药物动力学和/或安全性优异的含有特立帕肽的液态药物组合物的制作方法
本发明涉及含有特立帕肽或其盐的皮下给药用液态药物制剂。
背景技术:
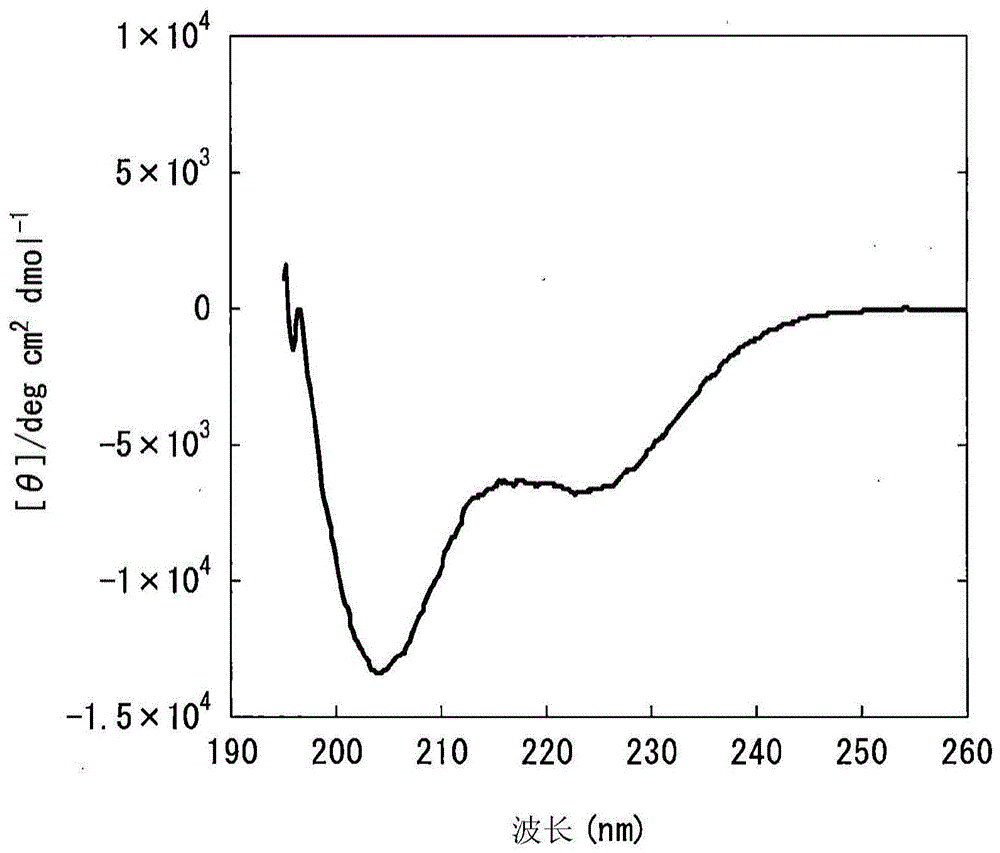
:pth(甲状旁腺激素)与降钙素类、维生素d类均是参与血中钙浓度调节的激素。关于作为天然型pth的生理活性等价物的pth肽,已知有含有pth肽的冻干制剂、含有pth肽的液态制剂。现有技术文献专利文献专利文献1:日本特开平5-306235号公报专利文献2:日本特开2004-10511号公报专利文献3:日本特开2007-186466号公报专利文献4:日本特表2001-525372号公报专利文献5:国际公开第2006/22301号专利文献6:国际公开第2012/169435号专利文献7:日本特表2015-504087号公报专利文献8:日本特开昭63-57527号公报专利文献9:日本特开平2-96533号公报专利文献10:日本特表2004-513069号公报专利文献11:日本特开2005-213158号公报专利文献12:国际公开第2011/139838号专利文献13:日本特表2014-507484号公报非专利文献非专利文献1:teribone(注册商标)皮下注射用56.5μg包装说明书(2015年11月修订(第6版使用上的注意事项等的修订))非专利文献2:forteo(注册商标)皮下注射试剂盒600μg包装说明书(2014年7月修订(第7版))非专利文献3:sungetal.,journalofbiologicalchemistry,(1991),vol.266,no.5,pp.2831-2835非专利文献4:takeietal.,peptidechemistry1979,(1980),pp.187-192非专利文献5:merrifield,advancesinenzymology,(1969),vol.32,pp.221-296非专利文献6:猪川和朗等,计量生物学,(2015),36,specialissue,s3-s18非专利文献7:machetal.,therapeuticdelivery,(2011),vol.2,no.6,pp.727-736非专利文献8:kinnunenetal.,journalofcontrolledrelease,(2014),vol.182,pp.22-32非专利文献9:“医薬品開発における薬物動態研究の重要性と今後の展開(医药品开发中的药物动力学研究的重要性和今后的开展)”、住友化学ii(26~34)非专利文献10:chenetal.,biochem.biophys.res.commun.,(1971),vol.44,no.6,pp.1285-1291非专利文献11:greenfield,natureprotocols,(2006),vol.1,no.6,pp.2876-2890非专利文献12:leeetal.,biopolymers,(1989),vol.28,pp.1115-1127非专利文献13:stricklandetal.,biochemistry,(1993),vol.32,pp.6050-6057非专利文献14:日本药学会年会要旨集第118年会、1998年,4,34非专利文献15:izutsuetal.,journalofpharmaceuticalsciences,(2006),vol.95,no.4,pp.781-789非专利文献16:平松弘嗣(东北大学-大学院药学研究科)、“赤外吸収スペクトルを用いた二次構造解析法(使用红外吸收光谱的二级结构分析法)”、蛋白質科学会アーカイブ(蛋白质科学会档案)、2009年,2,e054非专利文献17:伊豆津健一等,“タンパク質医薬品の非破壊評価に向けた水溶液と凍結乾燥固体中の二次構造検討(面向蛋白质医药品的非破坏评价的水溶液和冷冻干燥固体中的二级结构研究”、第21回近赤外フォーラム講演要旨集(第21次近红外论坛演讲要旨集)、2005年,59非专利文献18:armstrongetal.,proc.natl.acad.sci.usa,(1993),vol.90,pp.11337-11340非专利文献19:chakrabarttyetal.,biochemistry,(1993),vol.32,no.21,pp.5560-5565非专利文献20:wuetal.,proc.natl.acad.sci.usa,(1979),vol.76,no.8,pp.3656-3659非专利文献21:alojetal.,archivesofbiochemistryandbiophysics,(1972),vol.150,no.2,p.782-785非专利文献22:salginetal.,internationaljournalofelectrochemicalscience,(2012),vol.7,pp.12404-12414非专利文献23:yamamotoetal.,eurjpharmacol.(2015),vol.764,pp.457-462非专利文献24:teribone(注册商标)皮下注射用56.5μg申请资料概要(http://www.pmda.go.jp/drugs/2011/p201100155/index.html)非专利文献25:宫泽光博、“特集にあたって:タンパク質的立体構造解析法(专刊:蛋白质的立体结构分析法)”、蚕糸·昆虫バイオテック、2012年、81(2)、pp.105~106非专利文献26:日本药学会编スタンダード薬学シリーズ7製剤化のサイエンス(标准药学系列7制剂的科学)第1版第1次印刷2006年2月10日发行、pp.12~13非专利文献27:小阪谷典子他、“閉経期日本人女性における腰椎骨密度の5年間の減少に対する関連因子(闭经期日本女性中的腰椎骨密度5年减少的关联因子”、日本栄養·食糧学会誌(日本营养/粮食学会志)、1999年、第52卷、第5号、pp.307~313非专利文献28:水野仁贵等,“膜透過性ペプチドのアミノ酸配列改変によるph応答性の評価(由膜透过性肽的氨基酸序列改变所致的ph响应性的评价)”、日本大学生产工学部第48次学术演讲会演讲概要(2015-12-5)、pp.543~544非专利文献29:timjetal.,proteinscience,(2007),vol.16,pp.1193-1203非专利文献30:leonidk.,drugmetab.dispos.,(2014),vol.42,pp.1890-1905技术实现要素:发明所要解决的课题本发明的课题在于提供药物动力学良好(例如生物利用度高)和/或安全性高(例如消化道副作用出现频率得以抑制)的含有特立帕肽或其盐的皮下给药用液态药物制剂。用于解决课题的手段在本发明的皮下给药用液态药物制剂的一个方式中,特立帕肽或其盐中的α-螺旋含量为特定范围内(例如13.0%以上)。在本发明的皮下给药用液态药物制剂的一个方式中,特立帕肽或其盐中形成α-螺旋结构的氨基酸残基数为特定范围内(例如4.5个以上)。在本发明的皮下给药用液态药物制剂的一个方式中,制剂所显示出的基于圆偏振光二色性(cd)光谱测定(测定波长222nm)的平均残基摩尔椭圆率[θ]222为特定范围内(例如-6300(deg.cm2/dmol)以下)。利用这些皮下给药用液态药物制剂可得到优异的药物动力学(例如生物利用度高)。另外,在本发明的皮下给药用液态药物制剂的一个方式中,其每一次的特立帕肽或其盐的给药量为特定量(例如28.2μg)。