可用作诊断探针,以识别诸如帕金森病、路易体痴呆和多系统萎缩的疾病中的蛋白质即α-突触核蛋白的病理性聚集种子。关于这点,本发明的实施方案包括观察生物样品中蛋白质即α-突触核蛋白的聚集种子的方法,该方法包括使生物样品与结合α-突触核蛋白的残基68-78的肽接触,然后观察如果存在于该生物样品中,所述肽是否与蛋白质即α-突触核蛋白的聚集种子结合。通常在这些方法中,异源肽标签与抑制肽偶联以便于对肽进行观察。任选地,在这些方法中,生物样品来自疑似患有帕金森病、路易体痴呆或多系统萎缩的个体。
如本文所用,除非上下文另外明确指出,否则单数形式“一”、“一种(个)”和“所述(该)”包括复数指示物。例如,在前述情况中,药物组合物可包含一种或多种可以相同或不同的本发明的抑制肽分子。
本发明的另一方面是包含α-突触核蛋白分子(例如α-突触核蛋白单体、小聚集体、寡聚体或原纤维)和本发明抑制肽的复合物。它们可以彼此结合、偶联或以其他方式缔合。α-突触核蛋白和抑制肽可以共价或非共价连接。
本发明的其他方面包括编码本发明抑制肽的多核苷酸;包含所述多核苷酸的表达载体;用所述多核苷酸或表达载体转染的细胞;和制备所述肽的方法,其包括使所述肽在转染的细胞中表达,培养所述细胞并收获由此产生的肽。
本发明的另一方面是一种抑制(防止、终止)α-突触核蛋白分子(例如,α-突触核蛋白的单体、小聚集体、寡聚体或原纤维)聚集的方法,其包括使α-突触核蛋白分子与有效量的本发明的抑制肽或药物组合物接触。α-突触核蛋白分子可以处于溶液中或细胞中,细胞处于培养物中或受试者中。在一个实施方案中,接触作为单体、寡聚体或小聚集体的α-突触核蛋白分子防止α-突触核蛋白分子的聚集(寡聚化、进一步寡聚化和/或原纤维形成)。在另一个实施方案中,接触聚集形式的α-突触核蛋白或原纤维防止聚集形式或原纤维的进一步聚集(原纤化)。
在该方法的一个实施方案中,接触的α-突触核蛋白蛋白分子是在患有因存在原纤化的α-突触核蛋白而介导的疾病或病状(有时在本文中称为α-突触核蛋白介导的疾病或病状,或突触核蛋白病),诸如帕金森病(pd)、路易体痴呆和多系统萎缩的受试者中。α-突触核蛋白病理也见于其他相关的神经退行性疾病中,例如,散发性和家族性阿尔茨海默氏病(alzheimer’sdisease)。
本发明的另一方面是治疗患有α-syn介导的疾病或病状,例如帕金森病、路易体痴呆或多系统萎缩的受试者的方法,其包括向受试者施用有效量的本发明的抑制肽或药物组合物。该治疗可导致受试者中α-突触核蛋白聚集和/或α-突触核蛋白细胞毒性以及病理扩散(接种)的阻断(防止)或抑制。
本发明的另一方面是用于鉴定如本文所述的抑制α-突触核蛋白聚集和/或α-突触核蛋白细胞毒性的肽的计算机实施方法。本发明的另一方面是包含本发明的抑制肽的试剂盒,抑制肽任选地包装在容器中。本发明的另一方面是制备本发明抑制肽的方法,其包括化学合成抑制肽或重组产生抑制肽。
本发明的又一个实施方案是观察生物样品中α-突触核蛋白淀粉样蛋白原纤维的存在或不存在的方法,其包括将生物样品与结合α-突触核蛋白的残基68-78的肽组合,使肽结合可能存在于生物样品中的α-突触核蛋白淀粉样蛋白原纤维,然后监测该组合在α-突触核蛋白淀粉样蛋白原纤维与肽之间形成的复合物的存在;其中所述复合物的存在表明该生物样品中存在α-突触核蛋白淀粉样蛋白原纤维。任选地,在该方法中,使用与所述肽偶联的可检测标记(例如异源肽标签)监测在α-突触核蛋白淀粉样蛋白原纤维与所述肽之间形成的复合物的存在。在本发明的说明性实施方案中,肽包含序列gavvwgvtavkk(seqidno:3)或ravvtgvtavae(seqidno:4)。通常,对从疑似患有帕金森病的个体获得的生物样品进行该方法。本发明的此类实施方案可用于例如经设计用于观测观察pd的存在或状态的诊断方法中,例如用于检测在临床症状之前开始的疾病,并且跟踪治疗性治疗的有效性(或有效性的缺乏)。
本发明的肽抑制剂特异性(选择性,优先)结合α-突触核蛋白而不是非预期的蛋白质。肽抑制剂结合的蛋白质可以是例如单体、小聚集体、寡聚体或原纤维。例如,所述结合可以强2倍、5倍、10倍、100倍或200倍,或者根本不能检测到与非预期靶标的结合。可以使用常规方法测定结合特异性,例如竞争性结合测定或其他合适的分析方法。
还包括上述抑制肽的活性变体。“活性变体”是保持本文所述抑制肽的至少一种性质的变体(例如,结合α-突触核蛋白和/或阻断、抑制或防止α-突触核蛋白原纤维形成(聚集)和/或α-突触核蛋白细胞毒性的能力)。如本文所用,原纤化是指形成纤维或原纤维,例如淀粉样蛋白原纤维。
合适的活性变体包括拟肽化合物(含有非肽结构元件的任何化合物,其能够模拟天然模拟肽的生物化学和/或生物学作用,包括例如根据本文公开的方法经设计用于模拟所述肽的结构和/或结合活性(例如,氢键和疏水性包装相互作用的那些化合物)。本发明的抑制肽,包括其活性变体,在本文中有时称为“肽化合物”或“化合物”。
在一个实施方案中,抑制肽的活性变体在原始抑制肽的n-末端、c-末端或两处缩短1-3个(例如,1、2或3个)氨基酸。在另一个实施方案中,活性变体在原始抑制肽的c-末端延长(延伸)1、2、3或4个氨基酸,例如氨基酸残基在α-突触核蛋白中它们出现的位置。
包括各种其他类型的活性变体。在一些实施方案中,取代除上述氨基酸之外的氨基酸。这些氨基酸可以帮助保护肽抑制剂免于蛋白水解或以其他方式使肽稳定,和/或以其他方式促进所需的药效学特性。在一些实施方案中,非天然氨基酸允许抑制剂与靶标更紧密地结合,因为侧链优化了氢键和/或与其的非极性相互作用。另外,非天然氨基酸提供了引入可检测标志物,例如可以使用的强荧光标志物的机会,例如,以测量诸如抑制常数的值。还包括肽模拟物,例如类肽、β氨基酸、n-乙基化氨基酸和小分子模拟物。
在一个实施方案中,非天然氨基酸取代序列中的氨基酸。可商购获得超过100种非天然氨基酸。这些包括,例如,
可取代leu的非天然氨基酸:
可取代thr的非天然氨基酸:
thr(tbu)-oh71989-35-0
(rs)-2-氨基-3-羟基-3-甲基丁酸105504-72-1
可取代ile的非天然氨基酸:
别-ile-oh251316-98-0
n-me-别-ile-oh136092-80-3
同型leu-oh180414-94-2
可取代arg的非天然氨基酸:
nω-硝基-l-精氨酸58111-94-7
l-瓜氨酸133174-15-9
可取代tyr的非天然氨基酸:
可取代lys的非天然氨基酸:
在另一个实施方案中,一个或多个(例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个)l-氨基酸被d氨基酸取代。
在另一个实施方案中,肽中包括一个或多个(例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16或17个)n-甲基化残基。
本发明的抑制肽可包含例如,l-氨基酸、d-氨基酸、非天然氨基酸或其组合。
活性变体包括在肽的n-末端或c-末端包含各种标签的分子。例如,本发明的抑制肽可在其n-末端和/或其c-末端包含作为标签的:1、2、3、4、5、6、7、8、9或10个或更多个赖氨酸残基;1、2、3、4、5、6、7、8、9或10个或更多个精氨酸残基;1、2、3、4、5、6、7、8、9或10个或更多个谷氨酸残基;1、2、3、4、5、6、7、8、9或10个或更多个天冬氨酸残基;这些氨基酸残基的组合;或者对技术人员而言显而易见的其他极性标签。其他活性变体包括α-突触核蛋白序列的突变,其增加抑制肽对α-突触核蛋白的亲和力。
在一个实施方案中,抑制剂是通过jiang等人描述的方法63(其通过引用并入本文,特别是关于该方法),使用本文所述的α-突触核蛋白的纤维形成区段的原子结构作为设计抑制剂的基础而设计的小分子。可通过jiang等人的这种方法鉴定的合适小分子对技术人员来说将是显而易见的。
在本发明的一个实施方案中,修饰本发明的肽,使其1、2或3个氨基酸被具有非天然存在的侧链的氨基酸取代,诸如上面讨论的非天然氨基酸,或被具有通过由jiang等人63设计的小分子交联(例如,通过lys残基的ε氨基)修饰的侧链的氨基酸取代。一些代表性的纤维结合分子如下所示。这些活性变体不但封端α-突触核蛋白的生长聚集体,而且可以通过经修饰的侧链结合(夹紧)立体拉链的侧面,从而增强肽的抑制活性。
由jiang等人63设计的纤维结合化合物包括:
在本发明的一个实施方案中,使用常规技术诸如本文所述的方法分离或纯化本发明的抑制肽。“分离”意指与通常与之缔合的组分,例如,合成肽后存在的组分分开。分离的肽可以是含有肽序列的蛋白质裂解产物。“纯化”抑制肽的纯度可以例如,高于90%、95%、98%或99%。
在一个实施方案中,为了增强本发明抑制肽的细胞渗透性,使肽与多种细胞穿透肽(cpp)中的任何一种融合。cpp通常具有下述氨基酸组成,其含有高相对丰度的带正电荷的氨基酸诸如赖氨酸或精氨酸,或者具有含极性/带电荷氨基酸和非极性疏水氨基酸的交替模式的序列。这两种类型的结构分别称为聚阳离子性或两亲性。第三类cpp是仅含非极性残基,净电荷较低的疏水肽,或具有对细胞摄取至关重要的疏水性氨基酸基团。表2中提供了可以与本发明的抑制肽融合的一些典型cpp。
表2
·名称序列
参考–原始或综述
·聚argnr其中4<n<17(例如,n=5、6、7、8、9、10、11、12、13、14、15或16)(seqidno:8)
wender,p.a.,mitchell,d.j.,pattabiraman,k.,pelkey,e.t.,steinman,l.和rothbard,j.b.(2000).thedesign,synthesis,andevaluationofmoleculesthatenableorenhancecellularuptake:peptoidmoleculartransporters.proc.natl.acad.sci.u.s.a.97,13003-8。
·聚lysnk其中4<k<17(例如,k=5、6、7、8、9、10、11、12、13、14、15或16)
·d-聚argnr其中4<n<17(例如,n=5、6、7、8、9、10、11、12、13、14、15或16)
·d-聚lysnk其中4<k<17(例如,k=5、6、7、8、9、10、11、12、13、14、15或16)
·synb1rggrlsysrrrfststgr(seqidno:9)
·synb3rrlsysrrrf(seqidno:10)
·穿膜肽rqikiwfqnrrmkwkk(seqidno:11)
derossi,d.,joliot,a.h.,chassaing,g.和prochiantz,a.(1994).thethirdhelixoftheantennapediahomeodomaintranslocatesthroughbiologicalmem-branes.j.biol.chem.269,10444-50。
·penargrqiriwfqnrrmrwrr(seqidno:12)
·penlyskqikiwfqnkkmkwkk(seqidno:13)
·tatp59wgrkkrrqrrrpwq(seqidno:14)
·tat(48-60)grkkrrqrrrppq(seqidno:15)
vives,e.,brodin,p.和lebleu,b.(1997).atruncatedhiv-1tatproteinbasicdomainrapidlytranslocatesthroughtheplasmamembraneandaccumulatesinthecellnucleus.j.biol.chem.272,16010-7。
·r9-tatgrrrrrrrrrppq(seqidno:16)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·tatygrkkrrqrrr(seqidno:17)
