一种探测基因组小片段插入缺失的方法及装置与流程
本发明涉及核酸序列分析
技术领域:
,具体涉及一种探测基因组小片段插入缺失的方法及装置。
背景技术:
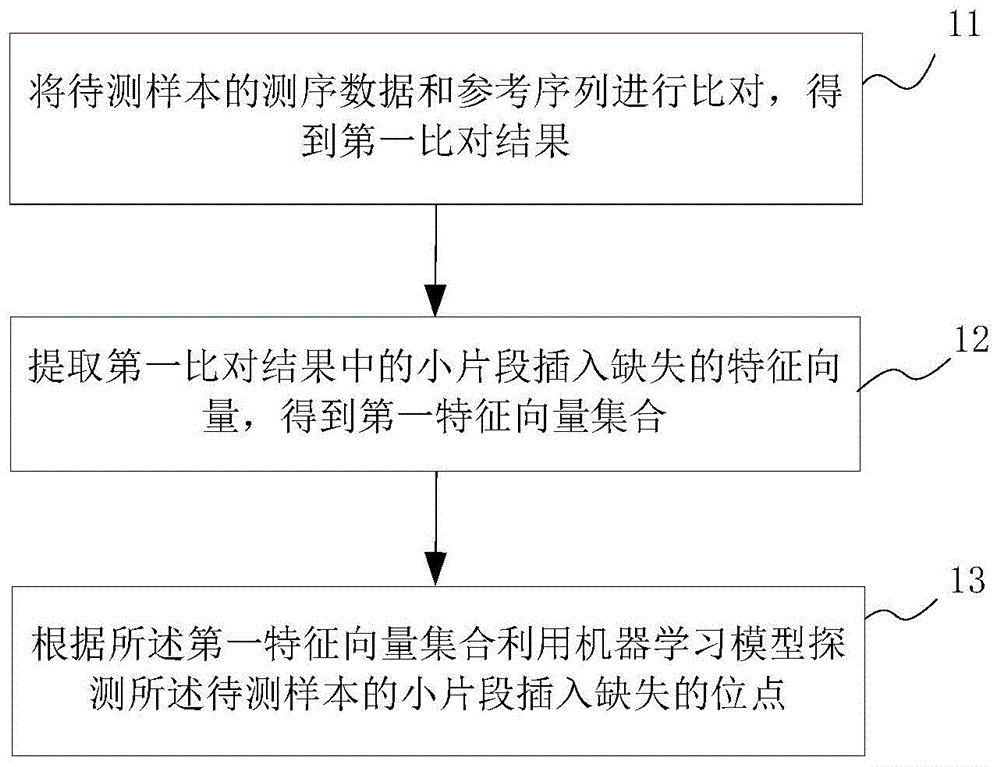
:随着二代测序技术的发展,随着二代测序技术的发展,在高通量全基因组或全外显子组测序数据分析中,小片段插入缺失(insertion-deletion,indel)的探测是一个关键且要求高精准度的问题。遗传性indel的探测可以用来发现物种基因型和表型的遗传规律;在肿瘤或病变组织数据中,体细胞indel检测在此基础上又给小片段插入缺失indel突变探测问题提出了更高的要求。现有技术步骤复杂,预处理较多。会对包含indel的位置进行重新比对,此外还有碱基质量校准等步骤,步骤多,耗时长。现有技术没采用机器学习的方法,性能提升有限。只通过统计信息,和根据统计信息计算的基因型质量值,这些信息对精度提升有限。总之,目前主流的技术探测indel的速度慢,同一套数据各个算法得到的结果一致性低。技术实现要素:本发明实施例的目的在于提供一种探测基因组小片段插入缺失的方法及装置,用以解决现有技术中探测indel的速度慢,同一套数据各个算法得到的结果一致性低的问题。为实现上述目的,本发明实施例第一方面提供了一种探测基因组小片段插入缺失的方法,包括以下步骤:步骤(11)将待测样本的测序数据和参考序列进行比对,得到第一比对结果。步骤(12)提取第一比对结果中的小片段插入缺失的特征向量,得到第一特征向量集合;其中,所述第一特征向量集合中含有待测样本的测序数据相对参考序列的插入缺失碱基长度、待测样本的测序数据相对参考序列的突变位点库长度均值、待测样本的测序数据相对参考序列的突变位点库长度方差、待测样本的测序数据相对参考序列的突变位点覆盖splitreads个数、待测样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;步骤(13)根据所述第一特征向量集合利用机器学习模型探测所述待测样本的小片段插入缺失的位点;其中,所述机器学习模型通过以下步骤构建:步骤(21)将标准样本的测序数据和所述参考序列进行比对,得到第二比对结果;步骤(22)提取第二比对结果中的小片段插入缺失的特征向量,得到第二特征向量集合;其中,所述第二特征向量集合中含有标准样本的测序数据相对参考序列的插入缺失碱基长度、标准样本的测序数据相对参考序列的突变位点库长度均值、标准样本的测序数据相对参考序列的突变位点库长度方差、标准样本的测序数据相对参考序列的突变位点覆盖splitreads个数、标准样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;步骤(23)根据所述第二特征向量集合在标准样本的可靠值序列中标注小片段插入缺失位点,得到标准样本的小片段插入缺失位点集合;步骤(24)利用机器学习方法对所述标准样品的小片段插入缺失位点集合进行模型训练,得到所述机器学习模型。在一种可能的实现方式中,所述步骤(11)还包括:将待测样本的对照样本的测序数据和所述参考序列进行比对,得到第三比对结果;在所述步骤(12)中,根据第三比对结果提取第一比对结果中的小片段插入缺失的特征向量,得到所述第一特征向量集合;所述步骤(21)还包括:将标准样本的对照样本的测序数据和所述参考序列进行比对,得到第四比对结果;在所述步骤(22)中,根据所述第四比对结果提取第二比对结果中的小片段插入缺失的特征向量,得到所述第二特征向量集合。