或者,在本发明的皮下给药用液态药物制剂的一个方式中,通过其单次给药而得到的特立帕肽或其盐的达到最高血浆浓度的时间(tmax)为特定范围内(例如小于0.7hr)。或者,本发明的皮下给药用液态药物制剂的一个方式中,其单次给药后的特立帕肽或其盐的血浆中浓度为特定阈值(例如250pg/ml)以上的状态的经过时间为特定范围内(例如小于1.0hr)。利用这些皮下给药用液态药物制剂可得到优异的安全性(例如消化道副作用出现频率得到抑制)。即,本发明涉及以下的发明等。[1]一种人皮下给药用液态药物制剂,其是作为每一次的给药量以特立帕肽换算计含有28.2μg成分1的人皮下给药用液态药物制剂,其中,成分1的浓度为80μg/ml~240μg/ml。·成分1)特立帕肽或其盐。[2]如上述[1]中所述的人皮下给药用液态药物制剂,其中,成分1的浓度为100~200μg/ml。[3]如上述[1]或[2]中所述的人皮下给药用液态药物制剂,其中,通过不依赖于药物动力学模型的分析(nca(noncompartmentalanalysis);非房室模型分析)而计算出的单次给药时的tmax为0.5~0.7(1/hr)。[4]如上述[1]~[3]中所述的人皮下给药用液态药物制剂,其中,单次给药后的成分1的血浆中浓度为100pg/ml或其以上的状态的经过时间小于2.1(hr),且单次给药后的成分1的血浆中浓度为250pg/ml或其以上的状态的经过时间小于1.0(hr)。[5]如上述[1]~[4]中所述的人皮下给药用液态药物制剂,其用于对闭经后女性给药。[6]如上述[1]~[5]中所述的人皮下给药用液态药物制剂,其中,成分1中形成α-螺旋结构的氨基酸残基数为4.5个以上5.5个以下。[7]如上述[6]中所述的液态药物制剂,其中,氨基酸残基数是由平均残基摩尔椭圆率的数值a使用以下的推算式1而推算出的基于α-螺旋含量的氨基酸残基数,该平均残基摩尔椭圆率的数值a是通过满足以下的测定条件1~4的圆偏振光二色性(cd)光谱测定而得到的;测定条件1:测定波长为222nm;测定条件2:试样浓度(成分1浓度)为0.1~0.3mg/ml;测定条件3:测定温度为20℃;测定条件4:比色皿长为1~2mm;推算式1:α-螺旋含量=-(数值a+2340)/30300。[8]如上述[1]~[7]中所述的人皮下给药用液态药物制剂,其中,成分1为特立帕肽醋酸盐。[9]如上述[1]~[8]中所述的人皮下给药用液态药物制剂,其中,人皮下给药用液态药物制剂为人皮下给药用水性药物制剂(不包括冻干制剂的重新构成物)。[10]如上述[1]~[9]中所述的人皮下给药用液态药物制剂,其中,人皮下给药用液态药物制剂为人皮下给药用水性药物制剂,其溶剂为注射用水。发明的效果根据本发明,提供药物动力学和/或安全性优异的含有特立帕肽或其盐的液态药物制剂。附图说明图1a示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方a作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1b示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方b作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1c示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方c作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1d示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方d作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1e示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方e作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1f示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方f作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1g示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方g作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1h示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方h作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图1i合并示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方a~h作为测定对象,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定而得到的测定结果。横轴“波长(nm)”表示测定波长(nm)(210~230nm),纵轴“[θ]/deg.cm2dmol-1”表示平均残基摩尔椭圆率[θ]。图2合并示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方a~h(合计8个配方)作为对象,实施圆偏振光二色性(cd)光谱测定试验和人药物动力学试验(实施例3;人药物动力学试验(2))而得到的结果。将圆偏振光二色性(cd)光谱测定试验结果作为该试验的测定2的测定结果(平均残基摩尔椭圆率[θ]222),将人药物动力学试验结果作为auclast(截至最终观察时间为止的血浆中浓度-时间曲线下面积)相关的各配方相对于对照配方2之比即auclastratio。图3合并示出了以“供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方a~h(合计8个配方)作为对象,实施圆偏振光二色性(cd)光谱测定试验和人药物动力学试验(实施例3;人药物动力学试验(2))而得到的结果。将圆偏振光二色性(cd)光谱测定试验结果作为该试验的测定2的测定结果(α-螺旋含有比例),将人药物动力学试验结果作为auclast(截至最终观察时间为止的血浆中浓度-时间曲线下面积)相关的各配方相对于对照配方2之比即auclastratio。图4合并示出了以配方a~h(合计8个配方)作为对象,实施圆偏振光二色性(cd)光谱测定试验和猴子药物动力学试验(实施例2猴子药物动力学试验)而得到的结果。将圆偏振光二色性(cd)光谱测定试验结果作为该试验的测定2的测定结果(平均残基摩尔椭圆率[θ]222),将猴子药物动力学试验结果作为auclast(截至最终观察时间为止的血浆中浓度-时间曲线下面积)相关的各配方相对于对照配方1之比即auclastratio。图5合并示出了以配方a~h(合计8个配方)作为对象,实施圆偏振光二色性(cd)光谱测定试验和猴子药物动力学试验(实施例2猴子药物动力学试验)而得到的结果。将圆偏振光二色性(cd)光谱测定试验结果作为该试验的测定2的测定结果(α-螺旋含有比例),将猴子药物动力学试验结果作为auclast(截至最终观察时间为止的血浆中浓度-时间曲线下面积)相关的各配方相对于对照配方1之比即auclastratio。