vives,e.,brodin,p.和lebleu,b.(1997).atruncatedhiv-1tatproteinbasicdomainrapidlytranslocatesthroughtheplasmamembraneandaccumulatesinthecellnucleus.j.biol.chem.272,16010-7。
·d-tatgrkkrrqrrrppq(seqidno:18)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·bmvgag(7-25)kmtraqrraaarrnrwtar(seqidno:19)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·fhvcoat(35-49)rrrrnrtrrnrrrvr(seqidno:20)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·htlv-iirex(4-16)trrqrtrrarrnr(seqidno:21)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·p22n-(14–30)naktrrherrrklaier(seqidno:22)
·pveclliilrrrirkqahahsk(seqidno:23)
elmquist,a.,lindgren,m.,bartfai,t.和langel,
·transportangwtlnsagyllgkinlkalaalakkil(seqidno:24)
pooga,m.,
·tp10agyllgkinlkalaalakkil(seqidno:25)
soomets,u.,lindgren,m.,gallet,x.,
·ptd-4pirrrkklrrlk(seqidno:26)
·ptd-5rrqrrtsklmkr(seqidno:27)
·pep-1ac-ketwwetwwtewsqpkkkrkv-cya(seqidno:28)
·pep-2ac-ketwfetwftewsqpkkkrkv-cya(seqidno:29)
morris,mc,chaloin,l,choob,m,archdeacon,j,heitz,f和divita,g(2004).combinationofanewgenerationofpnaswithapeptide-basedcarrierenablesefficienttargetingofcellcycleprogression.genether11:757–764。
·pep-3ac-kwfetwftewpkkrk-cya(seqidno:30)
morris,mc,gros,e,aldrian-herrada,g,choob,m,archdeacon,j,heitz,f等人(2007).anon-covalentpeptide-basedcarrierforinvivodeliveryofdnamimics.nucleicacidsres35:e49。
·en(1–22)mdaqtrrrerraekqaqwkaan(seqidno:31)
·b21n-(12–29)taktrykarraeliaerr(seqidno:32)
·u2af(142–153)sqmtrqarrlyv(seqidno:33)
·prp6(129–144)trrnkrnriqeqlnrk(seqidno:34)
·mapklalklalklalalkla(seqidno:35)
·sbpmglglhllvlaaalqgawsqpkkkrkv(seqidno:36)
·fbpgalflgwlgaagstmgawsqpkkkrkv(seqidno:37)
·mpgac-galflgflgaagstmgawsqpkkkrkv-cya(seqidno:38)
morris,mc,vidal,p,chaloin,l,heitz,f和divita,g(1997).anewpeptidevectorforefficientdeliveryofoligonucleotidesintomammaliancells.nucleicacidsres25:2730–2736。
·mpg(δnls)ac-galflgflgaagstmgawsqpkskrkv-cya(seqidno:39)
·rev(34-50)trqarrnrrrrwrerqr(seqidno:40)
futaki,s.(2002)arginine-richpeptides:potentialforintracellulardeliveryofmacromoleculesandthemysteryofthetranslocationmechanisms.int.j.pharm.245,1–7。
·来自jiang等人,pnas2004的acpp-小写表示d-aa。这些序列中的一些序列中的符号“__”表示可以插入多种本领域公认的蛋白酶裂解位点中的任一种的位置:
eeeeeddddk_axrrrrrrrrrxc(seqidno:41)
eeeeeddddk_arrrrrrrrrxc(seqidno:42)
eddddk_axrrrrrrrrrxc(seqidno:43)
eeddddk_arxrrxrrxrrxrrxc(seqidno:44)
ddddddk_arrrrrrrrrxc(seqidno:45)
在另一个实施方案中,cpp是聚d(1-16)。
通常,适宜的是cpp的长度相当短,例如小于约30个氨基酸,以便一旦分子进入细胞就提高稳定性和药效学特性。
在一些实施方案中,cpp与本发明的肽直接连接(融合)。在其他实施方案中,期望用接头将高度带电的cpp与抑制剂肽分离,以使抑制剂保持其活性。可以使用各种接头中的任何一种。接头的尺寸范围可以是例如1-7个或甚至更多个氨基酸(例如1、2、3、4、5、6或7个氨基酸)。例如,接头可以是n-末端的qvtnvg和c-末端的qktveg或其截短形式,截短形式与抑制肽连接的n-末端具有1、2、3、4或5个连续氨基酸。
在本发明的实施方案中,抑制肽经可检测地标记。可以使用标记的肽,例如,以更好地了解抑制肽的作用机制和/或细胞定位。使得检测成为可能(例如,提供可检测的信号,或可以被检测到)的合适标记是常规的并且是本领域技术人员公知的。合适的可检测标记包括例如放射性活性剂、荧光标记等。将此类标记连接到蛋白质上的方法,或用于检测其存在和/或量的测定是常规的并且是公知的。
本发明的抑制肽可以通过多种本领域公认的方法中的任一种合成(例如,化学合成或通过在合适的宿主细胞中重组表达)。为了产生足够量的用于本发明方法的抑制肽,从业者可以,例如,使用常规技术,产生编码肽的核酸(例如,dna)并将其插入表达中载体,其中序列受表达控制序列诸如启动子或增强子的控制,然后表达控制序列可以指导肽的合成。例如,可以(a)从头合成dna,在末端具有合适的接头以将其克隆到载体中;(b)将整个dna序列克隆到载体中;或者(c)以重叠寡核苷酸开始,通过常规的基于pcr的基因合成方法将其连接,并将所得dna插入载体中。合适的表达载体(例如,质粒载体、病毒(包括噬菌体)、载体、人工载体、酵母载体、真核生物载体等)对于技术人员将是显而易见的,如用于制备载体,目标插入序列,表达由核酸编码的蛋白质,并分离或纯化表达的蛋白质的方法也是如此。
本发明的另一方面是药物组合物,其包含一种或多种抑制肽和药学上可接受的载体。药物组合物的组分可以被可检测地标记,例如具有放射性或荧光标记或具有标记,例如适于通过正电子发射光谱(pet)或磁共振成像(mri)检测的标记。例如,本发明的肽可以与选自放射性标记、不透射线的标记、荧光染料、荧光蛋白、比色标记等可检测标记偶联。在一些实施方案中,抑制肽以有效量存在以达到所需目的。
“药学上可接受的”意指可用于制备通常安全、无毒,在生物学上和其他方面都不是不合需要的药物组合物,并且包括兽医以及人药物用途可接受的药物组合物。例如,化合物的“药学上可接受的盐”意指如本文所定义的药学上可接受,并且具有母体化合物的所需药理学活性的盐。
本发明的另一方面是编码本发明抑制肽的多核苷酸。在本发明的实施方案中,多核苷酸与调节性控制序列(例如,启动子或增强子)可操作地连接,以利于在引入(例如,通过转染)到合适细胞中后产生编码的蛋白质。其他实施方案包括包含表达载体的细胞;及制备本发明抑制肽的方法,其包括培养细胞并收获由此产生的肽。
如本申请全篇所用,“约”意指加上或减去值的5%。
本发明的另一方面是用于实施本文所述任何方法的试剂盒。试剂盒可包含适量的本发明的抑制肽;用于产生所述肽的试剂;用于测定其功能或活性的测定的试剂;等等。本发明的试剂盒可包括用于执行方法的说明书。本发明试剂盒的其他任选要素包括合适的缓冲液、培养基组分等;提供本文所述晶体结构的结构示意图的计算机或计算机可读介质;容器;或包装材料。还可以包括用于执行合适对照的试剂。试剂盒的试剂可以处于容器中,其中试剂是稳定的,例如呈冻干形式或稳定的液体。试剂也可以呈一次性使用形式,例如,呈单次反应形式施用给受试者。
本发明的候选抑制肽的表征可以通过多种常规方法中的任何一种进行。例如,可以测定所述肽降低或抑制α-突触核蛋白聚集或细胞毒性或细胞间扩散的能力。测定可以在体外或体内进行。合适的测定对于技术人员将是显而易见的;本文描述了一些合适的测定。
本发明的一个方面是减少或抑制α-突触核蛋白聚集的方法,其包括使α-突触核蛋白原丝与有效量的一种或多种本发明的抑制肽接触。此类方法可以在溶液中或在细胞(例如培养的细胞或受试者体内的细胞)中进行。
本发明的另一方面是治疗患有因存在原纤化的α-突触核蛋白而介导的疾病或病状(有时在本文中称为α-突触核蛋白介导的疾病或病状)的受试者的方法,其包括向受试者施用有效量的本发明的抑制肽或药物组合物。在此类疾病或病状中有例如,帕金森病(pd)、路易体痴呆或多系统萎缩。本发明的另一方面是防止此类疾病或病状(例如,pd)发作,或治疗处于此类疾病或病状的早期,或正在发展此类疾病或病状的受试者,以便防止或抑制病状或疾病发展的方法。
本发明的抑制肽或药物组合物在本文中有时称为“抑制剂”。
本发明抑制剂的“有效量”是可以引起可测量量的所需结果,例如抑制α-突触核蛋白聚集或细胞毒性的量;对于诊断测定而言,是可以检测目标靶标,诸如α-突触核蛋白聚集体的量;或者在治疗方法中,是可以以可测量的量减轻或改善正在治疗的疾病或病状的症状的量。
“受试者”可以是具有与可以通过本发明的方法治疗的病状或疾病相关的聚集的(原纤化的)α-突触核蛋白分子的任何受试者(患者)。在本发明的一个实施方案中,受试者患有pd。典型的受试者包括脊椎动物,诸如哺乳动物,包括实验动物、狗、猫、非人灵长类动物和人。
本发明的抑制剂可以配制成适合于所选施用途径的各种形式的药物组合物,施用途径例如口服、经鼻、腹膜内或胃肠外,通过静脉内、肌肉内、局部或皮下途径,或通过注射进入组织。
用于施用抑制剂的合适口服形式包括锭剂、糖锭、片剂、胶囊、泡腾片剂、口服崩解片剂、经设计用于增加胃滞留时间的漂浮片剂、口腔贴剂和舌下片剂。
本发明的抑制剂可以全身施用,例如,与药学上可接受的媒介物诸如惰性稀释剂或可同化的食用载体组合口服,或通过吸入或吹入施用。它们可以封装在有包衣或无包衣的硬壳或软壳明胶胶囊中,可以压缩成片剂,或者可以直接掺入患者饮食的食物中。对于口服治疗施用,化合物可以与一种或多种赋形剂组合,并以可摄入片剂、口含片、糖锭、胶囊、酏剂、悬浮液、糖浆、糯米纸囊剂等形式使用。对于适合施用给人的组合物,术语“赋形剂”意在包括但不限于remington:thescienceandpracticeofpharmacy,lippincottwilliams&wilkins,第21版(2006)(下文remington's)中描述的那些成分。
抑制剂可以与惰性细粉状载体组合,并由受试者吸入或吹入。此类组合物和制剂应含有至少0.1%的化合物。当然,组合物和制剂的百分比可以变化,并且可以方便地介于给定单位剂型重量的约2%至约60%之间。
