一种钆基金属富勒烯水溶性氮宾衍生物及其制备方法与应用与流程
本发明属于碳纳米材料领域,涉及一类顺磁性的钆基金属富勒烯水溶性氮宾衍生物及其制备方法和应用,该类钆基金属富勒烯水溶性氮宾衍生物可用作磁共振成像(mri)造影剂。
背景技术:
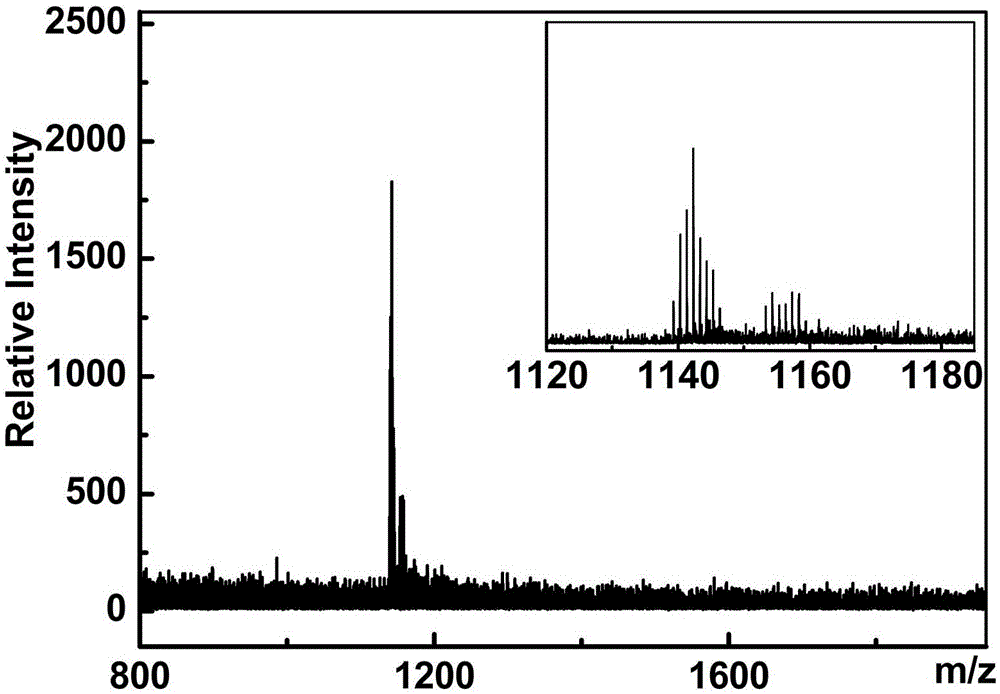
:具有高纵向弛豫率(r1)、低生物毒性的顺磁性金属富勒烯类t1型mri造影剂已发展近20年。其中,钆基金属富勒烯类mri造影剂作分子影像探针最常见于文献报道。这些mri探针均为羟基化、氨基化、bingel-hirsch或prato等反应单步或多步修饰的gd@c60、gd@c82、scxgd3-xn@c80、gd3n@c80、gd3n@c84衍生物。这些衍生物中,gd@c82(oh)40首次标记细胞作分子探针应用于mri细胞成像;gd@c82o6(oh)16(nhch2ch2cooh)8首次偶联受体蛋白作识别荧光蛋白的分子探针;结合识别脑胶质瘤小肽il-13的gd3n@c80(oh)26(ch2ch2cooh)16和gd3n@c80o12(oh)10(nh2)7(no2)2先后被报道作诊断与治疗功能一体化的肿瘤诊疗试剂。此外,gd@c82o14(oh)14(nh2)6免标记荧光染料就能构成mri-荧光双模态分子影像探针,其还可作抗氧化剂清除ros类自由基,具备肿瘤抑制潜力。当前,顺磁性钆基金属富勒烯衍生物作纳米诊疗试剂的应用研究存在瓶颈,关键就在于缺乏经济合理大规模化制备新型顺磁性钆基金属富勒烯水溶性衍生物的合成方法,导致难于提供充足产品应用于后续的生物效应研究。本发明的目的就在于采用免溶剂策略的反应技术,直接以呈液态的过量叠氮化合物分散固态的顺磁性钆基金属富勒烯,经氮宾反应,从而发明出一种顺磁性的钆基金属富勒烯水溶性衍生物及其制备方法。这为发展新型顺磁性的钆基金属富勒烯水溶性氮宾衍生物用作mri造影剂提供一条崭新的研究思路。技术实现要素:本发明要解决的第一个技术问题是提供一种碳笼外接侧链末端键联亲水性官能团的钆基金属富勒烯水溶性氮宾衍生物,该衍生物具有良好的水溶性及顺磁性能,并且其侧链末端存在具有反应活性的基团。本发明要解决的第二个技术问题是提供一种制备所述顺磁性的钆基金属富勒烯水溶性氮宾衍生物的方法。本发明要解决的第三个技术问题是提供所述顺磁性的钆基金属富勒烯水溶性氮宾衍生物作为磁共振成像造影剂的应用。下面对本发明为解决上述技术问题所采取的技术方案做具体说明。一方面,本发明提供了一种顺磁性的钆基金属富勒烯水溶性氮宾衍生物,是由顺磁性钆基金属富勒烯gdanb@c2n与叠氮物n3(ch2)mnh2或n3(ch2)m-1cooh通过氮宾反应制得,其结构通式如下:gdanb@c2n[n(ch2)mnh2]x或gdanb@c2n[n(ch2)m-1coom]x上述结构通式中,a=1~3;b=0~3;n=30~45;m=2~6;x=8~30;m为h、nh4、na或k。本发明所述的顺磁性的钆基金属富勒烯水溶性氮宾衍生物分子以顺磁性钆基金属富勒烯碳笼为核心,碳笼外至少带有8个亲水性侧链作连接臂,每个连接臂都通过氮桥与碳笼键联,连接臂为数个碳原子构成的短直饱和碳链且末端还键联有亲水性官能团-nh2或者-coom,由此确保整个衍生物分子具备水溶性。连接臂的数目可根据不同的叠氮物及钆基金属富勒烯品种确定,当连接臂数目低于8个时,其水溶性极差甚至完全不溶于水;当连接臂的数目高于30个时,产物的碳笼结构很不稳定,甚至造成开笼导致内包金属泄漏;故连接臂的数目宜在8~30个之间,对于gd@c82衍生物来说,特别优选14~24个。