在一种可能的实现方式中,小片段插入缺失的特征向量包括以下任一种或多种:总覆盖度、支持参考序列覆盖度、支持插入缺失的覆盖度、突变频率、小片段插入缺失人群数据库、gc含量、噪音的个数统计、噪音的频率统计、突变左右参考序列上数量最多的碱基比例、正链支持参考序列的数目、正链支持小片段插入缺失突变的数目、负链支持参考序列的数目、负链支持小片段插入缺失变异的数目、正链支持小片段插入缺失突变数目和负链支持小片段插入缺失突变数目的比值、小片段插入缺失位点在参考序列上距头尾最短距离的均值、小片段插入缺失位点在参考序列上距头尾最短距离的方差、小片段插入缺失位点在距头尾最短距离的均值,小片段插入缺失位点在碱基距头尾最短距离的方差、建库长度均值、建库长度方差、支持参考序列碱基基因序列比对质量平均值、支持参考序列碱基基因序列比对质量方差、支持参考序列碱基质量平均值、支持参考序列碱基质量方差、支持变异碱基基因序列比对质量平均值、支持变异碱基基因序列比对质量方差、支持变异碱基质量平均值、支持变异碱基质量方差、一致性质量、单样本rms质量、单样本校正的p值、有对照样本两两费歇尔检验的p值、有对照样本两两体细胞变异探测分数、信噪比;所述机器学习方法包括以下任一种:朴素贝叶斯法、逻辑回归法、线性回归法、最近邻近法、决策树法、boosting方法及其变种、svm支持向量机法、人工神经网络算法;其中,boosting方法及其变种包含adaptiveboosting、gradientboosting等;所述步骤(24)还包括:采用十折交叉验证法测试所述机器学习模型。在一种可能的实现方式中,第一比对结果存放在bam文件中;第二比对结果存放在bam文件中;待测样本的测序数据的测序平台和标准样本的测序数据的测序平台一致,且待测样本的测序数据的测序方法和标准样本的测序数据的测序方法一致。在一种可能的实现方式中,在所述步骤(12)中,根据特征设定条件提取第一比对结果中的小片段插入缺失的特征向量;在所述步骤(22)中,根据特征设定条件提取第二比对结果中的小片段插入缺失的特征向量;特征设定条件包括以下任一种或多种:测序质量、测序深度、对照样本中小片段插入缺失出现的频率。本发明实施例第二方面提供了一种探测基因组小片段插入缺失的装置,其特征在于,包括:比对单元、提取单元、探测单元、标注单元、训练单元;所述比对单元用于将待测样本的测序数据和参考序列进行比对,得到第一比对结果;所述提取单元用于提取第一比对结果中的小片段插入缺失的特征向量,得到第一特征向量集合;其中,所述第一特征向量集合中含有待测样本的测序数据相对参考序列的插入缺失碱基长度、待测样本的测序数据相对参考序列的突变位点库长度均值、待测样本的测序数据相对参考序列的突变位点库长度方差、待测样本的测序数据相对参考序列的突变位点覆盖splitreads个数、待测样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;所述探测单元用于根据所述第一特征向量集合利用机器学习模型探测所述待测样本的小片段插入缺失的位点;所述比对单元还用于将标准样本的测序数据和所述参考序列进行比对,得到第二比对结果;所述提取单元还用于提取第二比对结果中的小片段插入缺失的特征向量,得到第二特征向量集合;其中,所述第二特征向量集合中含有标准样本的测序数据相对参考序列的插入缺失碱基长度、标准样本的测序数据相对参考序列的突变位点库长度均值、标准样本的测序数据相对参考序列的突变位点库长度方差、标准样本的测序数据相对参考序列的突变位点覆盖splitreads个数、标准样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;所述标注单元用于根据所述第二特征向量集合在标准样本的可靠值序列中标注小片段插入缺失位点,得到标准样本的小片段插入缺失位点集合;所述训练单元用于利用机器学习方法对所述标准样品的小片段插入缺失位点集合进行模型训练,得到所述机器学习模型。在一种可能的实现方式中,所述比对单元还用于将待测样本的对照样本的测序数据和所述参考序列进行比对,得到第三比对结果;所述提取单元还用于根据第三比对结果提取第一比对结果中的小片段插入缺失的特征向量,得到所述第一特征向量集合;所述比对单元还用于将标准样本的对照样本的测序数据和所述参考序列进行比对,得到第四比对结果;所述提取单元还用于根据所述第四比对结果提取第二比对结果中的小片段插入缺失的特征向量,得到所述第二特征向量集合。