片剂、糖锭、丸剂、胶囊等也可含有下列物质:粘合剂诸如黄蓍胶、阿拉伯树胶、玉米淀粉或明胶;赋形剂诸如磷酸二钙;崩解剂诸如玉米淀粉、马铃薯淀粉、海藻酸等;润滑剂诸如硬脂酸镁;并且可以添加甜味剂诸如蔗糖、果糖、乳糖或阿斯巴甜(aspartame)或调味剂诸如薄荷、冬青油或樱桃调味剂。当单位剂型为胶囊时,除了以上类型的物质外,还可含有液体载体,诸如植物油或聚乙二醇。糖浆或酏剂可含活性化合物,作为甜味剂的蔗糖或果糖,作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯,染料和调味剂诸如樱桃或橙子调味剂。
各种其他物质可以作为包衣存在或以其他方式改变固体单位剂型的物理形式。例如,片剂、丸剂或胶囊可以包被有明胶、蜡、虫胶或糖等。当然,用于制备任何单位剂型的任何物质均应是药学上可接受的并且在所采用的量下基本上无毒。
另外,抑制剂可以掺入缓释制剂和装置中。例如,抑制剂可以掺入定时释放胶囊、定时释放片剂和定时释放丸剂中。在一些实施方案中,使用选自泡腾片剂、口腔崩解片剂、经设计用于增加胃滞留时间的漂浮片剂、口腔贴剂和舌下片剂的剂型施用所述组合物。
抑制剂也可以通过输注或注射经静脉内或腹膜内施用。抑制剂的溶液可以在水中制备,任选地与无毒表面活性剂混合。也可以在甘油、液体聚乙二醇、三醋精及其混合物和油中制备分散体。在普通储存和使用条件下,这些制剂可含有防腐剂以防止微生物生长。
适于注射或输注的药物剂型可包括无菌水溶液或分散体或无菌粉末,无菌粉末包含化合物,适于临时制备无菌注射液或输注溶液或分散体,任选地封装在脂质体中。在所有情况下,最终剂型应在生产和储存条件下是无菌、流动且稳定的。液体载体或媒介物可以是溶剂或液体分散介质,包括例如水、乙醇、多元醇(例如甘油、丙二醇、液体聚乙二醇等)、植物油、无毒甘油酯及其合适的混合物。例如,通过形成脂质体,通过在分散体的情况下保持所需粒度或通过使用表面活性剂,可以保持适当的流动性。
通过将所需量的化合物与上面列举的各种其他成分(根据需要)一起掺入适当的溶剂中,然后过滤除菌,来制备无菌注射液。在用于制备无菌注射液的无菌粉末的情况下,优选的制备方法是真空干燥和冷冻干燥技术,产生先前无菌过滤溶液中存在的活性成分加上任何其他所需成分的粉末。
对于局部施用,化合物可以呈纯净形式施用。然而,通常希望将它们作为组合物或制剂与皮肤病学可接受的载体一起施用至皮肤,所述载体可以是固体或液体。
有用的固体载体包括细碎固体,诸如滑石、粘土、微晶纤维素、二氧化硅、氧化铝等。其他固体载体包括常规的无毒聚合物纳米颗粒或微粒。有用的液体载体包括水、醇或二醇或水/醇/二醇共混物,其中化合物可以有效水平溶解或分散,任选地借助于无毒表面活性剂。可添加佐剂诸如香料和其他抗微生物剂以优化对于给定用途的性质。所得液体组合物可以从吸收垫施加,用于浸渍绷带和其他敷料,或使用泵型或气溶胶喷雾器喷洒到受影响的区域上。
可通过比较它们在动物模型中的体外活性和体内活性来测定本发明的肽或药物组合物的有用剂量。用于将小鼠和其他动物中的有效剂量外推至人的方法是本领域已知的。
例如,液体组合物诸如洗剂中化合物的浓度可以为约0.1-25重量%或约0.5-10重量%。半固体或固体组合物诸如凝胶或粉末中的浓度可为约0.1-5重量%或约0.5-2.5重量%。
本发明剂的有效剂量和施用途径是常规的。剂的确切量(有效剂量)将因受试者而异,取决于例如受试者的物种、年龄、体重和一般或临床状况,所治疗的任何病症的严重程度或机制,使用的特定剂或载体,施用的方法和时间安排等。治疗有效剂量可以通过本领域技术人员已知的常规方法凭经验测定。参见,例如thepharmacologicalbasisoftherapeutics,goodman和gilman编辑,macmillanpublishingco.,newyork。例如,最初可以在细胞培养测定中或在合适的动物模型中估计有效剂量。动物模型也可用于测定合适的浓度范围和施用途径。然后可将此类信息用于测定在人中施用的有用剂量和途径。治疗剂量也可以类似于同等治疗剂的剂量来选择。
主治临床医生将考虑病例的详细情况(例如,受试者、疾病、所涉及的疾病状态以及治疗是否是预防性的)来选择特定的施用模式和给药方案。治疗可包括在数天至数月或甚至数年的时间内日剂量或多个日剂量的化合物。
然而,一般而言,合适的剂量将在约0.001至约100mg/kg的范围内,例如,每天约0.01至约100mg/kg体重,诸如高于约0.1mg/kg,或在每天约1至约10mg/kg受者体重的范围内。例如,合适的剂量可以是每天约1mg/kg、5mg/kg、10mg/kg、20mg/kg或30mg/kg体重。
抑制剂可方便地呈单位剂型施用;例如,每单位剂型含有0.05至10000mg、0.5至10000mg、5至1000mg或约100mg活性成分。在一些实施方案中,剂量单位含有约0.1mg、约0.5mg、约1mg、约10mg、约25mg、约50mg、约75mg或约100mg的活性成分。
实施例
实施例i-帕金森病的α-突触核蛋白的毒性核:来自不可见晶体的结构
我们对nac结构域和相邻区域内的肽区段的晶体进行了筛选,寻求关于α-syn聚集和毒性的分子基础的结构信息。
对两个区段即nacore,残基68gavvtgvtava78(seqidno:46)和prenac,47gvvhgvttva56(seqidno:47)的广泛晶体筛选,表面上产生了非晶体、无定形聚集体。但是在通过电子显微镜术检查时,我们发现聚集体是细长纳米晶体簇,其横截面仅为50-300nm,因此通过传统光学显微镜不可见。我们在sacla和lcls自由电子激光器上确认了nacore的有序结晶度。我们还发现nacore中9个残基的片段,我们称之为subnacore,69avvtgvtav77(seqidno:48),产生体积是nacore纳米晶体1,000-10,000倍的晶体。因此,我们能够将同步加速器方法18,19应用于这些较大的晶体,以确定类淀粉样蛋白原纤维的结构。尽管与nacore相比,这9个残基的片段仅缺失两个残基,但它没有相同的毒性20,提供了下文所述的对α-syn毒性的了解。
为了确定不可见的nacore和prenac晶体的结构,我们转向microed21。在microed中,极低剂量电子束在低温条件下在透射电子显微镜内被引导到纳米晶体上,产生衍射图。在我们的实验中在200kev下使用的波长非常小
对于nacore和prenac,我们从纳米晶体收集电子衍射图,这些纳米晶体优先取向为平铺在多孔碳quantifoil格栅的表面上,呈冷冻水合状态。首先筛选格栅以获得适当尺寸的晶体,并筛选候选晶体进行衍射。我们使用显示强衍射的晶体通过围绕固定轴连续单向旋转来收集数据,以固定的时间间隔获取一系列衍射帧5。针状晶体通常超过衍射所需的长度;未弯曲且100至300nm宽的那些产生最佳衍射图。整合、缩放来自多个晶体的数据并合并在一起。
分别使用subnacore的原子模型和理想的β链模型作为探针通过分子置换为多晶nacore和prenac数据集进行定相。由subnacore探针结构和nacore结构因子计算的衍射相产生差分密度图,其清楚地揭示了在后续细化之后缺失的残基的位置、两个水分子和几个氢原子。针对microed数据对nacore和prenac的完整模型进行细化,产生了
nacore肽链的结构是几乎完全延伸的β-链。如通过定点自旋标记25,26预测的那样,这些nacore链互相对准堆积成β-折叠。如淀粉样蛋白棘中常见的那样,折叠是成对的,并且成对的折叠形成典型的立体拉链原丝,先前被视为由原纤维形成蛋白的短区段形成的许多类淀粉样蛋白原纤维中的棘。这种立体拉链不寻常的特征是拉链的11个残基宽度比我们先前观察到的更长,并且每对折叠在界面内含两个各自与苏氨酸侧链缔合的水分子,而不是完全干燥的。此外,在我们的nacore晶体中,每个折叠形成两个紧密界面:界面a,每个链具有
nacore的结构与全长α-syn原纤维的相关性通过其衍射图的相似性来确定。具体地,全长和n-端乙酰化的27α-syn蛋白对齐的原纤维的纤维衍射图显示出与对齐的nacore纳米晶的衍射相同的主峰。三种原纤维全部在其衍射图中显示出
nacore和prenac的组合结构允许我们构建a53t早发型突变体α-syn的大部分有序区段的推测模型。该模型的实验支持来自其模拟纤维衍射与测量的α-syn和n-乙酰基α-syn原纤维的衍射图以及对齐的nacore纳米晶体的一致性。上面我们提到nacore的较弱界面b模拟prenac与nacore的假设相互作用。实际上,较弱的nacore界面b中的相互作用侧链(g73、t75和v77)与prenac和nacore假设界面中相互作用的侧链(g51、t53、v55)相同。
由α-syn形成的细胞毒性淀粉样蛋白的身份和结构仍然是深入研究的主题17,28,29,30,31,32。在过去十年中,证据权重已经使来自作为毒性实体的路易体中发现的完全发育的淀粉样蛋白原纤维的科学观点倾向于较小的瞬时淀粉样蛋白寡聚体。然而,最近,提出了有利于原纤维的定量论证33。不希望受任何特定机理的束缚,据建议我们nacore对神经母细胞瘤细胞的细胞毒性的实验与原纤维有毒的观点一致:我们发现振荡和聚集72小时的nacore显示出丰富的原纤维,比新鲜溶解的nacore更具毒性,并且与类似聚集的完整α-syn具有相当的毒性。我们还发现nacore的细胞毒性比subnacore更强,后者缩短了两个残基。这与nacore比subnacore更快形成原纤维一致。这些观察结果并不排除在我们的nacore和α-syn聚集样品中存在但不可见的非纤维状寡聚体组装物的形成。当然,nacore仅仅是全长α-syn的片段,并且缺少与膜破坏相关的蛋白质n末端的大多数膜结合基序34,35。然而很明显,nacore是概括全长a-syn聚集和毒性的所有特征的最小实体。
nacore的极小尺寸是淀粉样蛋白晶体以及各种其他目标生物晶体的特征。对于淀粉样蛋白晶体,我们的推测是,微小尺寸是形成原纤维原丝的β-折叠自然扭曲的结果。晶格抑制扭曲,在这些晶体中产生随晶体生长而增加的应变。最终这种应变防止了β-链的进一步添加,限制了针状晶体的厚度。根据我们的经验,区段越长(例如,11个残基与9个残基相比)限制晶体生长甚至越多;在11-残基nacore和10-残基prenac的情况下,应变产生通过光学显微镜不可见的纳米晶体。这些晶体太小,无法进行固定和传统的同步加速器数据采集,但非常适合microed分析。我们的nacore和prenac结构证明microed能够以原子分辨率确定新的和准确的生物材料结构。这种发现为将microed应用于其中只能生长纳米晶体的其他重要的生物物质铺平了道路。在我们的特定应用中,我们已经能够了解关键nac结构域的核的原子排列。这为基于结构的α-syn的淀粉样蛋白形成抑制剂的设计提供了机会36。
方法
结晶
subnacore69avvtgvtav77(seqidno:48)的微晶由购自csbio的合成肽生长。通过悬滴蒸发使晶体在室温下生长。将冻干肽以于48mm氢氧化锂中2.9mg/ml的浓度溶于水中。将肽以2:1的比率与含有0.9m磷酸铵和0.1m乙酸钠ph4.6的储库混合。
nacore68gavvtgvtav78(seqidno:46)的纳米晶体由购自csbio的合成肽生长。将10批于无菌水中浓度为1mg/ml的合成肽(csbio)在37℃下在torreypines轨道混合板上以速度设定9振荡过夜。将不溶物质在30%(w/v)甘油中洗涤,然后在室温下储存于水中,之后进行衍射。样品含有原纤维和晶体的混合物。
prenac47gvvhgvttva56(seqidno:47)的纳米晶体由购自innopep的合成肽生长。分批准备合成肽的结晶试验。称取肽并按5mg/ml的浓度溶于含有0.1%dmso的经无菌过滤的50mm磷酸盐缓冲液ph7.0中。将该溶液在37℃下在torreypines轨道混合板上以速度设定9下振荡过夜。
数据收集和处理
使用先进光子源、东北协作访问团队(advancedphotonsource,northeastcollaborativeaccessteam)微聚焦光束线24-id-e的同步加速器辐射收集来自subnacore微晶的x射线衍射数据。光束线配备有adscquantum315ccd检测器。在