本发明所述的顺磁性钆基金属富勒烯为每个分子由60~90个碳原子组成的具有球状或类球状结构并且内部包含1-3个gd原子或者是gdanb原子团簇,所述的顺磁性钆基金属富勒烯优选为gd@c82、gd@c60、gd2@c80、gd3n@c80或gd3n@c84,更优选gd@c82、gd3n@c80或gd3n@c84,并以gd@c82为最优。本发明所述的叠氮物,其分子结构为碳链含碳原子数目2~6个的饱和脂肪直碳链且一端键联叠氮基(-n3)而另一末端键联亲水性的氨基(-nh2)或羧基(-cooh),结构通式为n3(ch2)mnh2或n3(ch2)m-1cooh,优选为2-叠氮基乙胺、2-叠氮基乙酸、3-叠氮基丙胺、3-叠氮基丙酸、4-叠氮基丁酸或6-叠氮基己酸。富勒烯可与含叠氮基团的化合物发生环加成反应是公知的技术,但鲜见有关顺磁性钆基金属富勒烯发生氮宾反应制备水溶性衍生物的报道。另一方面,本发明提供了一种经氮宾反应制备顺磁性的钆基金属富勒烯水溶性氮宾衍生物的方法。所述的顺磁性的钆基金属富勒烯水溶性氮宾衍生物结构通式为gdanb@c2n[n(ch2)mnh2]x或gdanb@c2n[n(ch2)m-1cooh]x,所述的制备方法包括如下步骤:(1)将钆基金属富勒烯gdanb@c2n与叠氮物n3(ch2)mnh2或n3(ch2)m-1cooh分别投料于反应器,在惰性气体氛围下搅拌,所述钆基金属富勒烯与叠氮物的物质的量之比为1:100~1000;(2)将步骤(1)得到的固液反应体系首先由室温以2-5℃/min升温速率升至40~80℃,保温0.5~2小时,然后再以0.5-2℃/min升温速率升至110~150℃,保温10~48小时,停止加热,整个反应过程由tlc跟踪监控;(3)反应产物添加有机溶剂a洗涤、过滤,去除大部分未反应的叠氮物,残留物用水进行提取,提取液过滤、干燥,得固化的粗产品;(4)粗产品复溶于水,使用mw=500-10000的透析袋透析,冷冻干燥,得到纯净的顺磁性的钆基金属富勒烯水溶性氮宾衍生物的固态产品。进一步,所述步骤(1)中,叠氮物相对于钆基金属富勒烯的投料比必须介于指定范围之内,即物质的量之比为100~1000:1。低于该指定投料比100:1下限时,反应体系易迅速固化而难于持续进行且衍生物加成数目少,导致难以获得水溶性产品;超过1000:1上限时,后续处理繁琐,且不易获得纯化的产品;在指定投料比范围内可相对低成本、高收率的获得水溶性的顺磁性钆基金属富勒烯氮宾衍生物。进一步,所述步骤(2)中,控温操作极为关键,低于110℃时,反应难于发生并形成水溶性产物,超过150℃时,反应过于剧烈,存在极大失控风险,优选控温范围110~150℃,更优选110~120℃。进一步,所述步骤(3)中,洗涤可采用常规试剂,如乙醚、二氯甲烷、三氯甲烷、乙酸乙酯等,去除大部分未反应的叠氮物。进一步,所述步骤(4)中,精制方法采用透析后冷冻干燥,如此纯化,处理方式步骤少且操作简单。本发明还提供了另一种经氮宾反应制备顺磁性的钆基金属富勒烯水溶性氮宾衍生物的方法,所述的顺磁性钆基金属富勒烯氮宾衍生物结构通式为gdanb@c2n[n(ch2)m-1coom]x,所述的m为na或k或nh4,所述的制备方法包括如下步骤:(a)将钆基金属富勒烯gdanb@c2n与叠氮物n3(ch2)m-1cooh分别投料于反应器,在惰性气体氛围下搅拌,所述钆基金属富勒烯与叠氮物的物质的量之比为1:100~1000;(b)将步骤(1)得到的固液反应体系首先由室温以2-5℃/min升温速率升至40~80℃,保温0.5~2小时,然后再以0.5-2℃/min升温至110~150℃,保温10~48小时,停止加热,整个反应过程由tlc跟踪监控;(c)反应产物添加有机溶剂b洗涤、过滤,去除大部分未反应的叠氮物,残留物复溶于碱性溶液,所述的碱性溶液为naoh或koh水溶液或氨水,使用mw=500-10000透析袋透析、干燥,得到纯净的顺磁性的钆基金属富勒烯水溶性氮宾衍生物的固态产品。