在一种可能的实现方式中,小片段插入缺失的特征向量包括以下任一种或多种:总覆盖度、支持参考序列覆盖度、支持插入缺失的覆盖度、突变频率、小片段插入缺失人群数据库、gc含量、噪音的个数统计、噪音的频率统计、突变左右参考序列上数量最多的碱基比例、正链支持参考序列的数目、正链支持小片段插入缺失突变的数目、负链支持参考序列的数目、负链支持小片段插入缺失变异的数目、正链支持小片段插入缺失突变数目和负链支持小片段插入缺失突变数目的比值、小片段插入缺失位点在参考序列上距头尾最短距离的均值、小片段插入缺失位点在参考序列上距头尾最短距离的方差、小片段插入缺失位点在距头尾最短距离的均值,小片段插入缺失位点在碱基距头尾最短距离的方差、建库长度均值、建库长度方差、支持参考序列碱基基因序列比对质量平均值、支持参考序列碱基基因序列比对质量方差、支持参考序列碱基质量平均值、支持参考序列碱基质量方差、支持变异碱基基因序列比对质量平均值、支持变异碱基基因序列比对质量方差、支持变异碱基质量平均值、支持变异碱基质量方差、一致性质量、单样本rms质量、单样本校正的p值、有对照样本两两费歇尔检验的p值、有对照样本两两体细胞变异探测分数、信噪比;所述机器学习方法包括以下任一种:朴素贝叶斯法、逻辑回归法、线性回归法、最近邻近法、决策树法、boosting方法及其变种、svm支持向量机法、人工神经网络算法;其中,boosting方法及其变种包含adaptiveboosting、gradientboosting等;所述训练单元还用于采用十折交叉验证法测试所述机器学习模型。在一种可能的实现方式中,第一比对结果存放在bam文件中;第二比对结果存放在bam文件中;待测样本的测序数据的测序平台和标准样本的测序数据的测序平台一致,且待测样本的测序数据的测序方法和标准样本的测序数据的测序方法一致。在一种可能的实现方式中,所述提取单元还用于根据特征设定条件提取第一比对结果中的小片段插入缺失的特征向量;所述提取单元还用于根据特征设定条件提取第二比对结果中的小片段插入缺失的特征向量;特征设定条件包括以下任一种或多种:测序质量、测序深度、对照样本中小片段插入缺失出现的频率。本发明实施例具有如下优点:基于特征提取、模型训练的机器学习方法来探测高置信度的indel,并且可以根据不同测序平台和测序方法定制不同的模型,以提高计算的速度和结果的精度;本发明实施例提供的探测基因组小片段插入缺失的方法及装置运行效率高;其他基因变异探测方法或装置需要数以天计的计算任务,本发明实施例提供的探测基因组小片段插入缺失的方法及装置可以在短时间内给出探测结果,可以最大限度利用计算资源,降低时间成本;并且通过使用机器学习的方法提高了探测单核苷酸突变的精度。附图说明图1为本发明实施例1提供的探测基因组小片段插入缺失的方法流程图。图2为本发明实施例1提供的机器学习模型的构建方法流程图。图3为本发明本实施3提供的探测基因组小片段插入缺失位点的装置的结果示意图。图中:11-13.步骤,21-24.步骤,31.比对单元,32.提取单元,33.探测单元,34.标注单元,35.训练单元。具体实施方式以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。须知,本说明书所附图式所绘示的结构、比例、大小等,均仅用以配合说明书所揭示的内容,以供熟悉此技术的人士了解与阅读,并非用以限定本发明可实施的限定条件,故不具技术上的实质意义,任何结构的修饰、比例关系的改变或大小的调整,在不影响本发明所能产生的功效及所能达成的目的下,均应仍落在本发明所揭示的技术内容得能涵盖的范围内。同时,本说明书中所引用的如“上”、“下”、“左”、右”、“中间”等的用语,亦仅为便于叙述的明了,而非用以限定本发明可实施的范围,其相对关系的改变或调整,在无实质变更技术内容下,当亦视为本发明可实施的范畴。