在线性相干光源(lcls)-slac的cxi仪器(相干x射线成像)上使用xfel辐射收集来自nacore纳米晶体的x射线衍射数据。x射线脉冲的光子能量为8.52kev
使用microed技术收集来自nacore和prenac纳米晶体的电子衍射数据5,6。这些纳米晶体通常凝块在一起。为了破碎凝块,将大约100μl体积的纳米晶体置于超声浴中30分钟。在适当稀释后,将纳米晶体以2-3μl的液滴沉积在quantifoil多孔碳em格栅上,这针对格栅上的晶体密度进行了优化。然后通过使用vitrobotmarkiv(fei)插入液体乙烷中使所有格栅沾染并玻璃化,然后转移至液氮中进行储存。使用gatan626低温保持器将冷冻的水合格栅转移至低温tem。使用在200kv下工作的配备feg的feitecnaif20tem收集衍射图和晶体图像,并使用底部安装的tvipsf416cmos相机记录,其传感器尺寸为4000平方像素,每平方尺寸大小各为15.6μm。通过使检测器在滚动快门模式下以2x2像素面元工作来记录衍射图,产生尺寸为2000平方像素的最终图像。使用照射光斑尺寸大约1微米的选定区域孔径,以每个图像3-4秒的曝光时间拍摄单独的图像帧。这种几何形状相当于每秒小于
使用多晶体黄金标准并通过参考电子衍射实验中的显著反射和xfel数据中的相应反射来完成样品到检测器距离的校准。通过cctbx.xfel通过针对嗜热菌蛋白数据集39细化光学测量的瓦片位置来执行64-瓦片cspad检测器的x/y位置校准。
为了获得与传统x射线数据处理程序的兼容性,衍射图像从tiff或tvips格式转换为smv晶体格式。我们使用xds为衍射图像编索引41,并使用xscale将来自不同晶体的数据集一起合并和缩放。对于nacore,合并来自四种晶体的数据,而对于prenac,合并来自三种晶体的数据以汇编最终数据集。
结构测定
使用程序phaser42获得subnacore的分子置换溶液。搜索模型由几何学上理想的由9个丙氨酸残基组成的β-链组成。用程序refmac43进行晶体学细化。使用程序phaser42获得nacore的分子置换溶液。搜索模型由先前确定的subnacore结构组成。用程序phenix44进行晶体学细化。使用程序phaser42获得prenac的分子置换溶液。搜索模型由几何学上理想的β-链组成,β-链由具有序列gvttva的六个残基组成。用程序phenix44进行晶体学细化。
使用coot45进行所有区段的建模。最终模型的坐标和结构因子已经保存在蛋白质数据库(proteindatabank)中,pdb代码4rik表示subnacore,4ril表示nacore,4znn表示prenac。使用pymol46说明结构。
α-突触核蛋白
先前表征了人野生型α-syn构建体47(prk172,氨苄青霉素,t7启动子),其序列为:
mdvfmkglskakegvvaaaektkqgvaeaagktkegvlyvgsktkegvvhg
vatvaektkeqvtnvggavvtgvtavaqktvegagsiaaatgfvkkdqlgkneegapqegiledmpvdpdneayempseegyqdyepea(seqidno:1)。
α-突触核蛋白纯化
根据公开的方案31纯化全长α-syn。将α-syn构建体转化到大肠杆菌表达细胞系bl21(de3)gold(agilenttechnologies,santaclara,ca)中,用于野生型α-syn蛋白表达。将单个菌落孵育到100ml补充有100μg/ml氨苄青霉素(fisherscientific,pittsburgh,pa)的lbmiller肉汤(fisherscientific,pittsburgh,pa)中,并在37℃下生长过夜。将1升补充有100μg/ml氨苄青霉素的lbmiller在2l摇瓶中与10ml过夜培养物一起孵育并在37℃下生长直至通过biophotometeruv/vis光度计(eppendorf,westbury,ny)所测量培养物达到od600~0.6-0.8。添加iptg(异丙基β-d-1-硫代半乳吡喃糖苷)至终浓度为0.5mm,并在30℃下生长4-6小时。通过在4℃下以5,500xg离心10分钟来收获细胞。将细胞团块冷冻并储存在-80℃下。
将细胞团块在冰上解冻并重新重悬于裂解缓冲液(100mmtris-hclph8.0、500mmnacl、1mmedtaph8.0)中并通过超声处理裂解。通过在4℃以15,000xg离心30分钟使粗制细胞裂解物澄清。将澄清的细胞裂解物煮沸并通过离心去除细胞碎片。边搅拌边通过滴定向冰上的蛋白质溶液添加hcl,使上清液中的蛋白质在ph3.5的酸中沉淀,然后在4℃下以15,000xg再离心30分钟。将上清液用缓冲液a(20mmtris-hcl,ph8.0)透析。透析后,将溶液通过0.45μm注射器(corning,ny14831)过滤,之后装载到20mlhiprepqhp16/10柱(gehealthcare,piscataway,nj)上。用5倍柱体积的缓冲液a洗涤q-hp柱,并在5倍柱体积的缓冲液b(20mmtris-hcl,1mnacl,ph8.0)中使用达到100%的线性梯度洗脱蛋白质。蛋白质在50-70%左右的缓冲液b中洗脱;汇集峰值级分。使用amiconultra-15离心过滤器将汇集的样品浓缩大约10倍。将大约5ml的浓缩样品装载到用过滤缓冲液(25mm磷酸钠、100mmnacl,ph7.5)平衡的hiprep26/60sephacryls-75hr柱上。汇集来自凝胶过滤柱的峰值级分,并用5mmtris-hcl(ph7.5)透析,浓缩至3mg/ml。将这些通过0.2μm孔径的过滤器(corning,ny14831)过滤并储存在4℃下。
基于der-sarkissian等人的方案25,以下列方式制备并纯化具有n-端乙酰化的重组表达的全长α-syn。
α-syn质粒与来自粟酒裂殖酵母(s.pombe)的异二聚体蛋白质乙酰化复合物共表达,以使n-端(pacyc-duet,氯霉素,t7启动子)乙酰化48。使用含有氨苄青霉素和氯霉素的培养基将两种载体共转化到大肠杆菌bl21(de3)中。使细胞培养物在含有氨苄青霉素和氯霉素的tb培养基中生长,并在25℃下用0.5mmiptg诱导以表达α-syn过夜。通过离心收获细胞,然后将细胞团块重新悬浮于裂解缓冲液(100mmtris-hclph8.0、500mmnacl、1mmedtaph8.0和1mm苯甲基磺酰氟)中,并使用emulsiflex匀浆器(avestin)裂解细胞。将裂解物煮沸并通过离心去除碎片。还通过在冰上在低ph下沉淀,然后离心去除蛋白质级分。通过滴定对剩余上清液进行ph调节,并用缓冲液a(20mmtris-hclph8.0、1mmdtt、1mmedtaph8.0)透析。将所得蛋白质溶液装载到用缓冲液a平衡的5mlq-sepharoseff柱(gehealthcare)上,并用线性梯度的缓冲液b(1mnacl、20mmtris-hclph8.0、1mmdtt、1mmedtaph8.0)洗脱。使用sds-page鉴定含有α-syn的级分,收集、浓缩并通过尺寸排阻法(sephacryls-10016/60,gehealthcare)在20mmtrisph8.0、100mmnacl、1mmdtt、1mmedta中进一步纯化。通过sds-page评估级分的纯度。
通过lc-ms2749表征乙酰化蛋白质。预期平均质量:对于α-突触核蛋白为14460.1da,对于乙酰化α-突触核蛋白为14502.1。观察到的平均质量:对于α-突触核蛋白为14464.0da,对于乙酰化α-突触核蛋白为14506.0。观察到的和预期的平均质量之间4da的偏移是由于仪器误差造成的。
抑制剂合成
抑制剂从genscriptinc.商购获得,纯度高于98%,并以10mm浓度溶解在100%dmso中。将溶解的抑制剂用0.1μm过滤器过滤,并在-20℃下分成20μl等分试样储存,直至进一步使用。
硫磺素t测定
在与用于聚集蛋白质进行接种测定的相同条件下(但添加了tht),以50μm蛋白质浓度进行原纤维形成测定。所有测定均在黑色nunc96孔光学底板(thermoscientific)中进行。将板在varioskan酶标仪(thermo)中在37℃下以3mm旋转直径以600rpm摇晃。使用λex=444nm,λem=482nm,每30分钟记录一次荧光测量,积分时间为200μs。
聚集测定
将纯化的α-突触核蛋白在0.1m硫酸钠、25mm磷酸钠中透析,并通过在37℃下在torreypine振荡器中在50μm下以速度9振荡使其聚集5-6天。
从pd人脑组织中提取肌氨酰不溶性蛋白丝
人冷冻脑组织获自ucla脑肿瘤转译资源中心(bttr)。使用先前描述的方案提取肌氨酰不溶性蛋白质。简言之,使用杜恩斯匀浆器以200mg/ml将冷冻组织在冰冷pbs中匀化。然后将匀浆在缓冲液a(10mmtris7.4、800mmnacl、1mmegta、10%蔗糖)中稀释至50mg/ml,总体积为1ml,并在4℃下以20,700g离心20分钟。将上清液收集在超速离心管中,将团块重新悬浮在0.5ml缓冲液a中,然后在4℃下以20,700g离心20分钟。将上清液汇集在一起。然后添加150μl于millipore水中的10%肌氨酰(w/v)并在室温下在平面旋转振荡器上以700rpm孵育1小时。然后使用sw55转子(beckmancoulter)将溶液在4℃下以100,000g离心1小时。弃去上清液并用5ml缓冲液a洗涤团块,并在4℃下以100,000g离心20分钟。将团块重新悬浮于100μl50mmtrisph7.4中。
hek293t细胞培养
稳定表达yfp标记的wtα-syn和a53tα-syn的hek293t细胞是来自marcdiamond博士的慷慨礼物。细胞在37℃、5%co2的潮湿培养箱中,在补充有10%胎牛血清(hyclone)、1%青霉素/链霉素(gibco)和1%glutamax(gibco)的杜尔贝科改良伊格尔培养基(gibco)中生长。
在hek293t细胞中接种
将90μl培养基中的10,000个细胞铺板在96孔黑壁板(cat#3660)中并使其粘附过夜。以125nm的最终单体浓度转染α-syn。将lipofectamine2000在optimem培养基(2.5+17.5μl)中稀释,并在室温下孵育5分钟。将蛋白质聚集体在optimem培养基(1:20)中稀释,并在水浴超声波仪中以低脉冲超声处理3分钟。然后将稀释的lipofectamine和蛋白质样品以1:1混合并在室温下孵育20分钟,之后向每个孔添加10μl。以一式三份添加所有样品,并重复实验最少两次。为了用抑制剂共转染α-syn原纤维,将原纤维和抑制剂在optimem中稀释并孵育3小时,然后超声处理。
细胞中细胞内斑点的测量
使用允许无偏测量的celigo成像细胞计数器测量斑点形成和细胞生长。使用荧光gfp通道对孔进行成像,并使用celigo分析软件测量融合度。拍摄整个孔的图像并用imagej通过粒子分析对颗粒计数。在所有天使用相同的设置分析一个板的孔。将每个孔中计数的总颗粒相对于融合度归一化并报告为每孔的颗粒数。
原纤维形成和检测
在37℃下在toreypine振荡器中振荡500μm浓度的于50mmtris、150mmkclph7.5中的纯化α-syn。为了形成subnacore和nacore的纤维状样品,将冻干肽溶于5mm氢氧化锂、20mm磷酸钠ph7.5和0.1mnacl中达到500μm的最终浓度。所有样品在37℃下在toreypine振荡器中振荡72小时。通过溶解冻干肽来制备新鲜溶解的样品,之后立即加入细胞中用于测定。
浊度测量用于比较nacore和subnacore聚集。将肽样品新鲜溶解在含有5mmlioh和1%dmso的样品缓冲液中至1.6mm,然后通过pvdf过滤器(millipore,0.1μm)过滤。使用具有200μl样品/孔的黑色nunc96孔板进行测量(每个样品3-4个重复)。将板在varioskan酶标仪(thermo)中在37℃下以3mm旋转直径以300rpm摇晃。在340nm下每3-15分钟记录一次吸光度读数。