进一步,所述步骤(a)中,叠氮物相对于钆基金属富勒烯的投料比必须介于指定范围之内,即物质的量之比为100~1000:1。低于该指定投料比100:1下限时,反应体系易迅速固化而难于持续进行且衍生物加成数目少,导致难以获得水溶性产品;超过1000:1上限时,后续处理繁琐,且不易获得纯化的产品;在指定投料比范围内可相对低成本、高收率的获得水溶性的顺磁性钆基金属富勒烯氮宾衍生物。进一步,所述步骤(b)中,控温操作极为关键,低于110℃时,反应难于发生并形成水溶性产物,超过150℃时,反应过于剧烈,存在极大失控风险,优选控温范围110~150℃,更优选110~120℃。进一步,所述步骤(c)中,洗涤可采用常规试剂,如乙醚、二氯甲烷、三氯甲烷、乙酸乙酯等,去除大部分未反应的叠氮物。本发明将步骤(3)和(c)中的有机溶剂用有机溶剂a和有机溶剂b表示,其目的仅在于区分不同的步骤,并不表示有机溶剂a和有机溶剂b必须采用不同的溶剂,事实上两者各自独立,本领域技术人员可以根据需要进行选择。进一步,所述步骤(c)中,碱性溶液优选为浓度>0.1mol/l且≤1mol/l的naoh或koh水溶液或氨水,更优选0.5mol/l的naoh或koh水溶液。进一步,步骤(c)所制得的顺磁性的钆基金属富勒烯水溶性氮宾衍生物,可通过叠氮物与顺磁性钆基金属富勒烯品种,来调控侧链数目的多寡及分布范围的宽窄。第三方面,本发明提供了所述顺磁性的钆基金属富勒烯水溶性氮宾衍生物作为磁共振成像造影剂的应用。本发明得到的顺磁性的钆基金属富勒烯水溶性氮宾衍生物分子中,碳笼表面通过氮原子桥联多个末端键联-nh2或-coom的亲水性侧链,使得整个笼型分子具备水溶性,同时亦表现出良好顺磁性质,故可用作磁共振成像造影剂。另外,借助于侧链末端具有反应活性-nh2或-coom官能团,其可作为反应中间体,通过酰胺化偶联反应键联诸如顺磁、光、酶标记等各类标记物以及糖类、核酸、多肽、抗体等生物大分子,由此构建功能化的纳米诊疗试剂,以应用于纳米诊疗
技术领域:
。与现有技术相比,本发明的有益效果在于:(a)本发明提供的化合物为一系侧链碳链长短、多寡及链末端烯亲水基互异的顺磁性的钆基金属富勒烯水溶性氮宾衍生物,这可大大扩展金属富勒烯水溶性衍生物的产品库。(b)本发明提供的大规模制备顺磁性的钆基金属富勒烯水溶性氮宾衍生物新方法的特点:合成工艺简便、高效,且纯化分离方法简单,易于实现大规模工业化制备。(c)本发明提供的固液相反应法的反应体系无需添加有机溶剂分散顺磁性钆基金属富勒烯,作为反应物的液的叠氮物自身与衍生物极性相近,而有利于其分散于液相反应体系,并最终产生更高加成数目的衍生物,直至产生水溶性衍生产物。(d)本发明所获得顺磁性的钆基金属富勒烯水溶性氮宾衍生物的碳笼外接侧链末端存在具有反应活性的基团-nh2或-coom,可通过酰胺化缩合反应偶合诸如磁、光、酶等各类标记物以及糖类、核酸、多肽、抗体等生物功能分子。(e)本发明所获得顺磁性的钆基金属富勒烯水溶性氮宾衍生物具备良好的水溶性,亦表现出良好顺磁性质,可作为磁共振成像造影剂,用于mri影像成像时,造影增强效果非常显著,在医学诊疗尤其是在mri影像引导的诊断治疗方面具有重要的应用前景。附图说明图1.实施例1中gd@c82(nch2ch2cooh)22水溶液的maldi-tofms谱图。图2.实施例1中gd@c82(nch2ch2cooh)22粉末的ft-ir谱图。