图6是示出了将实施例中提供的配方a、b、e、f以及h配方和参考例(成分1的tmax为特定范围内的发明所涉及的参考例)中提供的28.2μg制剂和56.5μg制剂分别对人进行给药而得到的血浆中特立帕肽醋酸盐浓度的时间推移的图。图7是实施例6和7中使用的药物动力学模型(1-房室模型)的示意图。此处,ka是指吸收速度常数,ke是指消失速度常数。具体实施方式以下根据具体实施方式详细地说明本发明。但本发明并不受以下实施方式的束缚,可以在不脱离本发明主旨的范围内以任意方式实施。1.皮下给药用液态药物制剂:本发明中,作为一个方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,上述制剂中的成分1的α-螺旋含量为特定范围内。本发明中,作为一个方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,上述制剂中的成分1中的形成α-螺旋结构的氨基酸残基数为特定范围内。本发明中,作为一个方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,制剂所显示出的基于圆偏振光二色性(cd)光谱测定(测定波长222nm)的平均残基摩尔椭圆率[θ]222为-6300(deg.cm2/dmol)以下。另外,本发明中,作为另一方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,其每一次的特立帕肽或其盐的给药量为特定量。此外,本发明中,作为另一方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,通过其单次给药而得到的成分1的tmax为特定范围内。或者,本发明中,作为另一方式,提供一种皮下给药用液态药物制剂,其是含有特立帕肽或其盐作为成分1的皮下给药用液态药物制剂,其中,单次给药后的特立帕肽或其盐的血浆中浓度为特定阈值以上的状态的经过时间为特定范围内。(1)液态药物制剂:本发明的液态药物制剂只要是含有后述的特立帕肽或其盐(成分1)的液态的皮下给药用药物制剂,对其形态就没有特别限定。作为本发明的液态药物制剂,可例示出皮下注射剂、皮下插入用胶囊剂。本发明的液态药物制剂只要为皮下给药用制剂,对其容器、针、包装等没有特别限定。此处的“药物制剂”是指用于对哺乳动物(人、猴、大鼠等)进行任意疾病的预防/治疗/诊断的药剂。作为药物制剂,可优选例示出人用药物制剂。给药对象为人的情况下,对其性別、年龄、罹患疾病的有无、种类等没有特别限定,例如可以为闭经后女性。本发明的液态药物制剂中使用的溶剂没有特别限定,可以为水性溶剂,也可以为非水性溶剂,优选包含水性溶剂,溶剂可以实质上仅由水性溶剂构成。本发明优选为水性药物制剂。液态药物制剂或溶剂(水性溶剂等)可以在不脱离本发明的主旨的范围内含有无机盐、有机盐、缓冲剂、添加剂等各种成分。例如,可以利用注射用水、生理盐水等制备本发明的液态药物制剂。作为本发明的液态药物制剂,可优选例示出人皮下给药用水性药物制剂,可最优选例示出人皮下注射用水性药物制剂。此处,本发明的液态药物制剂为皮下给药用制剂的情况下,对皮下给药的部位没有特别限定,优选神经和血管的分布少、皮下脂肪多、无骨的部位。作为这样的部位,可优选举出腹部、上臂部、大腿部、臀部,优选腹部。(2)特立帕肽或其盐(成分1):本发明中,人pth(1-34)是作为人甲状旁腺激素的人pth(1-84)的氨基酸序列中的由从n末端侧看的第1位到第34位的氨基酸残基构成的部分氨基酸序列所表示的肽。本发明中,特立帕肽是指游离体的人pth(1-34)。特立帕肽也可以为盐的形态。本发明中,作为特立帕肽的盐,可以举出由特立帕肽以及1种或2种以上的挥发性有机酸形成的任意的盐。作为挥发性有机酸,可例示出三氟乙酸、甲酸、乙酸等。关于游离体的特立帕肽与挥发性有机酸形成盐时的两者的比例,只要可形成该盐就没有特别限定。其中,作为挥发性有机酸,优选乙酸。即,作为本发明中的特立帕肽的盐,可优选例示出特立帕肽醋酸盐。由于特立帕肽或其盐为肽,因此其具有等电点(pi)。关于pi的测定,可利用本身公知的方法(例如使用hplc、电泳等的方法)来实施。通常已知特立帕肽或其盐的pi为8.3~8.4。特立帕肽或其盐(成分1)可通过本身公知的方法(例如非专利文献3~5等中记载的方法)来制造。(3)特立帕肽或其盐(成分1)的含量、用量、浓度:本发明的液态药物制剂中含有的特立帕肽或其盐(成分1)的量没有特别限定,可适宜地例示出以下的量。即,作为制剂中的成分1的量,优选为10μg以上,更优选为20μg以上、25μg以上、27μg以上、进而为28μg以上。另外,作为制剂中的成分1的量,优选为100μg以下,更优选为50μg以下、40μg以下、35μg以下、进而为30μg以下。其中,成分1的含量以特立帕肽计优选为28.2μg或29.2μg。在所使用的特立帕肽为醋酸盐的情况下,还可例示出加上乙酸量后的量。例如,为特立帕肽五醋酸盐的情况下,以特立帕肽五醋酸盐计,优选成分1的含量为30.3μg或31.3μg。本发明的液态药物制剂中含有的特立帕肽或其盐(成分1)每一次给药的给药量没有特别限定,可适宜地例示出以下的给药量。即,作为制剂中的成分1的每一次给药的给药量,更优选为25μg以上、27μg以上、进而为28μg以上。另外,作为制剂中的成分1的每一次给药的给药量,更优选为35μg以下、30μg以下、进而为29μg以下。其中,成分1每一次给药的给药量以特立帕肽计优选为28.2μg。特别是通过使成分1的每一次给药的给药量为上述上限值以下,可优选得到伴随单次给药的优异的安全性。另外,还可例示出使成分1每一次给药的给药量为56.5μg的方式。本发明的液态药物制剂中含有的特立帕肽或其盐(成分1)的浓度没有特别限定,可适宜地例示出以下的浓度。即,作为制剂中的成分1的浓度,优选为50μg/ml以上,更优选为70μg/ml以上、80μg/ml以上、100μg/ml以上、大于100μg/ml、110μg/ml以上、进而为120μg/ml以上。另外,作为制剂中的成分1的浓度,优选为500μg/ml以下,更优选为250μg/ml以下、小于250μg/ml、240μg/ml以下、200μg/ml以下、180μg/ml以下、进而为160μg/ml以下。其中,可最优选例示出141μg/ml。通过使成分1的浓度处于上述范围,可优选得到成分1的高吸收速度、伴随本制剂的单次给药的优异的安全性。需要说明的是,此处,成分1为特立帕肽盐的情况下,优选将成分1的浓度换算为其游离体(特立帕肽)浓度。(4)特立帕肽或其盐(成分1)的α-螺旋含量和形成α-螺旋的氨基酸残基数:本发明中,成分1(特立帕肽或其盐)的α-螺旋含量是指,相对于本发明的液态药物制剂中含有的成分1所具有的全部氨基酸残基数(全部残基数:即34个),形成α-螺旋结构的平均氨基酸残基数(对应残基数)的比例。比例可以由对应残基数除以全部残基数而得到的值(0~1)来表示,也可以换算为百分数(0~100(%))。例如,成分1的α-螺旋含量为13%是指成分1的34个氨基酸残基数之中平均约4.42(=0.13×34)个氨基酸残基形成了α-螺旋结构。此处,关于本发明的液态药物制剂中含有的成分1的α-螺旋结构的形成部位和其量,可以存在多种分子且其间可以呈动态平衡,本发明的液态药物制剂中含有的多种成分1也可以显示出实质上相同的α-螺旋结构的形成部位和其量。