负染色透射电子显微镜术
通过电子显微镜术评价细胞毒性样品中原纤维的存在。简言之,将5μl样品直接点在新辉光放电的碳涂覆的电子显微镜格栅(tedpella,redding,ca)上。孵育4分钟后,用5-μl蒸馏水冲洗格栅两次,并用2%乙酸双氧铀染色1分钟。在feit12电子显微镜上检查样本。
原纤维衍射
由纯化的α-syn形成的有和无n-端乙酰化的原纤维通过离心而浓缩,洗涤并在两根玻璃毛细管之间干燥的同时定向。同样,nacore纳米晶体也经浓缩、洗涤并使其在两根玻璃毛细管之间干燥的同时定向。将对齐的原纤维或纳米晶体固定在cryopin上,使用配备有htc成像板和内置x-stream低温冷却装置的rigaku旋转阳极发生器产生的
细胞毒性测定
粘附pc12细胞在atcc配制的补充有10%马血清和5%胎牛血清的rpmi1640培养基(atcc;目录号30-2001)中培养,并以每孔10,000铺板,最终体积为90μl。所有mtt测定均用celltiter96水性非放射性细胞增殖试剂盒(mtt,promega目录号4100)进行。将细胞在96孔板中在37℃、5%co2中培养20小时,然后添加样品(costar目录号3596)。向含有90μl培养基的每个孔中添加10μl样品,并在37℃、5%co2中孵育24小时。然后,然后向每孔添加15μl染料溶液(promega目录号4102),然后在37℃、5%co2中孵育4小时。在这之后向每孔添加100μl增溶溶液/终止混合物(promega目录号4101)。在室温下孵育12小时后,在570nm下测量吸光度。在700nm下记录本底吸光度。用1%(w/v)sds处理的细胞将数据归一化至0%降低,用样品缓冲液处理的细胞归一化至100%降低。
根据制造商的说明,使用cytotox-onetmhomogeneousmembraneintegrity(promega,目录号g7890)进行乳糖脱氢酶测定。简言之,将细胞以每板10,000个细胞铺板于96孔黑壁、透明底(fishercat#07-200-588)组织培养板中至90μl的最终体积。在添加样品之前,将细胞在37℃、5%co2中再孵育20小时。接下来,向每孔添加10μl样品,然后将细胞再孵育24小时。向每孔添加100μl试剂并在室温下孵育15分钟。添加50μl终止溶液终止反应。分别使用560nm和590nm的激发和发射波长测量荧光。使用经缓冲液处理的细胞作为0%释放和0.1%tritonx-100处理的细胞作为100%释放将数据归一化。
下面的nacore:gavvtgvtava(seqidno:46)的结构的原子坐标在表3中。下面的prenac的原子坐标在表4中。
nacore的原子结构揭示了由自我互补的成对β-折叠形成的淀粉样蛋白原纤维,β-折叠紧密配合形成立体拉链。通过基于rossetta的计算建模,将每种抑制剂设计成与立体拉链的尖端结合,从而‘封端’原纤维,防止α-突触核蛋白的其他分子添加到原纤维中。我们设计了3种候选抑制剂;有利地结合拉链一端或两端的s37、s61和s71。计算出的三种抑制剂中的每一种与原纤维的结合能和形状互补性也有利于结合。每种抑制剂均保留nacore天然序列的大多数残基,但是也含有一个或多个经修饰的残基。rodriquez等人表明nacore[69-77]中较小的9残基区段聚集比nacore慢,并且结构与nacore相似。为了防止我们设计的抑制剂自聚集,我们使用较短的区段连同一个或多个修饰。s37在thr72处具有w突变并且在c端具有另外的聚赖氨酸标签以诱导电荷-电荷排斥。预期它会结合立体拉链原纤维的两个尖端(即顶部和底部)。s61和s62保留与s37相同的抑制剂序列,但是代替聚赖氨酸标签,添加tat标签以帮助溶解并防止自聚集。s71在gly73处具有弱化沿β-折叠的氢键的甲基化甘氨酸和为了溶解和细胞穿透的附加tat标签。
实施例ii:计算设计
基于α-突触核蛋白的纤维形成区段的原子结构,我们设计了一系列抑制聚集的抑制剂。我们设计了肽抑制剂以特异性“封端”α-突触核蛋白的生长聚集体。使用zipperdb算法(goldschmidt等人55),鉴定了nac结构域中可结晶的淀粉样蛋白形成区段。据报道,该结构域对于α-突触核蛋白的聚集和毒性是必要且充分的(bodles等人20)。鉴定的区段(由残基69-77和残基68-78组成的α-突触核蛋白区段)经化学合成并结晶;其三维结构通过微观晶体学和微电子衍射(microed)确定。然后我们按其步骤应用基于rosetta的方法(sievers等人36)以使用α-突触核蛋白69-77结构或α-突触核蛋白68-78结构作为模板,设计破坏α-突触核蛋白聚集的抑制剂。鉴定并选择假定抑制剂用于实验表征。
实施例iii-第二轮设计-基于序列的合理设计
在另一轮设计中,我们从几个α-突触核蛋白淀粉样蛋白形成区段开始:47-56、68-78和84-94。在天然氨基酸序列中的两个独立残基处引入n-甲基化。在这些n-甲基氨基酸中,另外的甲基置换了胺氢。因此,阻断了氢键并防止在其生长末端添加肽单体。除n-甲基化之外,认为从天然α-突触核蛋白序列的额外极性/带电氨基酸延伸会改善肽抑制剂的总体溶解度。
表3
标题帕金森病毒性核的电子衍射结构
标题2α-突触核蛋白淀粉样蛋白的电子衍射结构,gavvtgvtava
化合物mol_id:1;
化合物2分子:α-突触核蛋白;
化合物3链:a;
化合物4异名:ad淀粉样蛋白的非aβ组分,
化合物5淀粉样蛋白前体的非a4组分nacp;
化合物6工程化:是
来源mol_id:1;
来源2合成的:是;
来源3生物_科学:智人;
来源4生物_常见:人;
来源5生物_taxid:9606;
来源6其他_详情:合成肽gavvtgvtava对应于
来源7人α-突触核蛋白的区段68-78
关键词淀粉样蛋白、α-突触核蛋白、帕金森病、毒性核、nacore,
关键词2脂质接合蛋白
expdta电子晶体学
作者j.a.rodriguez,m.ivanova,m.r.sawaya,d.cascio,f.reyes,d.shi,l.johnson,
作者2e.guenther,s.sangwan,j.hattne,b.nannenga,a.s.brewster,
作者3m.messerschmidt,s.boutet,n.k.sauter,t.gonen,d.s.eisenberg
期刊作者j.a.rodriguez,m.ivanova,m.r.sawaya,d.cascio,f.reyes,d.shi,
期刊作者2l.johnson,e.guenther,s.sangwan,j.hattne,b.nannenga,
期刊作者3a.s.brewster,m.messerschmidt,s.boutet,n.k.sauter,t.gonen,
期刊作者4d.s.eisenberg
期刊标题α-突触核蛋白毒性核的微观结构
期刊标题2淀粉样蛋白,
期刊标题3与帕金森病发展相关的蛋白质
待公开的期刊参考文献
期刊参考文献
备注2
备注2分辨率。1.43埃。
备注3
备注3细化。
备注3程序:buster
备注3作者:bricogne,blanc,brandl,flensburg,keller,
备注3:paciorek,roversi,smart,vonrhein,womack,
备注3:matthews,teneyck,tronrud
备注3
备注3用于细化的数据。
备注3高分辨率范围(埃):1.43
备注3低分辨率范围(埃):16.43
备注3数据截止值(∑(f)):0.000
备注3范围完整性(%):87.9
备注3反射次数:1073
备注3
备注3适合用于细化的数据。
备注3交叉验证法:从头到尾
备注3自由r值测试集选择:随机
备注3r值(工作+测试集):0.251
备注3r值(工作集):0.248
备注3自由r值:0.275
备注3自由r值测试集尺寸(%):11.840
备注3自由r值测试集计数:127
备注3自由r值估计误差:零
备注3
备注3适合最高分辨率库。
备注3所用面元的总数:5
备注3高分辨率库范围(埃):1.43
备注3低分辨率库范围(埃):1.60
备注3面元完整性(工作+测试)(%):87.88备注3面元反射(工作+测试集):286
备注3面元r值(工作+测试集):0.2642
1
备注3面元反射(工作集):245
备注3面元r值(工作集):0.2532
备注3面元自由r值:0.3310
备注3面元自由r值测试集尺寸(%):14.34
备注3面元自由r值测试集计数:41
备注3面元自由r值估计误差:零
备注3
备注3用于细化的非氢原子数
备注3蛋白质原子:66
备注3核酸原子:0
备注3异型杂种原子:0
备注3溶剂原子:2
备注3
备注3b值。
备注3来自威尔逊标绘法(a**2):10.33
备注3平均b值(总体,a**2):12.75
备注3总体各向异性b值。
备注3b11(a**2):-3.98760
备注3b22(a**2):3.49080
备注3b33(a**2):0.49680
备注3b12(a**2):0.00000
备注3b13(a**2):-1.55090
备注3b23(a**2):0.00000
备注3
备注3估计坐标误差。
备注3来自luzzati标绘法的估计值(a):0.305
备注3dpi(bloweq-10)基于r值(a):零
备注3dpi(bloweq-9)基于自由r值(a):零
备注3dpi(cruickshank)基于r值(a):0.108
备注3dpi(cruickshank)基于自由r值(a):零
备注3
备注3参考文献:blow,d.(2002)actacrystd58,792-797
备注3cruickshank,d.w.j.(1999)actacrystd55,583-601
备注3
备注3相关系数。
备注3相关系数fo-fc:0.913
备注3自由相关系数fo-fc:0.900
备注3
备注3定义的几何函数项数:15
备注3项计数加权函数。
备注3键长:65;2.000;谐波
备注3键角:90;2.000;谐波
备注3扭转角度:16;2.000;正弦
备注3三角型碳平面:1;2.000;谐波
备注3一般平面:10;5.000;谐波
备注3各向同性热力因子:65;20.000;谐波
备注3差的非键合接触:零;零;零
备注3不当扭转:零;零;零
备注3假回转角度:零;零;零
备注3手性不当扭转:11;5.000;半谐波
备注3占用总和:零;零;零
备注3效用距离:零;零;零
备注3效用角:零;零;零
备注3效用扭转:零;零;零
备注3理想距离接触项:76;4.000;半谐波
备注3
备注3与理想值的rms偏差。
备注3键长(a):0.010
备注3键角(度):1.65
备注3肽ω扭转角度(度):4.08
备注3其他扭转角度(度):6.49
备注3
备注3tls详情
备注3tls群的数量:零
2
备注3
备注3其他细化备注:无
备注4
备注44ril遵照格式v.3.30,2011年7月13日
备注100
备注100该条目已于2014年10月9日经rcsb处理。
备注100rcsbid代码为rcsb087390。
备注240
备注240实验细节
备注240重构方法:无
备注240样品类型:无
备注240样本类型:无
备注240数据获取
备注240数据收集日期:2014年8月28日
备注240温度(开尔文):100.0
备注240ph:零
备注240所用晶体数量:4
备注240显微镜型号:tecnaif20tem
备注240检测类型:tvipsf416cmos相机
备注240加速电压(kv):零
备注240唯一反射数:1073
备注240高分辨率范围(a):1.430
备注240低分辨率范围(a):16.430
备注240数据换算软件:xscale
备注240范围完整性(%):89.9
备注240数据冗余:4.400
备注240处于最高分辨率壳体内