实施例1本实施例提供了一种探测基因组小片段插入缺失的方法,如图1所示,该方法包括如下步骤。步骤11将待测样本的测序数据和参考序列进行比对,得到第一比对结果。在步骤11之前,选取待测样本进行测序文库建立。利用测序平台(例如illumina、bgiseq、iontorrent等)进行测序。具体可以为全基因组测序,也可以为全外显子测序,也可以为靶向测序。测序结果生成原始fastq文件,从而得到待测样本的测序数据。利用基因组比对软件(如bwa、bowtie、stampy等)对fastq文件进行参考基因组(如人类基因组grch37等)比对,得到比对结果,比对结果可以以二进制序列比对格式(binarysequencealignmentmapformat,bam)文件形式存在。在一个示例中,待测样本可以为健康人的组织细胞、生殖细胞等。在一个示例中,待测样本可以为病人的病变组织细胞,例如肿瘤患者的肿瘤细胞。当待测样本为病人的病变组织细胞时,在本实施例中,进行待测样本的对照样本的测序数据和参考序列进行比对,得到第三比对结果。待测样本的对照样本可以为该病人的非病变组织。例如,待测样本为病人的肿瘤细胞,该待测样本的对照样本可以为癌旁组织细胞或者为血液中的细胞。对照样本的测序、比对,可参照待测样本进行,此处不再赘述。步骤12提取第一比对结果中的小片段插入缺失的特征向量,得到第一特征向量集合;其中,所述第一特征向量集合中含有待测样本的测序数据相对参考序列的插入缺失碱基长度、待测样本的测序数据相对参考序列的突变位点库长度均值、待测样本的测序数据相对参考序列的突变位点库长度方差、待测样本的测序数据相对参考序列的突变位点覆盖splitreads个数、待测样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;测序序列分为两个或多个部分,每个部分分别比对到参考基因组上多个位置,这种测序序列,称为splitreads。通过bam文件提取小片段插入缺失的特征向量,并根据特征设定条件进行初步筛选。特征设定条件可以为测序质量和/或测序深度。如果有对照样本,则通过对照样本的bam文件和待测样本的bam文件统计待测样本和对照样本两两比较的特征向量。特征设定条件则包括以下任一种或多种:测序质量、测序深度、对照样本中小片段插入缺失出现的频率。从第一比对结果提取得到的小片段插入缺失的特征向量具体可以为如下任一种或多种:总覆盖度、支持参考序列覆盖度、支持插入缺失的覆盖度、突变频率、小片段插入缺失人群数据库、gc含量、噪音(与参考序列和变异序列都不一致的序列视为噪音)的个数统计、噪音的频率统计、突变左右参考序列上数量最多的碱基比例、正链支持参考序列的数目、正链支持小片段插入缺失突变的数目、负链支持参考序列的数目、负链支持小片段插入缺失变异的数目、正链支持小片段插入缺失突变数目和负链支持小片段插入缺失突变数目的比值、小片段插入缺失位点在参考序列上距头尾最短距离的均值、小片段插入缺失位点在参考序列上距头尾最短距离的方差、小片段插入缺失位点在距头尾最短距离的均值,小片段插入缺失位点在碱基距头尾最短距离的方差、建库长度均值、建库长度方差、支持参考序列碱基基因序列比对质量平均值、支持参考序列碱基基因序列比对质量方差、支持参考序列碱基质量平均值、支持参考序列碱基质量方差、支持变异碱基基因序列比对质量平均值、支持变异碱基基因序列比对质量方差、支持变异碱基质量平均值、支持变异碱基质量方差、一致性质量、单样本rms(rootmeansquare,比对质量均方根)质量、单样本校正(binomialtest)的p值、有对照样本两两费歇尔检验(fishertest)的p值、有对照样本两两体细胞变异探测分数(somaticscore)、信噪比。步骤13根据所述第一特征向量集合利用机器学习模型探测所述待测样本的小片段插入缺失的位点。