在任一情况下,α-螺旋含量均是指成分1中的形成α-螺旋结构的氨基酸残基数相对于成分1所具有的全部氨基酸残基数的比例。本发明中,液态药物制剂中含有的成分1的α-螺旋含量可通过例如圆偏振光二色性(cd)光谱测定法进行推算(参照非专利文献10~11等)。例如,优选将含有成分1的液态药物制剂作为试样,获得222nm测定波长下的圆偏振光二色性(cd)光谱测定值([mdeg]),将该测定值换算为平均残基摩尔椭圆率([deg.cm2/dmol]),使用所得到的平均残基摩尔椭圆率的数值a由以下的数学式推算出成分1的α-螺旋含量。[数1]α-螺旋含量=-(数值a+2340)/30300(非专利文献10)测定条件没有特别限定,例如可以在以下的条件下进行测定。1)测定波长222nm2)试样浓度(成分1浓度)0.1~0.3mg/ml3)温度20℃4)比色皿长1~2mm试样容量可以适当选择,例如可以为0.5ml左右。用于测定cd光谱的装置没有特别限定,例如可以利用圆二色性分散计(日本分光株式会社销售;j-720)。另外,在液态药物制剂中包含高浓度的氨基酸等作为添加物的情况下,背景增高,结果可能使利用圆偏振光二色性(cd)光谱测定法进行的α-螺旋含量的测定变得困难。这样的情况下,也可以使用例如核磁共振法(nmr)来代替cd光谱测定法。但是,通常在由圆偏振光二色性(cd)光谱测定结果来推算成分1的α-螺旋含量的情况下,根据该推算中使用的推算式,α-螺旋含量的推算值可能不同。另外,即使在以同一液态药物组合物作为对象的情况下,基于nmr法的α-螺旋含量的推算值和基于cd法的α-螺旋含量的推算值也可能不同,例如,根据基于cd法的α-螺旋含量推算时所使用的推算式,前者可能比后者更高。因此,在使用nmr法的情况下,优选以利用cd光谱测定法推算出α-螺旋含量的液态药物制剂作为对照品,对于该对照品获得由nmr得到的cα的化学位移,采用基于两测定法的含量的背离来进行数值校正。此外,还可以使用atr-ftir(傅利叶转换红外分光的全反射测定法)、ir(红外分光法、参照非专利文献16)、拉曼分光法等方法对液态药物制剂中含有的成分1的α-螺旋含量进行测定。但是,在应用这些测定法的情况下,需要按照以至少1%(w/v)以上的浓度含有成分1的方式来制备作为测定对象的被检组合物。在利用nmr法进行成分1的α-螺旋含量测定时,优选将供于试验的液态药物制剂中的成分1的浓度适当调整为适于测定的浓度(非专利文献25)。例如,可以按照使成分1的浓度为0.5~4mm的方式适当调节液态药物制剂中的成分1的浓度以利用nmr法实施测定。本发明的液态药物制剂中含有的成分1的α-螺旋含量没有特别限定,优选为13%以上。其中可更优选例示出13.5%以上、或13.8%以上。通过使液态药物制剂中含有的成分1的α-螺旋含量为上述下限值以上,可得到显示出优异的药物动力学的液态药物制剂。本发明的液态药物制剂中含有的成分1的α-螺旋含量通常满足上述下限值(13%以上、13.5%以上、13.8%以上等)即可,其上限没有特别限制,可优选例示出例如100%以下、80%以下、60%以下、50%以下、40%以下、30%以下、25%以下、20%以下、18%以下、16%以下、或15.8%以下。本发明的液态药物制剂中含有的成分1的形成α-螺旋的氨基酸残基数没有特别限定,可以从4个以上的范围中选择,可以为4.2个以上、4.4个以上、4.42个以上、4.5个以上。其中,可更优选例示出4.59个以上、4.6个以上、4.69个以上、4.7个以上。通过使液态药物制剂中含有的成分1的形成α-螺旋的氨基酸残基数为上述下限值以上,可得到显示出良好的药物动力学的皮下给药用液态药物制剂。本发明的液态药物制剂中含有的成分1的形成α-螺旋的氨基酸残基数通常满足上述下限值(4.2个以上、4.5个以上等)即可,其上限没有特别限制,例如可以为34个以下、30个以下、25个以下、20个以下、18个以下、16个以下、15个以下、12个以下、10个以下、9个以下、8个以下、7个以下、6.8个以下、6.5个以下、6.1个以下、5.5个以下、5.44个以下、5.4个以下、5.37个以下。本发明的液态药物制剂所显示的基于圆偏振光二色性(cd)光谱测定(测定波长222nm)的平均残基摩尔椭圆率[θ]的上限没有特别限定,例如可以为-6000以下、-6100以下、-6300以下、-6400以下,其中可优选例示出-6300以下。同样地,对其下限也没有特别限定,例如可优选例示出-8000以上、-7500以上、-7300以上、-7200以上、或-7100以上。通过使液态药物制剂所显示的基于圆偏振光二色性(cd)光谱测定(测定波长222nm)的平均残基摩尔椭圆率[θ]为上述上限以下,可得到显示出良好的药物动力学的皮下给药用液态药物制剂。需要说明的是,本发明中,对于液态药物制剂中的成分1的α-螺旋含量或形成α-螺旋的氨基酸残基数进行调整或使其增加的手段没有特别限制,可例示出:使本发明的液态药物制剂实质上不含有缓冲剂;适当添加离子性化合物或离子性物质(氯化钠等);调节ph;等等(还可参照后述的实施例1中“(2)供于人药物动力学试验的液态药物制剂的制备”和后述的实施例3~4、非专利文献18、非专利文献20等)。或者,可以通过降低本发明的液态药物制剂的极性的手段(具体地说通过向组合物中添加各种醇类)而增加组合物中的成分1的α-螺旋含量和形成α-螺旋的氨基酸残基数。作为α-螺旋形成能力强的醇类,已知有三氟乙醇(tfe)(非专利文献19),但也可以通过向本发明的液态药物制剂中添加作为医药品添加物使用的异丙醇或乙醇来代替tfe,由此增加组合物中的成分1的α-螺旋含量和形成α-螺旋的氨基酸残基数。另外,也可以通过向本发明的液态药物制剂中添加钙离子(ca2+)而增加组合物中的成分1的α-螺旋含量和形成α-螺旋的氨基酸残基数(非专利文献20)。添加量没有特别限定,优选添加成分1浓度的约100~1000倍左右的ca2+。需要说明的是,在专利文献5中公开了,在向药液中添加乙酸钠缓冲液时,与不添加时相比,可提高药液中的生理活性肽的生物学利用率(ba)(实施例2)。另一方面,本发明的液态药物制剂中,即使实质上不含有缓冲剂(更具体地说为乙酸缓冲剂),也可得到良好的药物动力学。(5)特立帕肽或其盐(成分1)的tmax:将本发明的液态药物制剂向皮下单次给药时所得到的成分1的达到最高血浆浓度的时间(tmax;hr)没有特别限定,可适宜地例示出以下时间。即,作为通过不依赖于药物动力学模型的分析(nca(noncompartmentalanalysis);非房室模型分析)计算出的tmax,优选为0.75(hr)以下,更优选为0.7(hr)以下、0.65(hr)以下、0.625(hr)以下、0.6(hr)以下或0.5(hr)以下。另外,作为通过不依赖于药物动力学模型的分析(nca(noncompartmentalanalysis);非房室模型分析)计算出的tmax,更优选为0.1(hr)以上、0.2(hr)以上、0.25(hr)以上、0.3(hr)以上、0.4(hr)以上或0.5(hr)以上。其中优选为0.5~0.7(hr)、0.5~0.625(hr)。通过使成分1的tmax处于上述范围内,可优选地显示出伴随单次给药的优异的安全性。或者,作为通过1-房室(药物动力学)模型分析计算出的tmax,优选为0.6(hr)以下,更优选为0.55(hr)以下或0.5(hr)以下。