备注240最高分辨率壳体,高范围(a):1.43
备注240最高分辨率壳体,低范围(a):1.60
备注240壳体完整性(%):82.5
备注240壳体数据冗余:4.40
备注240壳体r合并(i):0.56500
备注240用于测定结构的方法:分子置换
备注240所用软件:phaser
备注240启动模式:4rik
备注290
备注290晶体学对称性
备注290空间群的对称性算子:c121
备注290
备注290symop对称性
备注290nnnmmm算子
备注2901555x,y,z
备注2902555-x,y,-z
备注2903555x+1/2,y+1/2,z
备注2904555-x+1/2,y+1/2,-z
备注290
备注290nnn->算子数量时
备注290mmm->平移向量
备注290
备注290晶体学对称性转换
备注290以下转换作用于原子/hetatm
备注290该条目中的记录以结晶方式产生
备注290相关分子。
备注290smtry111.0000000.0000000.0000000.00000
备注290smtry210.0000001.0000000.0000000.00000
备注290smtry310.0000000.0000001.0000000.00000
备注290smtry12-1.0000000.0000000.0000000.00000
备注290smtry220.0000001.0000000.0000000.00000
备注290smtry320.0000000.000000-1.0000000.00000
备注290smtry131.0000000.0000000.00000035.40500
备注290smtry230.0000001.0000000.0000002.41000
备注290smtry330.0000000.0000001.0000000.00000
备注290smtry14-1.0000000.0000000.00000035.40500
备注290smtry240.0000001.0000000.0000002.41000
3
备注290smtry340.0000000.000000-1.0000000.00000
备注290
备注290备注:无
备注300
备注300生物分子:1、2
备注300参见所提供的作者和/或程序的备注350
备注300生成的该条目中
备注300结构的组件信息。备注也可提供有关
备注300隐藏表面区域的信息。
备注300备注:该生物单元是β-折叠的一部分。一个折叠是
备注300由链a和晶胞沿b平移组成
备注300尺寸。其他折叠由对称伴侣-x+1/2、
备注300y+1/2、-z和晶胞沿b平移组成。
备注350
备注350通过应用下面给出的biomt转换可以生成代表分子
备注350的已知生物学上重要的寡聚态的
备注350完整多聚体
备注350的坐标。同时给出了
备注350非晶体学和晶体学操作。
备注350
备注350生物分子:1
备注350作者确定的生物单元:四聚体
备注350对链应用以下:a
备注350biomt111.0000000.0000000.0000000.00000
备注350biomt210.0000001.0000000.0000000.00000
备注350biomt310.0000000.0000001.0000000.00000
备注350biomt121.0000000.0000000.0000000.00000
备注350biomt220.0000001.0000000.0000004.82000
备注350biomt320.0000000.0000001.0000000.00000
备注350biomt13-1.0000000.0000000.00000035.40500
备注350biomt230.0000001.0000000.0000002.41000
备注350biomt330.0000000.000000-1.0000000.00000
备注350biomt14-1.0000000.0000000.00000035.40500
备注350biomt240.0000001.0000000.0000007.23000
备注350biomt340.0000000.000000-1.0000000.00000
备注350
备注350生物分子:2
备注350作者确定的生物单元:单体
备注350对链应用以下:a
备注350biomt111.0000000.0000000.0000000.00000
备注350biomt210.0000001.0000000.0000000.00000
备注350biomt310.0000000.0000001.0000000.00000
dbref4rila111unpp37840syua_人6878
序列1a11glyalavalvalthrglyvalthralavalala
分子式2hoh*2(h2o)
晶体170.8104.82016.79090.00105.6890.00c1214
origx11.0000000.0000000.0000000.00000
origx20.0000001.0000000.0000000.00000
origx30.0000000.0000001.0000000.00000
比例10.0141220.0000000.0039640.00000
比例20.0000000.2074690.0000000.00000
比例30.0000000.0000000.0618610.00000
原子1nglya1-1.6642.3026.3491.0023.58n
原子2caglya1-1.1942.6055.0071.0023.02c
原子3cglya10.1051.9014.7021.0021.89c
原子4oglya10.1210.6764.5791.0020.14o
原子5nalaa21.1972.6664.5581.0016.28n
原子6caalaa22.5092.0944.2211.0015.41c
原子7calaa23.6642.7315.0051.0012.05c
原子8oalaa23.6683.9425.2271.0010.77o
原子9cbalaa22.7642.1862.7121.0015.67c
原子10nvala34.6401.9055.4171.005.98n
原子11cavala35.8522.3046.1291.003.93c
原子12cvala37.0231.7005.3441.005.93c
4
原子13ovala37.2170.4855.3881.006.66o
原子14cbvala35.8561.8917.6291.007.66c
原子15cg1vala37.1482.3378.3301.006.70c
原子16cg2vala34.6612.4948.3331.006.73c
原子17nvala47.7022.5224.5331.003.26n
原子18cavala48.7822.0873.6381.004.61c
原子19cvala410.0652.7434.0881.004.32c
原子20ovala410.1493.9734.0711.003.00o
原子21cbvala48.4582.4572.1711.008.53c
原子22cg1vala49.4861.8741.1881.009.58c
原子23cg2vala47.0412.0461.7901.009.20c
原子24nthra511.0661.9524.4381.003.32n
原子25cathra512.3602.4264.8961.003.88c
原子26cthra513.5041.7694.0921.005.43c
原子27othra513.5670.5394.0391.006.20o
原子28cbthra512.5402.0516.3511.006.84c
原子29og1thra511.4142.5037.1301.009.13o
原子30cg2thra513.8912.5246.9431.0010.98c
原子31nglya614.4172.5883.5461.006.04n
原子32caglya615.5712.1002.7931.006.46c
原子33cglya616.8742.6883.2921.007.38c
原子34oglya616.9593.8963.5011.006.32o
原子35nvala717.9131.8643.4361.004.11n
原子36cavala719.2232.3113.8841.005.04c
原子37cvala720.2431.7302.8701.004.68c
原子38ovala720.2440.5192.6781.005.69o
原子39cbvala719.5321.8025.2941.009.15c
原子40cg1vala721.0071.9915.6291.009.39c
原子41cg2vala718.6062.4316.3671.009.75c
原子42nthra821.1052.5702.2791.003.68n
原子43cathra822.2012.1691.3571.004.35c
原子44cthra823.4682.7831.9301.006.01c
原子45othra823.5304.0052.0311.006.48o
原子46cbthra821.8742.496-0.0831.0012.43c
原子47og1thra820.6011.921-0.3941.0014.76o
原子48cg2thra822.9291.954-1.0751.0013.32c
原子49nalaa924.4531.9772.3391.007.04n
原子50caalaa925.6282.5422.9941.009.57c
原子51calaa926.9671.8492.8491.0011.59c
原子52oalaa927.0520.6282.7561.008.18o
原子53cbalaa925.3282.7294.4781.0010.30c
原子54nvala1028.0172.6462.9561.0010.58n
原子55cavala1029.4052.2063.0431.0012.39c
原子56cvala1029.8962.8804.3141.0018.51c
原子57ovala1030.1714.0754.3011.0016.08o
原子58cbvala1030.2732.5611.8161.0017.83c
原子59cg1vala1031.7442.2432.0851.0018.89c
原子60cg2vala1029.7781.8310.5671.0017.39c
原子61nalaa1129.9162.1365.4271.0017.81n
原子62caalaa1130.3402.6476.7261.0023.47c
原子63calaa1131.6431.9887.0971.0051.68c
原子64oalaa1131.6220.9167.7341.0058.63o
原子65cbalaa1129.2892.3507.7781.0024.49c
原子66oxtalaa1132.6912.4856.6531.0074.50o
ter67alaa11
hetatm68ohoha1019.8304.9907.0851.0011.92o
hetatm69ohoha10218.5483.8730.2601.0020.36o
主要的203000000668101
结束
5
下面的prenac:gvvhgvttva(seqidno:47)的结构的原子坐标在表4中。
表4
表头----27-apr-15xxxx
化合物---
备注3
备注3细化。
备注3程序:refmac5.8.0073
备注3作者:murshudov,skubak,lebedev,pannu,
备注3steiner,nicholls,winn,long,vagin
备注3
备注3细化目标:最大似然率
备注3
备注3用于细化的数据。