本实施例提供的探测基因组小片段插入缺失的方法,可探测1个样本,也可以同时探测多个样本,每个样本的同一位置,也会放在一起比较,每个样本之间可互为对照。接下来,对所述机器学习模型的构建进行具体介绍。如图2所示,所述机器学习模型通过以下步骤构建。步骤21、将标准样本的测序数据和所述参考序列进行比对,得到第二比对结果。在步骤21之前,选取已知可靠值集合的标准样本进行测序文库建立。利用测序平台(例如illumina、bgiseq、iontorrent等)进行测序。具体可以为全基因组测序,也可以为全外显子测序,也可以为靶向测序。测序结果生成原始fastq文件,从而得到对照样本的测序数据。需要明确指出的是,此处的测序文库建立并非是指为了得到可靠值集合(如上所述,在进行本实施例中的标准样本测序文库建立之前,标准样本的可靠值集合已经是已知的了)的测序文库建立,而是为了训练模型,采用普通的、一般的待测样本的测序文库的构建方法进行测序文库构建。在一个示例中,构建测序文库的构建方法和待测样本的测序文库的构建方法一致,即采用相同的测序平台和测序方法。在本实施例中,可靠值是指测序深度较深(例如20000×及以上),从而测序结果比较可靠,可以认为是真实的碱基序列。利用基因组比对软件(如bwa、bowtie、stampy等)对fastq文件进行参考基因组(如人类基因组grch37等)比对,得到比对结果,比对结果可以以二进制序列比对格式(binarysequencealignmentmapformat,bam)文件形式存在。在一个示例中,标准样本可以为健康人的组织细胞、生殖细胞等。在一个示例中,标准样本可以为病人的病变组织细胞,例如肿瘤患者的肿瘤细胞。当标准本为病人的病变组织细胞时,在本实施例中,进行标准样本的对照样本的测序数据和参考序列进行比对,得到第四比对结果。待测样本的对照样本可以为该病人的非病变组织。例如,标准样本为病人的肿瘤细胞,该标准样本的对照样本可以为癌旁组织细胞或者为血液中的细胞。对照样本的测序、比对可参照标准样本进行,此处不再赘述。步骤22、提取第二比对结果中的小片段插入缺失的特征向量,得到第二特征向量集合。其中,所述第二特征向量集合中含有标准样本的测序数据相对参考序列的插入缺失碱基长度、标准样本的测序数据相对参考序列的突变位点库长度均值、标准样本的测序数据相对参考序列的突变位点库长度方差、标准样本的测序数据相对参考序列的突变位点覆盖splitreads个数、标准样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;测序序列分为两个或多个部分,每个部分分别比对到参考基因组上多个位置,这种测序序列,称为splitreads。通过bam文件提取小片段插入缺失的特征向量,并根据特征设定条件进行初步筛选。特征设定条件可以为测序质量和/或测序深度。如果有对照样本,则通过对照样本的bam文件和标准样本的bam文件统计标准样本和对照样本两两比较的特征向量。特征设定条件则包括以下任一种或多种:测序质量、测序深度、对照样本中小片段插入缺失出现的频率。从第二比对结果中提取得到的小片段插入缺失的特征向量具体可以为如下任一种或多种:总覆盖度、支持参考序列覆盖度、支持插入缺失的覆盖度、突变频率、小片段插入缺失人群数据库、gc含量、噪音(与参考序列和变异序列都不一致的序列视为噪音)的个数统计、噪音的频率统计、突变左右参考序列上数量最多的碱基比例、正链支持参考序列的数目、正链支持小片段插入缺失突变的数目、负链支持参考序列的数目、负链支持小片段插入缺失变异的数目、正链支持小片段插入缺失突变数目和负链支持小片段插入缺失突变数目的比值、小片段插入缺失位点在参考序列上距头尾最短距离的均值、小片段插入缺失位点在参考序列上距头尾最短距离的方差、小片段插入缺失位点在距头尾最短距离的均值,小片段插入缺失位点在碱基距头尾最短距离的方差、建库长度均值、建库长度方差、支持参考序列碱基基因序列比对质量平均值、支持参考序列碱基基因序列比对质量方差、支持参考序列碱基质量平均值、支持参考序列碱基质量方差、支持变异碱基基因序列比对质量平均值、支持变异碱基基因序列比对质量方差、支持变异碱基质量平均值、支持变异碱基质量方差、一致性质量、单样本rms(rootmeansquare,比对质量均方根)质量、单样本校正(binomialtest)的p值、有对照样本两两费歇尔检验(fishertest)的p值、有对照样本两两体细胞变异探测分数(somaticscore)、信噪比。