另外,作为通过1-房室(药物动力学)模型分析计算出的tmax,更优选为0.1(hr)以上、0.2(hr)以上、0.25(hr)以上、0.3(hr)以上或0.35(hr)以上。其中优选为0.3~0.6(hr)、0.35~0.5(hr)。通过使成分1的tmax处于上述范围内,可优选地显示出伴随单次给药的安全性。对于使将本发明的液态药物制剂向皮下单次给药时所得到的成分1的tmax处于上述范围内的方法没有特别限定。关于tmax,在表征药剂的药物动力学的药剂的吸收、分布、代谢、排泄(有时根据absorption(吸收)、distribution(分布)、metabolism(代谢)、elimination(排泄)各自的首字母称为adme)中,大致由药物的吸收速度常数(ka)和消失速度常数(kel)限定,若使用代表性的模型,则可由下述式计算出。[数2]tmax=ln(ka/kel)/(ka-kel)(其中ka≠kel)本发明的实施例中,与将公知的成分1制剂向皮下单次给药时所得到的成分1的tmax相比,将本发明的液态药物制剂向皮下单次给药时所得到的成分1的tmax显示出更小的值;与ka相比,据信kel对制剂的组成依赖性更低;考虑到这些方面,本发明中,作为用于使成分1的tmax处于上述范围内的方法,可优选例示出提高成分1的ka(即提高成分1的吸收速度)的方法。可认为本发明的液态药物制剂所含有的成分1的离子化具有对皮下给药时的成分1的吸收带来影响的可能性。因此,为了使成分1的tmax处于上述范围内,可以将本发明的液态药物制剂所含有的成分1制成与1种或2种以上的挥发性有机酸的盐,或者也可以参考后述的实例适当调节本发明的液态药物制剂的ph。另外,为了使成分1的tmax处于上述范围内,也可以参考后述的实例适当选择本发明的液态药物制剂的添加剂。另外,为了使成分1的tmax处于上述范围内,优选将本发明的液态药物制剂所含有的成分1的浓度在上述范围内适当调节,例如,可以使该浓度为80~240μg/ml、100~200μg/ml、109~190μg/ml或120~160μg/ml。通常已知药物的分子量、药剂中的添加剂、麻醉、温热、挤压等会对皮下施用药物的吸收速度、吸收量带来影响(非专利文献30)。另外,通过提高液态药物制剂所含有的成分1的浓度,在将液态药物制剂向给药对象的皮下给药时所得到的成分1的吸收速度常数(ka)增大(非专利文献26)。通过增大成分1的ka,可缩短成分1的tmax。另外,本发明中,在给药对象为人的情况下,为了使成分1的tmax处于上述范围内,也可以使施用本发明的液态药物制剂的人例如为闭经后女性。本发明的液态药物制剂向皮下单次给药时所得到的成分1的tmax可以通过本身公知的方法进行测定来确认。对皮下给药的部位没有特别限定,优选神经和血管的分布少、皮下脂肪多、无骨的部位。作为这样的部位,可优选举出腹部、上臂部、大腿部、臀部,最优选腹部。在对成分1的tmax进行测定时,优选确保充分的测定时刻数。例如,优选如后述的实施例中的各种评价步骤那样例如在给药前、给药5、15、30以及45分钟后以及1、1.5、2、3、4以及6小时后采集血液试样,对血浆中的成分1的浓度进行测定。(6)维持大于特定阈值的特立帕肽或其盐(成分1)浓度的时间:通常,药剂的血中浓度越高,该药剂的效果越倾向于增强。例如,在为时间依赖性的抗菌药的情况下,大于mic的时间(timeabovemic)(在高于最小发育浓度(mic)的血中浓度下推移的时间)对于其作用是很重要的。另一方面,已知特立帕肽参与体内的钙稳态,高血钙水平是伴随特立帕肽给药的恶心的原因之一(非专利文献23)。另外,通过特立帕肽的反复给药,由于这样的特立帕肽的生理活性而使高血钙水平得以维持和增强,作为结果,有时也要考虑高钙血症或高钙尿症等副作用风险。本发明中,作为一个方式,举出了向皮下单次给药后的特立帕肽或其盐的血浆中浓度为特定阈值以上的状态的经过时间呈特定范围内的液态药物制剂,作为特定阈值可以例示出2个方式。设一者为特定阈值a、另一者为特定阈值b时,特定阈值a和特定阈值b均没有特别限定,作为特定阈值a,优选为50(pg/ml)以上,可以为60(pg/ml)以上或80(pg/ml)以上,作为特定阈值a的上限,优选为200(pg/ml)以下、150(pg/ml)以下或120(pg/ml)以下。作为特定阈值a的适宜例,可优选举出100(pg/ml)。通过使血浆中成分1浓度为特定阈值a以上的状态的经过时间为特定范围内,可优选地抑制伴随单次给药的血中钙浓度的上升。血中钙浓度的上升抑制可有助于消化器官副作用发生频率和/或高钙血症/尿症发症风险的降低。此处,经过时间的特定范围没有特别限定,可以为3小时以内,优选为小于2.5小时、小于2.1小时、小于2.0小时、小于1.73小时、小于1.7小时、小于1.5小时、进而小于1.0小时。其下限也没有特别限定,可以为0.5小时以上、0.7小时以上、进而为0.8小时以上。其中,更优选为小于2.1小时、0.7~2.1小时、小于1.7小时或0.7~1.7小时。特定阈值b也如上所述没有特别限定,优选为100(pg/ml)以上,可以为150(pg/ml)以上或200(pg/ml)以上,作为上限,优选为500(pg/ml)以下、400(pg/ml)以下或300(pg/ml)以下。作为特定阈值b的优选例,可优选举出250(pg/ml)。通过血浆中成分1浓度为特定阈值b以上的状态的经过时间为特定范围内,可优选地显示出伴随单次给药的优异的安全性(特别是消化道副作用出现的频率得以抑制的安全性)。上述的经过时间没有特别限定,可以小于1.4小时,优选可以小于1.3小时、小于1.2小时、小于1.1小时、小于1.0小时、进而可以小于0.9小时、小于0.8小时或小于0.7小时。对其下限也没有特别限定,可以为0.0以上、进而为0.1小时以上。其中,更优选小于0.8小时、0.1小时以上。将本发明的液态药物制剂向皮下单次给药时,通常认为,伴随着每一次的成分1的给药量的增减,成分1的血浆中浓度也倾向于增减。因此,为了使血浆中成分1浓度为特定阈值以上的状态的经过时间为特定范围内,优选将每一次的成分1的给药量在上述的范围内适当调节,最优选以特立帕肽计为28.2μg。可认为本发明的液态药物制剂所含有的成分1的离子化具有对皮下给药时的成分1的吸收带来影响的可能性。因此,出于对血浆中成分1浓度为特定阈值以上的状态的经过时间进行调节的目的,可以将本发明的液态药物制剂所含有的成分1制成特立帕肽与1种或2种以上的挥发性有机酸的盐、或者可以参考后述的实例适当调节本发明的液态药物制剂的ph。另外,出于同样的目的,也可以参考后述的实例适当选择本发明的液态药物制剂的添加剂。另外,为了使成分1的从达到上述特定阈值的时刻起直至低于该值的时刻为止的经过时间为上述经过时间,优选将本发明的液态药物制剂所含有的成分1的浓度在上述的范围内适当调节,例如可以使该浓度为80~240μg/ml、100~200μg/ml、109~190μg/ml或120~160μg/ml。在成分1的特定阈值存在2个的方式(特定阈值a、b,此处设b>a)的情况下,尽管可通过进一步减小成分1的tmax从而进一步缩短从达到特定阈值a的时刻起直至低于该值的时刻为止的经过时间(经过时间a),但在过度减小tmax时,可能会进一步延长从达到特定阈值b的时刻起直至低于该值的时刻为止的经过时间(经过时间b)。因此,这样的情况下,优选通过兼顾着缩短经过时间a和经过时间b这两者的时间而使基于单次给药的安全性变得适宜,更具体地说,例如优选使本发明的液态药物制剂所含有的成分1的浓度处于上述的浓度范围、或者使成分1的tmax处于上述的时间范围。