备注3高分辨率范围(埃):1.41
备注3低分辨率范围(埃):32.94
备注3数据截止值(∑(f)):无
备注3范围完整性(%):86.73
备注3反射次数:1006
备注3
备注3适合用于细化的数据。
备注3交叉验证法:从头到尾
备注3自由r值测试集选择:随机
备注3r值(工作+测试集):0.26104
备注3r值(工作集):0.25243
备注3自由r值:0.33523
备注3自由r值测试集尺寸(%):10.0
备注3自由r值测试集计数:112
备注3
备注3适合最高分辨率库。
备注3所用面元的总数:20
备注3面元高分辨率范围:1.409
备注3低分辨率库范围:1.446
备注3库中的反射(工作集):49
备注3库完整性(工作+测试)(%):51.43
备注3面元r值(工作集):0.388
备注3面元自由r值集合计数:5
备注3面元自由r值:0.528
备注3
备注3用于细化的非氢原子数
备注3所有原子:68
备注3
备注3b值。
备注3来自威尔逊标绘法(a**2):无
备注3平均b值(总体,a**2):19.367
备注3总体各向异性b值。
备注3b11(a**2):-0.23
备注3b22(a**2):-0.23
备注3b33(a**2):0.49
备注3b12(a**2):0.00
备注3b13(a**2):-0.30
备注3b23(a**2):-0.00
备注3
备注3估计总体坐标误差。
备注3基于r值的esu(a):0.118
备注3基于自由r值的esu(a):0.133
备注3基于最大似然率的esu(a):0.155
备注3基于最大似然率的b值的esu(a**2):4.934
备注3
备注3相关系数。
备注3相关系数fo-fc:0.950
备注3自由相关系数fo-fc:0.895
备注3
备注3与理想值的rms偏差计算rms权重
备注3键长细化原子(a):66;0.017;0.019
备注3其他键长(a):69;0.010;0.020
备注3键角细化原子(度):91;1.876;1.905
1
备注3其他键角(度):155;0.648;3.000
备注3扭转角度,周期1(度):9;5.684;5.000
备注3扭转角度,周期2(度):1;75.981;20.000
备注3扭转角度,周期3(度):7;7.001;15.000
备注3手性中心限值(a**3):14;0.098;0.200
备注3一般平面细化原子(a):73;0.005;0.020
备注3其他一般平面(a):13;0.001;0.020
备注3
备注3各向同性热力因子限值。计算rms权重
备注3主链键细化原子(a**2):39;6.545;1.715
备注3主链键其他原子(a**2):38;3.948;1.607
备注3主链角细化原子(a**2):41;8.395;2.554
备注3主链角其他原子(a**2):42;8.555;2.605
备注3侧链键细化原子(a**2):27;5.989;2.165
备注3侧链键其他原子(a**2):27;5.243;2.077
备注3侧链角其他原子(a**2):44;8.091;3.056
备注3长范围b细化原子(a**2):54;9.624;14.765
备注3长范围b其他细化原子(a**2):55;11.399;15.221
备注3
备注3ncs限制统计数据
备注3ncs群的数量:零
备注3
备注3孪晶详情
备注3孪晶域数量:零
备注3
备注3
备注3tls详情
备注3tls群的数量:零
备注3
备注3
备注3散装溶剂建模。
备注3所用方法:掩蔽
备注3掩模计算参数
备注3vdw探针半径:1.20
备注3离子探针半径:0.80
备注3收缩半径:0.80
备注3
备注3其他细化备注:
备注3已将氢添加在安放位置
备注3u值:单独细化
备注3
晶体117.9304.71033.03090.0094.3390.00p1211
标度10.0557720.0000000.0042190.00000
标度2-0.0000000.2123140.0000000.00000
标度30.000000-0.0000000.0303620.00000
原子1nglya47-7.911-0.63918.2781.0020.19n
原子2caglya47-6.9010.23918.8391.0016.21c
原子3cglya47-5.632-0.52819.1111.0016.40c
原子4oglya47-5.600-1.72719.0111.0022.51o
原子5nvala48-4.5680.16919.4441.0017.59n
原子6cavala48-3.313-0.46019.7291.0015.77c
原子7cbvala48-2.269-0.08818.6371.0017.32c
原子8cg1vala48-0.855-0.46219.0561.0017.64c
原子9cg2vala48-2.659-0.73117.3261.0018.74c
原子10cvala48-2.9060.08021.0621.0013.82c
原子11ovala48-3.0401.26221.3271.0018.65o
原子12nvala49-2.365-0.78221.8951.0016.57n
原子13cavala49-1.779-0.34223.1361.0016.11c
原子14cbvala49-2.592-0.88724.3171.0016.48c
原子15cg1vala49-1.861-0.62225.6281.0020.39c
原子16cg2vala49-4.002-0.32424.3031.0020.73c
原子17cvala49-0.366-0.88223.2501.0013.58c
原子18ovala49-0.178-2.04323.1371.0020.25o
原子19nhisa500.614-0.06323.5571.0016.22n
2
原子20cahisa502.002-0.52423.5931.0015.91c
原子21cbhisa502.612-0.17622.2201.0018.19c
原子22cghisa504.036-0.54322.0241.0023.24c
原子23nd1hisa504.728-1.39022.8511.0032.41n
原子24ce1hisa505.964-1.53922.3991.0029.90c
原子25ne2hisa506.088-0.83921.2911.0034.03n
原子26cd2hisa504.892-0.21521.0281.0033.27c
原子27chisa502.6800.17124.7361.0014.51c
原子28ohisa502.6861.39624.7761.0021.97o
原子29nglya513.213-0.58025.6891.0015.69n
原子30caglya513.8770.02126.8291.0011.67c
原子31cglya512.9570.80027.7401.0012.93c
原子32oglya512.9672.02227.7861.0020.51o
原子33nvala522.1740.08128.4981.0015.10n
原子34cavala521.3150.69229.5141.0014.31c
原子35cbvala52-0.1620.39929.2781.0014.02c
原子36cg1vala52-1.0020.80330.4891.0016.11c
原子37cg2vala52-0.6261.14628.0771.0015.58c
原子38cvala521.7780.09430.8021.0013.92c
原子39ovala521.742-1.13330.9691.0020.14o
原子40nthra532.2530.95431.6981.0017.96n
原子41cathra532.8860.48132.9431.0018.30c
原子42cbthra534.3810.88533.0101.0020.92c
原子43og1thra535.0640.40931.8401.0029.94o
原子44cg2thra535.0630.25534.1751.0022.40c
原子45cthra532.1161.03734.1321.0014.85c
原子46othra531.7782.19334.1031.0021.57o
原子47nthra541.8400.20235.1511.0015.66n
原子48cathra541.2390.60736.4271.0014.08c
原子49cbthra54-0.1840.05936.6021.0018.69c
原子50og1thra54-1.0310.49135.5341.0035.75o
原子51cg2thra54-0.7680.52537.8721.0023.36c
原子52cthra542.061-0.04737.5001.0012.83c
原子53othra542.023-1.25837.6261.0022.40o
原子54nvala552.8210.72538.2621.0014.38n
原子55cavala553.6390.20339.3391.0013.58c
原子56cbvala555.0990.64139.1381.0015.65c
原子57cg1vala555.9150.34740.3581.0016.16c
原子58cg2vala555.696-0.07837.9391.0017.66c
原子59cvala553.1000.75640.6731.0014.57c
原子60ovala552.9161.96840.7801.0016.23o
原子61nalaa562.809-0.13041.6391.0017.51n
原子62caalaa562.4660.26843.0221.0026.29c
原子63cbalaa561.113-0.27943.4651.0021.51c
原子64calaa563.547-0.22243.9711.0038.92c
原子65oalaa563.757-1.42544.1111.0069.25o
原子66oxtalaa564.2490.54944.6341.0054.03o
hetatm67ohohb14.7152.48229.9911.0027.67o
hetatm68ohohb2-1.0343.06533.5001.0032.66o
3
参考文献
1.biere,a.l.etal.parkinson’sdisease-associatedalpha-synucleinismorefibrillogenicthanbeta-andgamma-synucleinandcannotcross-seeditshomologs.j.biol.chem.275,34574-34579(2000).
2.giasson,b.i.,murray,i.v.j.,trojanowski,j.q.&lee,v.m.-y.ahydrophobicstretchof12aminoacidresiduesinthemiddleofα-synucleinisessentialforfilamentassembly.j.biol.chem.276,2380-2386(2001).
3.du,h.-n.etal.apeptidemotifconsistingofglycine,alanine,andvalineisrequiredforthefibrillizationandcytotoxicityofhumanalpha-synuclein.biochemistry(mosc.)42,8870-8878(2003).
4.periquet,m.,fulga,t.,myllykangas,l.,schlossmacher,m.g.&feany,m.b.aggregatedα-synucleinmediatesdopaminergicneurotoxicityinvivo.j.neurosci.27,3338-3346(2007).