步骤23、根据所述第二特征向量集合在标准样本的可靠值序列中标注小片段插入缺失位点,得到标准样本的小片段插入缺失位点集合。标准样本的可靠值序列可以从数据库下载,也可以预先测序,该预先测序的深度较高,比如20000×及以上,可以认为可靠值序列是可靠的。该预先测序是指在本实施例步骤21中为训练模型而进行的测序文库构建之前进行的测序(该预先测序是为了在步骤21之前,获知标准样本的可靠值序列)。具体的,当在标准样本的可靠值序列中出现一个小片段插入缺失位点时,标记为1,反之标记为0,得到标注好的集合。相应地,在上文的步骤13中,探测到分类为1的小片段插入缺失位点做为待测样本的变异位点。步骤24、利用机器学习方法对所述标准样品的小片段插入缺失位点集合进行模型训练,得到所述机器学习模型。所述机器学习方法包括以下任一种:朴素贝叶斯法、逻辑回归法、线性回归法、最近邻近法、决策树法、boosting方法及其变种、svm支持向量机法、人工神经网络算法。boosting方法及其变种包含adaptiveboosting、gradientboosting等。根据不同的机器学习方法可以生成不同的机器学习模型。待测样本的测序数据的测序平台和测序方法,与,标准样本的测序数据的测序平台和测序方法一致;即待测样本的测序数据的测序平台和标准样本的测序数据的测序平台一致,且待测样本的测序数据的测序方法和标准样本的测序数据的测序方法一致。根据不同的测序平台和不同测序方法可以生成多种机器学习模型,在进行待测样本探测时,根据待测样本的测序平台和测序方法选择对应的机器学习模型,以获得更高的探测准确率。在一个示例中,在训练机器学习模型时,采用十折交叉验证法测试训练的机器学习模型。在本实施例中,可以进行多样品对比探测,对于每种变异分别进行正常样本和肿瘤样本的对比探测,最大限度保留低频变异。同时,可以将生殖细胞变异和体细胞变异探测同步进行。本实施例提供的探测基因组小片段插入缺失的方法基于特征提取、模型训练的机器学习方法来探测高置信度的indel,并且可以根据不同测序平台和测序方法定制不同的模型,以提高计算的速度和结果的精度;本实施例提供的探测基因组小片段插入缺失的方法及装置运行效率高;其他基因变异探测方法需要数以天计的计算任务,本实施例提供的探测基因组小片段插入缺失的方法可以在短时间内(对于数据深度30x的数据,32核心、512g内存节点运算15分钟就可以给出探测结果)给出探测结果,可以最大限度利用计算资源,降低时间成本;并且通过使用机器学习的方法提高了探测单核苷酸突变的精度。实施例2在本实施例中,提供了一种探测基因组小片段插入缺失位点的装置,如图3所示,该装置包括:比对单元31、提取单元32、探测单元33、标注单元34、训练单元35。所述比对单元31用于将待测样本的测序数据和参考序列进行比对,得到第一比对结果。所述提取单元32用于提取第一比对结果中的小片段插入缺失的特征向量,得到第一特征向量集合;其中,所述第一特征向量集合中含有待测样本的测序数据相对参考序列的插入缺失碱基长度、待测样本的测序数据相对参考序列的突变位点库长度均值、待测样本的测序数据相对参考序列的突变位点库长度方差、待测样本的测序数据相对参考序列的突变位点覆盖splitreads个数、待测样本的测序数据相对参考序列的错配碱基的个数中的一种或多种。所述探测单元33用于根据所述第一特征向量集合利用机器学习模型探测所述待测样本的小片段插入缺失的位点;所述比对单元31还用于将标准样本的测序数据和所述参考序列进行比对,得到第二比对结果。