通过提高液态药物制剂所含有的成分1的浓度,在将液态药物制剂向给药对象的皮下给药时所得到的成分1的吸收速度常数(ka)增大(非专利文献26)。通过增大成分1的ka,成分1的tmax缩短,作为结果,可使血浆中成分1的浓度的消失相的斜率增大(即,由于翻转(flip-flop)现象倾向于被消除,因而可使消失相的斜率接近消失速度常数)。通过缩短成分1的tmax和增大血浆中成分1的浓度的消失相的斜率,可使成分1的从达到上述特定阈值的时刻起直至该低于值的时刻为止的经过时间缩短。本发明中,在给药对象为人的情况下,被施用本发明的液态药物制剂的人的性别优选为女性,优选年龄为45岁以上(优选50岁以上)、体重为42~62kg(优选45~60kg)。另外,本发明中,在给药对象为人的情况下,为了对于血浆中成分1浓度为特定阈值以上的状态的经过时间进行调节,也可以使被施用本发明的液态药物制剂的人例如为闭经后女性(非专利文献27)。或者,本发明中,在给药对象为人的情况下,也可以根据被施用本发明的液态药物制剂的人的体重等基于医生等的判断适当调节给药量。本发明的液态药物制剂向皮下单次给药时所得到的血浆中成分1浓度可以利用本身公知的方法进行测定来确认(参照图6)。对皮下给药的部位没有特别限定,优选神经和血管的分布少、皮下脂肪多、无骨的部位。作为这样的部位,可优选举出腹部、上臂部、大腿部、臀部,最优选腹部。在对血浆中成分1浓度进行测定时,优选确保充分的测定时刻数。例如,优选如后述的实施例中的各种评价步骤那样例如在给药前、给药5、15、30以及45分钟后以及1、1.5、2、3、4以及6小时后采集血液试样,对血浆中的成分1的浓度进行测定。(7)ph、添加剂、缓冲剂:本发明中的液态药物制剂的ph没有特别限定,可优选例示出以下ph。即,液态药物制剂的ph例如优选为3.5以上、4.0以上、大于4.0、4.2以上或4.4以上。液态药物制剂的ph例如优选为6.0以下、5.5以下、5.0以下、小于5.0、4.9以下或4.8以下。其中优选为5.0以下、进而优选为4.0以上5.0以下、4.0以上且小于5.0、4.2以上且小于5.0,最优选为4.4以上4.9以下。通过使本制剂的ph为上述范围,可有效地得到优异的稳定性(例如成分1的脱酰胺体或切断体(31-34)的生成的生成抑制性等)和/或药物动力学。另外,也可以使本发明的液态药物制剂含有各种添加物。作为添加物,例如可以举出增溶剂、稳定剂、等渗剂、ph调节剂、防腐剂(保存剂)等。作为添加物,例如可例示出氯化钠、d-甘露醇、蔗糖、l-蛋氨酸。作为ph调节剂,例如可以举出盐酸、氢氧化钠。另外,在本发明的液态药物制剂中可以包含在医药领域中通常使用的缓冲剂。或者,本发明的制剂也可以为实质上不含有缓冲剂的液态药物制剂,其中,通过为实质上不含有乙酸缓冲剂的液态药物制剂,可优选地有效得到优异的药物动力学。本发明的液态药物制剂含有至少一种以上的无机盐和/或有机盐的情况下,对其浓度没有特别限定,优选为2mg/ml以上,更优选为3mg/ml以上,其中进一步优选为5.5mg/ml以上。另一方面,优选为25mg/ml以下,其中更优选为11mg/ml以下。在本发明的液态药物制剂含有至少一种以上的无机盐和/或有机盐的情况下,其相对于特立帕肽或其盐的质量比(成分1:成分2的质量比)没有特别限定,作为下限,例如优选为1:5以上,更优选为1:10以上或1:15以上,其中进一步优选为1:20以上,最优选为1:35以上。另一方面,作为上限,例如优选为1:500以下,更优选为1:300以下,最优选为1:80以下。本发明的液态药物制剂的ph可以通过本身公知的方法、例如使用缓冲剂或ph调节剂进行调整。另外,作为本发明的液态药物制剂的一个方式,可例示除了作为每一次的给药量含有以特立帕肽换算计为28.2μg或56.5μg的特立帕肽醋酸盐、进而将含有氯化钠和精制白糖的冻干制剂排除在外的液态药物制剂。此外,作为本发明的液态药物制剂的一个方式,还可以举出将含有冰乙酸、乙酸钠(可以为无水物)和d-甘露醇、ph为3.8~4.5(例如ph为4.1)的液态药物制剂排除在外的液态药物制剂。或者、作为本发明的液态药物制剂的一个方式,还可例示出将作为每一次的给药量含有以特立帕肽换算计为28.2μg或56.5μg的特立帕肽醋酸盐的冻干制剂排除在外的液态药物制剂。另外,作为本发明的液态药物制剂的一个方式,可例示出将含有成分1和单糖类(例如甘露醇、葡萄糖、山梨糖醇、肌醇)的冻干制剂排除在外的液态药物制剂。或者,作为发明的液态药物制剂的一个方式,可以例示出将含有成分1和木糖醇的液态药物制剂排除在外的液态药物制剂。(8)冷冻干燥:本发明的液态药物制剂可以包含由冻干制剂重新构成的液态药物制剂的方式,也可以不是由冻干制剂重新构成的液态药物制剂。以往已知含有特立帕肽或其盐的冻干制剂、在使用时被溶解(再溶解)在生理盐水等中进行制备而制成液态药物制剂,本发明的液态药物制剂可以为这样的冻干制剂的再溶解品(用时制备品),也可以为不经历冻干制剂的制剂(预先制成液体制剂的制剂)。本发明中可以提供及时不经历冻干制剂也具有良好的药物动力学的制剂。(9)药物动力学:在本发明的皮下给药用液态药物制剂的一个方式中,特立帕肽或其盐(成分1)的α-螺旋含量为特定范围内(例如,13.0%以上)。另外,在本发明的皮下给药用液态药物制剂的一个方式中,成分1中的形成α-螺旋的氨基酸残基数为特定范围内(例如4.5个以上)。利用这样的皮下液态药物制剂可得到优异的药物动力学。在将液态药物制剂对于人、猴子等哺乳动物进行给药时,有多少量会到达全身循环血而起到作用是个重要的问题。通常,在将液态药物制剂进行静脉给药的情况下,上述制剂中的药物大致完全被生物体利用;但在非静脉给药(经口、直肠、经皮、皮下等)给药时,并非全部会到达循环血。作为测定到达全身循环血的量的指标,多采用auc(血浆中浓度-时间曲线下面积)。另外,有时也将通过非静脉给药得到的auc相对于通过静脉给药得到的auc的比例作为(绝对)生物学利用率(%)来对药物的生物体利用度进行评价。改善基于非静脉给药的auc、生物学利用率等药物动力学参数对于提高药物所赋予的治疗效果、安全性等是很重要的。液态药物制剂的药物动力学可以以各种药物动力学参数作为指标进行评价。作为药物动力学参数的示例,可优选例示出达到最高血浆浓度的时间(tmax)、最高血浆中浓度(cmax)、血浆中浓度-时间曲线下面积(auc)以及生物学利用率(%)等。作为auc没有特别限定,可以举出例如aucinf(直至无限大时间为止的血浆中浓度-时间曲线下面积)、auclast(截至最终观察时间为止的血浆中浓度-时间曲线下面积)以及在反复给药时在每1次给药间隔所得到的aucτ(从时间为零时起直至给药间隔时间τ为止的血浆中浓度-时间曲线下面积)等。在进行药物动力学参数的评价时,对给药的部位没有特别限定,优选神经和血管的分布少、皮下脂肪多、无骨的部位。作为这样的部位,可优选举出腹部、上臂部、大腿部、臀部,最优选腹部。药物动力学参数的计算方法也没有特别限制,使用不依赖于药物动力学模型的分析和依赖于药物动力学模型的分析方法(例如1-房室模型)中的任一种均可进行计算(非专利文献6)。但优选通过不依赖于药物动力学模型的分析方法即nca(非房室模型,noncompartmentalanalysis)进行计算。作为nca中的auc计算方法,可以举出线性梯形法(lineartrapezoidalrule)、对数线性梯形法(loglineartrapezoidalrule)。