5.nannenga,b.l.,shi,d.,leslie,a.g.w.&gonen,t.high-resolutionstructuredeterminationbvcontinuous-rotationdatacollectioninmicroed.nat.methods11,927-930(2014).
6.shi,d.,nannenga,b.l.,iadanza,m.g.&gonen,t.thtee-dimensionalelectroncrystallographyofproteinmicrocrystals.elife2,e01345(2013).
7.spillantini,m.g.etal.alpha-synucleininlewybodies.nature388,839-840(1997).
8.goedert,m.,spillantini,m.g.,deltredici,k.&braak,h.100yearsoflewypathology.nat.rev.neurol.9,13-24(2013).
9.polymeropoulos,m.h.etal.mutationinthealpha-synucleingeneidentifiedinfamilieswithparkinson’sdisease.science276,2045-2047(1997).
10.krüger,r.etal.ala30promutationinthegeneencodingalpha-synucleininparkinson’sdisease.nat.genet.18,106-108(1998).
11.zarranz,j.j.etal.thenewmutation,e46k,ofalpha-synucleincausesparkinsonandlewybodydementia.ann.neurol.55,164-173(2004).
12.
13.singleton,a.b.etal.alpha-synucleinlocustriplicationcausesparkinson’sdisease.science302,841(2003).
14.uéda,k.etal.molecularcloningofcdnaencodinganunrecognizedcomponentofamyloidinalzheimerdisease.proc.natl.acad.sci.u.s.a.90,11282-11286(1993).
15.han,h.,weinreb,p.h.&lansbury,p.t.thecorealzheimer’speptidenacformsamyloidfibrilswhichseedandareseededbybeta-amyloid:isnacacommontriggerortargetinneurodegenerativedisease?chem.biol.2,163-169(1995).
16.el-agnaf,o.m.etal.aggregatesfrommutantandwild-typealpha-synucleinproteinsandnacpeptideinduceapoptoticcelldeathinhumanneuroblastomacellsbyformationofbeta-sheetandamyloid-likefilaments.febslett.440,71-75(1998).
17.conway,k.a.,harper,j.d.&lansbury,p.t.aceeleratedinvitrofibrilformationbyamutantalpha-synucleinlinkedtoearly-onsetparkinsondisease.nat.med.4,1318-1320(1998).
18.nelson,r.etal.structureofthecross-betaspineofamyloid-likefibrils.nature435,773-778(2005).
19.sawaya,m.r.etal.atomicstructuresofamyloidcross-betaspinesrevealvariedstericzippers.nature447,453-457(2007).
20.bodles,a.m.,guthrie,d.j.,greer,b.&irvine,g.b.identificationoftheregionofnon-abetacomponent(nac)ofalzheimer’sdiseaseamyloidresponsibleforitsaggregationandtoxicity.j.neurochem.78,384-395(2001).
21.nannenga,b.l.&gonen,t.proteinstructuredeterminationbymicroed.curr.opin.struct.biol.27c,24-31(2014).
22.nannenga,b.l.elife(2014).structureofcatalasedeterminedbymicroed.2014;3:e03600
23.yonekura,k.,kato,k.,ogasawara,m.,tomita,m.&toyoshima,c.electroncrystallographyofultrathin3dproteincrystals:atomicmodelwithcharges.proc.natl.acad.sci.u.s.a.112,3368-3373(2015).
24.doyle,p.a.&turner,p.s.relativistichartree-fockx-rayandelecttonscatteringfactors.actacrystallogr.sect.a24,390-397(1968).
25.der-sarkissian,a.,jao,c.c.,chen,j.&langen,r.structuralorganizationofalpha-synucleinfibrilsstudiedbysite-directedspinlabeling.j.biol.chem.278,37530-37535(2003).
26.chen,m.,margittai,m.,chen,j.&langen,r.investigationofalpha-synucleinfibrilstructurebysite-directedspinlabeling.j.biol.chem.282,24970-24979(2007).
27.sarafian,t.a.etal.impairrnentofmitochondriainadultmousebrainoverexpressingpredominantlyfull-length,n-terminallyacetylatedhumanα-synuclein.plosone8,e63557(2013).
28.caughey,b.&lansbury,p.t.protofibrils,pores,fibrils,andneurodegeneration:separatingtheresponsibleproteinaggregatesfromtheinnocentbystanders.annu.rev.neurosci.26,267-298(2003).
29.danzer,k.m.,schnack,c.,sutcliffe,a.,hengerer,b.&gillardon,f.functionalproteinkinasearraysrevealinhibitionofp-21-activatedkinase4byalpha-synucleinoligomers.j.neurochem.103,2401-2407(2007).
30.karpinar,d.p.etal.pre-fibrillaralpha-synucleinvariantswithimpairedbeta-structureincreaseneurotoxicityinparkinson’sdiseasemodels.emboj.28,3256-3268(2009).
31.winner,b.etal.invivodemonstrationthatalpha-synucleinoligomersaretoxic.proc.natl.acad.sci.u.s.a.108,4194-4199(2011).
32.chen,s.w.etal.structuralcharacterizationoftoxicoligomersthatarekineticallytrappedduringα-synucleinfibrilformation.proc.natl.acad.sci.u.s.a.112,e1994-2003(2015).
33.bousset,l.etal.structuralandfunctionalcharacterizationoftwoalpha-synucleinstrains.nat.commun.4,2575(2013).
34.auluck,p.k.,caraveo,g.&lindquist,s.d-synuclein:membraneinteractionsandtoxicityinparkinson’sdisease.annu.rev.celldev.biol.26,211-233(2010).
35.lee,j.c.,langen,r.,hummel,p.a.,gray,h.b.&winkler,j.r.alpha-synucleinstructuresfromfluorescenceenergy-transferkinetics:implicationsfortheroleoftheproteininparkinson’sdisease.proc.natl.acad.sci.u.s.a.101,16466-16471(2004).
36.sievers,s.a.etal.structure-baseddesignofnon-naturalamino-acidinhibitorsofamyloidfibrilformation.nature475,96-100(2011).
37.otwinowski,z.&minor,w.inmethodsinenzymology(ed.charlesw.carter,j.)volume276,307-326(academicpress,1997).
38.weierstall,u.,spence,j.c.h.&doak,r.b.injectorforscatteringmeasurementsonfullysolvatedbiospecies.rev.sci.instrum.83,035108(2012).
39.hattne,j.etal.accuratemacromolecularstructuresusingminimalmeasurementsfromx-rayfree-electronlasers.nat.methods11,545-548(2014).
40.sauter,n.k.,hattne,j.,grosse-kunstleve,r.w.&echols,n.newpython-basedmethodsfordataprocessing.actacrystallogr.dbiol.crystallogr.69,1274-1282(2013).
41.kabsch,w.xds.actacrystallogr.dbiol.crystallogr.66,125-132(2010).
42.mccoy,a.j.etal.phasercrystallographicsoftware.j.appl.crystallogr.40,658-674(2007).
43.murshudov,g.n.,vagin,a.a.&dodson,e.j.refinementofmacromolecularstructuresbythemaximum-likelihoodmethod.actacrystallogr.dbiol.crystallogr.53,240-255(1997).
44.afonine,p.v.etal.towardsautomatedcrystallographicstructurerefinementwithphenix.refine.actacrystallogr.dbiol.crystallogr.68,352-367(2012).
45.emsley,p.,lohkamp,b.,scott,w.g.&cowtan,k.featuresanddevelopmentofcoot.actacrystallogr.dbiol.crystallogr.66,486-501(2010).
46.delano,w.thepymolmoleculargraphicssystem.(
47.jakes,r.,spillantini,m.g.&goedert,m.identificationoftwodistinctsynucleinsfromhumanbrain.febslett.345,27-32(1994).
48.johnson,m.,coulton,a.t.,geeves,m.a.&mulvihill,d.p.targetedamino-terminalacetylationofrecombinantproteinsine.coli.plosone5,e15801(2010).
49.whitelegge,j.p.,zhang,h.,aguilera,r.,taylor,r.m.&cramer,w.a.fullsubunitcoverageliquidchromatographyelectrosprayionizzationmassspectrometry(lcms+)ofanoligomericmembraneprotein:cytochromeb(6)fcomplexfromspinachandthecyanobacteriummastigocladuslaminosus.mol.cell.proteomicsmcp1,816-827(2002).
50.rao,j.n.,jao,c.c.,hegde,b.g.,langen,r.&ulmer,t.s.acombinatorialnmrandeprapproachforevaluatingthestructuralensembleofpartiallyfoldedproteins.j.am.chem.soc.132,8657-8668(2010).
51.brunger,a.t.version1.2ofthecrystallographyandnmrsystem.nat.protoc.2,2728-2733(2007).
52.fabiola,f.,bertram,r.,korostelev,a.&chapman,m.s.animprovedhydrogenbondpotential:impactonmediumresolutionproteinstructures.proteinsci.publ.proteinsoc.11,1415-1423(2002).
53.comellas,g.etal.structuredregionsofα-synucleinfibrilsincludetheearly-onsetparkinson’sdiseasemutationsites.j.mol.biol.411,881-895(2011).
54.vilar,m.etal.thefoldofalpha-synucleinfibrils.proc.natl.acad.sci.u.s.a.105,8637-8642(2008).
55.goldschmidt,l.,teng,p.k.,riek,r.&eisenberg,d.identifyingtheamylome,proteinscapableofformingamyloid-likefibrils.proc.natl.acad.sci.107,3487-3492(2010).
56.read,r.j.improvedfouriercoefficientsformapsusingphasesfrompartialstructureswitherrors.actacrystallogr.a42,140-149(1986).
57.eisenberg,d.s.etal.structure-baseddesignofpeptideinhibitorsofamyloidfibrillation,uspatentno.8,754,034
58.eisenberg,d.s.etal.structure-basedpeptideinhibitorsofp53aggregationasanewapproachtocancertherapeutics;wo2014/182961(2014);us2014/037387.
59.theccp4suite:programsforproteincrystallography.actacrystallogrdbiolcrystallogr50,760-3(1994).
60.lawrence,m.c.&colman,p.m.shapecomplementarityatprotein/proteininterfaces.jmolbiol234,946-50(1993).
61.kytej,doolittlerf(may1983).″asimplemethodfordisplayingthehydropathiccharacterofaprotein″.j.mol.biol.157(1):105-32.
62.warrenl.delano″thepymolmoleculargraphicssystem.″delanoscientificllc,sanclos,ca,usa.http://www.pvmol.org
63.jiang,l.etal.(2013).structure-baseddiscoveryoffiber-bindingcompoundsthatreducethecytotoxicityofamyloidbeta.elife2013;2e00857
从前面的描述中,本领域技术人员可以容易地确定本发明的基本特征,并且在不脱离其精神和范围的情况下,可以对本发明进行改变和修改以使其适应各种使用和条件并最大程度地利用本发明。前述实施方案应解释为仅仅是说明性的,并且不以任何方式限制本发明的范围。本文和附图中引用的所有申请、专利和出版物的全部公开内容据此通过引用整体并入本文,特别是关于引用它们的信息。
- 还没有人留言评论。精彩留言会获得点赞!