所述提取单元32还用于提取第二比对结果中的小片段插入缺失的特征向量,得到第二特征向量集合。其中,所述第二特征向量集合中含有标准样本的测序数据相对参考序列的插入缺失碱基长度、标准样本的测序数据相对参考序列的突变位点库长度均值、标准样本的测序数据相对参考序列的突变位点库长度方差、标准样本的测序数据相对参考序列的突变位点覆盖splitreads个数、标准样本的测序数据相对参考序列的错配碱基的个数中的一种或多种;所述标注单元34用于根据所述第二特征向量集合在标准样本的可靠值序列中标注小片段插入缺失位点,得到标准样本的小片段插入缺失位点集合。所述训练单元35用于利用机器学习方法对所述标准样品的小片段插入缺失位点集合进行模型训练,得到所述机器学习模型。在一个示例中,所述比对单元31还用于将待测样本的对照样本的测序数据和所述参考序列进行比对,得到第三比对结果;所述提取单元32还用于根据第三比对结果提取第一比对结果中的小片段插入缺失的特征向量,得到所述第一特征向量集合;所述比对单元31还用于将标准样本的对照样本的测序数据和所述参考序列进行比对,得到第四比对结果;所述提取单元32还用于根据所述第四比对结果提取第二比对结果中的小片段插入缺失的特征向量,得到所述第二特征向量集合。在一个示例中,小片段插入缺失的特征向量包括以下任一种或多种:总覆盖度、支持参考序列覆盖度、支持插入缺失的覆盖度、突变频率、小片段插入缺失人群数据库、gc含量、噪音(与参考序列和变异序列都不一致的序列视为噪音)的个数统计、噪音的频率统计、突变左右参考序列上数量最多的碱基比例、正链支持参考序列的数目、正链支持小片段插入缺失突变的数目、负链支持参考序列的数目、负链支持小片段插入缺失变异的数目、正链支持小片段插入缺失突变数目和负链支持小片段插入缺失突变数目的比值、小片段插入缺失位点在参考序列上距头尾最短距离的均值、小片段插入缺失位点在参考序列上距头尾最短距离的方差、小片段插入缺失位点在距头尾最短距离的均值,小片段插入缺失位点在碱基距头尾最短距离的方差、建库长度均值、建库长度方差、支持参考序列碱基基因序列比对质量平均值、支持参考序列碱基基因序列比对质量方差、支持参考序列碱基质量平均值、支持参考序列碱基质量方差、支持变异碱基基因序列比对质量平均值、支持变异碱基基因序列比对质量方差、支持变异碱基质量平均值、支持变异碱基质量方差、一致性质量、单样本rms(rootmeansquare,比对质量均方根)质量、单样本校正(binomialtest)的p值、有对照样本两两费歇尔检验(fishertest)的p值、有对照样本两两体细胞变异探测分数(somaticscore)、信噪比。所述机器学习方法包括以下任一种:朴素贝叶斯法、逻辑回归法、线性回归法、最近邻近法、决策树法、boosting方法及其变种,包含adaptiveboosting、gradientboosting等、svm支持向量机法、人工神经网络算法;boosting方法及其变种包含adaptiveboosting、gradientboosting等;所述训练单元35还用于采用十折交叉验证法测试所述机器学习模型。在一个示例中,第一比对结果存放在bam文件中;第二比对结果存放在bam文件中;待测样本的测序数据的测序平台和测序方法,与,标准样本的测序数据的测序平台和测序方法一致。在一个示例中,所述提取单元32还用于根据特征设定条件提取第一比对结果中的小片段插入缺失的特征向量;所述提取单元32还用于根据特征设定条件提取第二比对结果中的小片段插入缺失的特征向量;特征设定条件包括以下任一种或多种:测序质量、测序深度、对照样本中小片段插入缺失出现的频率。本实施例提供的探测基因组小片段插入缺失位点可以参照实施例1实现,此处不再赘述。