例如也可以在直至达到最高血浆浓度的时间(tmax)为止的吸收相中使用线性梯形法计算出auc,在tmax以后的消失相中使用对数线性梯形法计算出auc。在计算药物动力学参数时,优选确保充分的测定时刻数。例如可以如后述的实施例中的各种评价步骤那样在例如给药前、给药5、15、30以及45分钟后以及1、1.5、2、3、4以及6小时后采集血液试样,对血浆中的特立帕肽或盐的浓度进行测定。为了计算出液态药物制剂的药物动力学参数,优选确保充分的例数。各药物动力学参数可以为通过将各病例所示出的数值相加所得到的和值除以例数而得到的平均值(mean)、或者可以为将各病例所示出的数值进行排序而得到的位于其中央的中央值(median)。为了得到两种以上的液态药物制剂的药物动力学参数,可以使用组间比较试验、交叉试验。特立帕肽比较容易洗脱,另外,从能够压缩例数的方面出发,为了得到两种以上的液态药物制剂的药物动力学参数的目的,优选应用交叉试验。作为药物动力学的指标,例如可以通过下述式计算出成分1的绝对生物学利用率(%)。[数3]成分1的绝对生物学利用率(%)=[{(基于皮下给药而得到的成分1的aucinf)×(基于静脉给药的成分1的给药量)}]/[{(基于静脉给药而得到的成分1的aucinf)×(基于皮下给药的成分1的给药量)}]×100(%)根据aucinf的测定误差等,有时会得到超过作为理论上限的100%的绝对生物学利用率(%)。成分1的绝对生物学利用率(%)没有特别限定,可适宜地例示出以下数值。即,例如优选为70%以上、80%以上、90%以上、95%以上、100%以上、110%以上。另外,作为上限,例如优选为180%以下、160%以下、150%以下。其中优选为90%以上且160%以下,最优选为100%以上且150%以下。成分1的cmax没有特别限定,可适宜地例示出以下数值。即,优选为230(pg/ml)以上、240(pg/ml)以上、250(pg/ml)以上。另外,作为上限,例如优选为380(pg/ml)以下、360(pg/ml)以下、350(pg/ml)以下。其中优选为250~350(pg/ml)。成分1的auclast没有特别限定,可适宜地例示出以下数值。即,优选为350(hr·pg/ml)以上、360(hr·pg/ml)以上、370(hr·pg/ml)以上、380(hr·pg/ml)以上、390(hr·pg/ml)以上。另外,作为上限,例如优选为600(hr·pg/ml)以下、580(hr·pg/ml)以下、570(hr·pg/ml)以下、550(hr·pg/ml)以下、530(hr·pg/ml)以下。其中优选为350~550(hr·pg/ml)。成分1的aucinf没有特别限定,可适宜地例示出以下数值。即,优选为380(hr·pg/ml)以上、390(hr·pg/ml)以上、400(hr·pg/ml)以上、420(hr·pg/ml)以上。另外,作为上限,例如优选为650(hr·pg/ml)以下、600(hr·pg/ml)以下、590(hr·pg/ml)以下、580(hr·pg/ml)以下、560(hr·pg/ml)以下。其中优选为400~600(hr·pg/ml)。优选成分1的绝对生物学利用率(%)、tmax、cmax、auclast、aucinf中的至少一者以上处于上述范围。需要说明的是,通过何种机理而表现出上述突破性的结果尚不明确。在药物动力学方面,例如在临床现场使用的皮下给药用抗体制剂仅有大概50~60%的生物学利用能力,据报告,可能诱导该低生物学利用能力的原因为:制剂中的蛋白质所显示出的电荷或疏水性、制剂中的添加剂成分、给药用量、给药深度呈多样化;带正电的抗体被皮下组织所吸附(非专利文献7),但其中对于制剂中的抗体的二级结构对生物学利用能力所带来的影响没有任何公开或暗示。其他报告中还公开了,将特立帕肽醋酸盐冻干制剂(添加物为精制白糖和氯化钠)利用生理盐水溶解并将所得到的药液(ph5.0~7.0)进行皮下给药时,在大约35~50分钟左右达到最高血中浓度,由aucinf计算出的绝对生物学利用率大致为100%(非专利文献1中的“[药物动力学]2.生物学的利用率”栏),但对于药液中的特立帕肽醋酸盐的二级结构对生物学利用能力所带来的影响没有任何暗示。另一方面,关于特立帕肽的二级结构的方面,例如有下述报告:在水溶液中,特立帕肽的主体柔软且伸长;作为其例外,在自n末端起第20~24位(arg-val-glu-trp-leu)存在非无规的部分结构;利用二维nmr测定几乎观察不到2级结构;等等(非专利文献12),但是,在该文献中根本未暗示特立帕肽的二级结构会对其吸收、代谢、排泄等药物动力学性带来影响。如此地,仅基于现有的报告难以考察特立帕肽的二级结构对其药物动力学特性所带来的影响。在这样的状况下,本发明人在下文中对于通过何种机理获得了上述的突破性的结果进行了考察。皮肤将体内与外界环境隔离而发挥出维持人体恒常性的重要作用,其具有为了发挥出其作用的各种功能,并具有用于实现该功能的复杂结构。若对皮肤进行截面观察,则可知其大致具有表皮-真皮-皮下组织的3层结构。皮下组织中,脂肪组织为主体,起到中性脂肪的贮留、保温功能、缓冲外力的作用等。有提案指出,由于向皮下给药的药物制剂的构成与进行给药的皮下组织的结构不同,因此在皮下给药后,在药物到达血管、淋巴管的期间,各种应力可能影响其稳定性、溶解性、功能(非专利文献8)。此处,如非专利文献8中所述,作为应力的示例,已知有:1)细胞外基质的立体结构障碍、静电相互作用、特定相互作用(表4);2)给药前后的ph变动对于药物制剂中分别包含的添加物针对药物的保护作用带来的影响(27~28页);3)因给药后的添加物的快速移动所致的药物的聚集和皮下组织的吸附(图5的d);4)给药前后的ph变动对药物稳定性带来的影响(29页);5)给药前后的药物附近的温度变动对药物吸收性带来的影响(29页);6)因给药所致的间质液静水压或胶质渗透压的变动对药物稳定性带来的影响(29~30页);等等。本发明人其并不拘泥于此,作为一个理论,认为特立帕肽或其盐中的α-螺旋含量与上述应力中的至少1者相关。关于此处的相关,只要为皮下给药的特立帕肽或其盐显示出优异的药物动力学的机理就没有特别限定,例如可提示特立帕肽或其盐中的α-螺旋或其量1)通过提高血管内皮的透过性而提高其生物学利用率(%)的方式作为一个思路。血管内皮细胞是在全身循环的血管的最内层的细胞,其起到炎症细胞与血管的粘接、血管透过性、凝固/纤溶系的调节等重要的作用。另一方面,已知该α-螺旋与肽的膜透过密切相关(非专利文献28)。因此,作为一个理论,在皮下给药的特立帕肽或其盐中存在一定程度以上的α-螺旋的情况下,与其他情况相比,据信还有下述机理:通过肽的血管内皮细胞的膜透过性上升而提高在血中的转移,结果可提高生物学利用率(%)。另外,例如还可提示特立帕肽或其盐中的α-螺旋或其量2)通过直接或间接地抑制细胞外基质中的各种障碍或相互作用而提高其生物学利用率(%)的方式作为一个思路。细胞外基质是细胞外所存在的超分子结构体,其具有骨架的作用并且提供细胞粘接的落脚点,参与信号传递等。细胞外基质由结构蛋白(胶原蛋白等)或蛋白多糖等构成。蛋白多糖是糖胺多糖(有时也被称为gag)共价键合至作为核心的蛋白质而成的复合体,作为gag,可例示出硫酸软骨素、透明质酸、肝素等。还已知胶原蛋白或gag可引起与皮下给药的药物的特异性相互作用(非专利文献8)。另外已知作为甲状旁腺激素的pth(1-84)通过与肝素或者各种多阴离子性物质的相互作用而诱导α-螺旋(非专利文献29)。