图3.实施例1中gd@c82(nch2ch2cooh)22粉末的热重分析谱图。图4.实施例3中gd@c82(nch2ch2nh2)16粉末的ft-ir谱图。图5.gd@c82(nch2cooh)24(a)和gd@c82(nch2ch2cooh)22(b)的质子纵向弛豫率。具体实施方式以下实施例有助于进一步理解本发明,但本发明保护范围并不限于列举的实施例。以下实施例中所述实验方法均为常规方法;所述试剂和材料均可从商业途径获得。其中,gd@c82来源于厦门福纳新材料科技有限公司,纯度高于99.0%。实施例1:制备gd@c82与叠氮丙酸的水溶性衍生物gd@c82(nch2cooh)22将10mggd@c82与液态的n3ch2ch2cooh按化学计量比1:500依次投料于三口反应瓶,氮气保护,搅拌;首先以2℃/min升温速率缓慢升温至60℃,保温1小时,然后再以0.5℃/min升温速率升至120℃,保温48小时,停止加热;添加乙酸乙酯洗涤、过滤,残留物用去离子水反复提取至无色,提取液过滤、干燥,得固化粗产品;复溶于去离子水,使用mw=3500透析袋透析,滤膜过滤、冷冻干燥,得到纯净的顺磁性gd@c82聚丙氨酸水溶性衍生物的固态产品。如图1所示,maldi-tofms中m/z为1142处离子峰归属于gd@c82碳笼母体单元,一般认为,金属富勒烯水溶性衍生物碳笼外接侧链在maldi-tofms实验过程中因激光解吸附作用从金属富勒烯碳笼上被剥离,而不出现衍生物的分子离子峰,仅存在金属富勒烯单元的离子峰。由于gd@c82不溶于水相,可判断该水溶性衍生物就是gd@c82的衍生物。如图2所示,红外光谱在3420cm-1处的强吸收峰归属为o-h伸缩振动、1720cm-1处吸收峰归属于c=o、1100cm-1归属于c-oh伸缩振动,在2930cm-1处的弱吸收峰归属于c-h伸缩振动,在1640cm-1处的强吸收峰归属于gd@c82碳笼中残余的c=c伸缩振动。ir谱图证明gd@c82聚丙氨酸水溶性衍生物分子中存在-cooh。如图3所示,tg-dtg-dsc同步分析结果表明,gd@c82聚丙氨酸水溶性衍生物分子在温度区间110-515℃百分比质量从93.89%减少到35.33%,此区间减少的百分比质量(57.80%)完全归因于gd@c82聚丙氨酸水溶性衍生物分子外接基团-nch2ch2cooh的全部丢失,而515℃时残留的质量则归属于gd@c82碳笼,其中碳笼式量为1142,全部外接侧链(-nch2ch2cooh)的式量为87x,计算x=21.47,据此得gd@c82聚丙氨酸水溶性衍生物平均分子式为gd@c82(nch2ch2cooh)22。实施例2:制备gd@c82与叠氮乙酸的水溶性衍生物gd@c82(nch2cooh)24将10mggd@c82与液态的n3ch2cooh依次投料于三口反应瓶,氮气保护,搅拌;首先以2℃/min升温速率缓慢升温至80℃,保温2小时,然后再以0.5℃/min升温速率升120℃,保温48小时,停止加热;添加乙醚洗涤,其余条件同本发明实施例1。得到纯净的顺磁性gd@c82聚甘氨酸水溶性衍生物固态产品。热重分析表征其平均分子式为gd@c82(nch2cooh)24。实施例3:制备gd@c82与叠氮乙胺的水溶性衍生物gd@c82(nch2ch2nh2)20将10mggd@c82与液态的n3ch2ch2nh2依次投料于三口反应瓶,氮气保护,搅拌;首先以2℃/min升温速率缓慢升温至60℃,保温2小时,然后再以0.5℃/min升温速率升至125℃,保温48小时,停止加热;添加乙酸乙酯洗涤;其余条件同本发明实施例1。