本实施例具有如下优点:基于特征提取、模型训练的机器学习装置来探测高置信度的indel,并且可以根据不同测序平台和测序方法定制不同的模型,以提高计算的速度和结果的精度;本实施例提供的探测基因组小片段插入缺失的装置运行效率高;其他基因变异探测装置需要数以天计的计算任务,本实施例提供的探测基因组小片段插入缺失的装置可以在短时间内(对于数据深度30x的数据,32核心、512g内存节点运算15分钟就可以给出探测结果)给出探测结果,可以最大限度利用计算资源,降低时间成本;并且通过使用机器学习的方法提高了探测单核苷酸突变的精度。实施例3在本实施例中,以标准样本为na12878样本为例,对本发明实施例提供的机器学习模型进行说明。na12878样本来自于瓶中基因组联盟(genomeinabottle),该联盟由美国国家标准技术研究所nist发起成立,该联盟包括来自产业界、学术界和政府部门的相关人员,旨在建立参考标准帮助人们评估测序仪器、试剂和数学算法的性能,推动人类基因组测序的临床应用。na12878该样本来自具有犹他州ceph血统的一位妇女,是研究的最多的基因组。测序平台为illuminahiseq2000,测序深度50×。由ebi下载,数据号是err194147。根据实施例1记载的内容得到初筛数据集合如表1所示。表1在训练模型时,将数据均分为10份,其中九份作为训练数据,剩余一份作为测试数据。采用十折交叉验证方法,使用机器学习方法(gradientboosting算法)将训练数据建模,并使用测试数据进行性能验证。最终性能为十折交叉验证结果均值。验证结果如表2所示。表2数据准确率(均值)召回率(均值)f1-score(均值)na12878.idf99.55%99.78%99.66%本实施例具有如下优点:基于特征提取、模型训练的机器学习装置来探测高置信度的indel,并且可以根据不同测序平台和测序方法定制不同的模型,以提高计算的速度和结果的精度;本实施例提供的探测基因组小片段插入缺失的方法及装置运行效率高;其他基因变异探测装置需要数以天计的计算任务,本实施例提供的探测基因组小片段插入缺失的方法及装置可以在短时间内(对于数据深度30x的数据,32核心、512g内存节点运算15分钟就可以给出探测结果)给出探测结果,可以最大限度利用计算资源,降低时间成本;并且通过使用机器学习的方法提高了探测单核苷酸突变的精度。实施例4在本实施例中,以标准样本为肿瘤患者的病变样本为例,对本发明实施例提供的机器学习模型进行说明。测序数据来源于theicgc-tcgadreamgenomicmutationcallingchallengeset5data(https://www.synapse.org/#!synapse:syn312572/wiki/62018)。根据实施例1记载的内容得到初筛数据集合如表3所示。表3在训练模型时,将数据均分为10份,其中九份作为训练数据,剩余一份作为测试数据。采用十折交叉验证方法,使用机器学习方法(gradientboosting算法)将训练数据建模,并使用测试数据进行性能验证。最终性能为十折交叉验证结果均值。验证结果如表4所示。表4数据准确率(均值)召回率(均值)f1-score(均值)set5.idf98.68%99.76%99.21%本实施例具有如下优点:基于特征提取、模型训练的机器学习装置来探测高置信度的indel,并且可以根据不同测序平台和测序方法定制不同的模型,以提高计算的速度和结果的精度;本实施例提供的探测基因组小片段插入缺失的方法及装置运行效率高;其他基因变异探测装置需要数以天计的计算任务,本实施例提供的探测基因组小片段插入缺失的方法及装置可以在短时间内(对于数据深度30x的数据,32核心、512g内存节点运算15分钟就可以给出探测结果)给出探测结果,可以最大限度利用计算资源,降低时间成本;并且通过使用机器学习的方法提高了探测单核苷酸突变的精度。虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1