得到纯净的顺磁性gd@c82聚乙二胺水溶性衍生物gd@c82(nch2ch2nh2)20的固态产品。如图4所示,红外光谱在3385cm-1处的强吸收峰归属为n-h伸缩振动、1388cm-1处吸收峰归属于nh弯曲振动、1037cm-1归属于c-nh2伸缩振动在2935、2850cm-1处的弱吸收峰归属于c-h对称与反对称伸缩振动,在1654cm-1处的强吸收峰归属于gd@c82碳笼中残余的c=c伸缩振动。ir谱图证明gd@c82聚乙二胺水溶性衍生物分子中存在-nh2。实施例4:制备gd@c82与叠氮乙酸的金属富勒烯基聚甘氨酸钠水溶性衍生物gd@c82(nch2coona)14将10mggd@c82与液态的n3ch2cooh按化学计量比1:100依次投料于三口反应瓶,氮气保护,机械搅拌;首先以2℃/min升温速率缓慢升温至40℃,保温0.5小时,然后再以0.5℃/min升温速率升至110℃,保温10小时,停止加热;添加乙醚洗涤、过滤,残留物复溶于0.5mol/lnaoh溶液,使用mw=3500透析袋透析,滤膜过滤、冷冻干燥,精制获得含甘氨酸钠的gd@c82水溶性氮宾衍生物的固态产品,热重分析表征其平均分子式为gd@c82(nch2coona)10。实施例5:制备gd@c82与叠氮己酸的金属富勒烯基聚己氨酸钠水溶性衍生物gd@c82(nch2ch2ch2ch2ch2coona)10将10mggd@c82与液态的n3ch2ch2ch2ch2ch2cooh按化学计量比1:250依次投料于三口反应瓶,氮气保护,机械搅拌;首先以2℃/min升温速率缓慢升温至80℃,保温2小时,然后再以0.5℃/min升温速率升至140℃,保温24小时,停止加热;添加乙酸乙酯洗涤、过滤,残留物复溶于0.5mol/lnaoh溶液,滤膜过滤,使用mw=3500透析袋透析,冷冻干燥,精制获得金属富勒烯基聚己氨酸钠水溶性衍生物,热重分析表征其平均分子式为gd@c82(nch2ch2ch2ch2ch2coona)10。对比例参照实施例1,区别在于:gd@c82与液态的n3ch2ch2cooh混合后,直接升温至回流温度,反应剧烈进行,约30min-60min后,体系迅速固化,反应被迫终止,停止加热;添加乙酸乙酯洗涤、过滤,残留物用去离子水反复提取3-5次,提取液过滤、浓缩,使用mw=3500透析袋透析,滤膜过滤、冷冻干燥后未得到gd@c82聚丙氨酸水溶性衍生物gd@c82(nch2ch2cooh)x的固态产品。实施例6:gd@c82氮宾衍生物作mri造影剂时弛豫性能测试的具体实施方式弛豫性能实验在200兆动物mri仪上进行。分别配置gd@c82(nch2cooh)24和gd@c82(nch2ch2cooh)22的一系列不同浓度样品。采用反转恢复自旋回波成像序列扫描,序列参数为:重复时间10s,回波时间13.5ms,成像面积3.5×3.5cm2,片厚1.0mm,数据矩阵64×64,在0.05~15s之间选择一系列合适的ti值。通过三参数单指数函数拟合,获得所有待测样品的纵向驰豫时间t1。通过其系列不同浓度时t1值拟合其质子纵向弛豫率即r1值。图5为gd@c82(nch2cooh)24和gd@c82(nch2ch2cooh)22纵向弛豫速率1/t1相对于其浓度变化趋势图,此变化趋势的斜率即为其r1值,结果示于表1。一般来说,连接臂中碳链越短,弛豫率越高,加成数目越多,弛豫率越高。表1样品名r1(mm-1s-1)gd@c82(nch2cooh)2426.29gd@c82(nch2ch2cooh)2217.34当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1