gag与各种蛋白质的相互作用被认为可调节病期等的多种生物学现象;肝素与肝素结合性蛋白质发生相互作用时,具有天然结构的该蛋白质更优先进行;基于上述机制等,提供了下述模型:pth(1-84)通过与gag的相互作用而发生α-螺旋等的结构变化,产生了这样的结构变化的pth(1-84)与受体结合(非专利文献29)。因此,作为一个理论,还可以考虑下述机理:在皮下给药的特立帕肽或其盐中存在一定程度以上的α-螺旋的情况下,与其他情况相比,与gag的相互作用减弱,结果可提高生物学利用率(%)。α-螺旋含量对于特立帕肽或其盐与gag的相互作用的影响的机理也没有特别限定,例如也许可以考虑特立帕肽或其盐中的极性/非极性的平衡变化。以前有报告指出,水溶液中的特立帕肽主体柔软且伸长(非专利文献12),因此本发明人推测,其3级结构不存在较大差异的可能性高。此外,本次未确认到水溶液中的特立帕肽的ζ电位与药物动力学之间存在明确关系。根据以上内容,发明人认为,水溶液中的特立帕肽中的α-螺旋含量和形成α-螺旋的氨基酸残基数与药物动力学的关系的确切性变得更明确。特立帕肽中,作为形成α-螺旋的氨基酸残基,可以为n末端的1~34位中的任一者,没有特别限定,例如可以为3~12位、17~26位等。这些氨基酸残基似乎容易形成螺旋结构。因此,本发明的制剂中,这些氨基酸残基数中的至少1者形成α-螺旋即可。特别是这些氨基酸残基(3~12位、17~26位)中,平均4个以上(例如4.2个以上、4.4个以上、4.42个以上、4.5个以上、4.59个以上、4.6个以上、4.69个以上、或4.7个以上)的氨基酸残基形成α-螺旋即可。另外,这些氨基酸残基(3~12位、17~26位)中,平均20个以下(例如18个以下、16个以下、15个以下、12个以下、11个以下、10个以下、9个以下、8个以下、7个以下、6.8个以下、6.5个以下、6.1个以下、5.5个以下、5.44个以下、5.4个以下、或5.37个以下)的氨基酸残基形成α-螺旋即可。另外,氨基酸残基中,选自自n末端起第13位(赖氨酸残基)、14位(组氨酸残基)以及27位(赖氨酸残基)中的至少1个氨基酸残基形成α-螺旋即可。这些残基均为碱性氨基酸残基,因此据推测在向皮下组织给药时带正电。这些氨基酸残基似乎对于上述应力中的任一者的影响均比较强,通过使这些氨基酸残基形成α-螺旋,可有效地得到良好的药物动力学。(10)安全性:在本发明的液态药物制剂的一个方式中,其每一次特立帕肽或其盐的给药量为特定量(例如28.2μg)。或者,在本发明的液态药物制剂的一个方式中,通过其单次给药得到的特立帕肽或其盐达到最高血浆浓度的时间(tmax)为特定范围内(例如小于0.7小时)。或者,在本发明的皮下给药用液态药物制剂的一个方式中,通过其单次给药得到的特立帕肽或其盐的血浆中浓度为特定阈值(例如100pg/ml或250pg/ml)以上的状态的经过时间为特定范围内(例如小于2.1小时、小于1.0小时)。利用这些皮下液态药物制剂可得到优异的安全性。此处,关于安全性,全部的作为不优选的医疗事件的有害现象以及不能否能有害现象与药剂的关系的因果关系的副作用均包含在内。作为严重的有害现象,可以举出死亡、残疾等,本发明中的安全性并不限定于此,其包括在与医药品的有效性(益处)的关系中可对其评价带来影响的全部风险。安全性的种类、程度也没有特别限定,例如为皮肤/皮肤附属器官、肌肉-骨骼、中枢/末梢神经、自主神经、视觉、嗅觉、精神、消化道、肝脏-胆管、代谢/营养障碍、内分泌、心脏/血管、呼吸器官、血细胞/血小板、泌尿器官、生殖器官、全身等所产生的障碍或不优选的症状,也并不限定其强度和频率。可优选例示出消化道副作用、血压降低风险,其中可最优选例示出恶心/呕吐/反胃的频率。医药品多数情况下作为生活习惯病治疗药等长期反复持续使用,该治疗持续对于得到良好的治疗是很重要的。但是,在医药品反复给药时,其谷值上升,由此可能使副作用增强,因这样的副作用增强所引起的治疗遗漏可能会对治疗的功效带来不良影响。或者,在每次进行医药品的给药时医药品的血中浓度一过性地上升,由此可能会频繁地或强烈地表现出副作用,在这样的情况下,作为结果也可能发生治疗遗漏这样的不优选的状况。如此地,在利用医药品时,优选在每次和持续使用的期间均考虑其安全性。换言之,优选医药品具备伴随单次给药的安全性和伴随反复持续给药的安全性这两方面的安全性。与现有的包含特立帕肽的药物制剂相比,本发明的液态药物制剂中,优选伴随单次给药的安全性得以改善。作为改善伴随单次给药的安全性的示例,可优选例示出但并不限于伴随单次给药的消化道副作用频率和/或血压降低风险的降低。(11)性状等:本发明的液态药物制剂优选至少在其制造时为无色澄明的,其相对于生理盐水的渗透压比可以为约1(例如1.0~1.4)。2.液态药物制剂的制法:本发明的液态药物制剂可以通过本身公知的各种制法来制造。通常,适宜地选择构成本发明的液态药物制剂的上述各种成分并与适当的溶剂混合使其溶解即可。在制造本发明的皮下给药用液态药物制剂的情况下,优选制成水性液态药物制剂。在水性液态药物制剂的情况下,优选在给药前实施了无菌处理。作为无菌处理采用无菌操作法的情况下,将所称量的各原料溶解在注射用水等中,将溶解液过滤灭菌,由此可制造出液态药物制剂。注射用水通常被理解为适合于发热性物质(内毒素)试验的灭菌纯净水,通过蒸馏法制造的注射用水有时也被称为注射用蒸馏水。可以将该注射用液态药物制剂进一步填充、密封在进行了清洗、灭菌处理的容器中,经过检査、包装等步骤,制造出填充注射用液态药物制剂而成的注射剂。此处,作为容器,例如可例示出安瓿、小瓶、预充式注射器、袋等。容器的材质没有特别限定,可以举出玻璃、塑料。从强度、处理容易性、安全性等方面出发,可优选例示出塑料作为容器的材质。3.改善药物动力学参数的方法:本发明中,作为一个方式,提供一种方法,其是在将包含成分1的液态药物制剂进行皮下给药时对于上述制剂所示的成分1的药物动力学参数进行改善的方法,其包括对于成分1的α-螺旋含量和/或成分1中的形成α-螺旋的氨基酸残基数进行调整(使其增加等)。本方法例如可通过依次实施以下的工序来实施。工序1)按照成分1的α-螺旋含量为上述的特定范围内(例如13.0%以上)和/或成分1中的形成α-螺旋的氨基酸残基数为上述的特定范围内(例如4.5个以上)的方式来制备含有成分1的液态药物制剂。工序2)将液态药物制剂向人皮下给药,从人采集给药前和给药后的复数个时间点的血液试样。工序3)对各时间点的血液试样所含有的成分1的浓度进行测定。工序4)由各时间点的成分1浓度计算出某一药物动力学参数a的数值a。工序5)将成分1的α-螺旋含量和/或成分1中的形成α-螺旋的氨基酸残基数处于上述特定范围外的含有成分1的液态药物制剂向人皮下给药,将此时得到的药物动力学参数a的数值b与数值a比较,判定数值a是否比数值b良好。在药物动力学参数为成分1的绝对生物学利用率(%)的情况下,该数值的增加意味着药物动力学参数的改善。药物动力学参数为成分1的auclast或aucinf的情况下,该数值的增加意味着药物动力学参数的改善。另外,本发明中,作为一个方式,提供一种改善药物动力学参数的方法,其是将包含特立帕肽或其盐作为成分1的液态药物制剂进行皮下给药时对于上述制剂所表现出的成分1的药物动力学参数进行改善的方法,其特征在于,实施下述操作中的至少一者:1)使成分1的每一次给药量为上述特定量(例如28.2μg);2)使成分1的浓度为特定范围内(例如120~160μg/ml);3)将成分1制成与1种或2种以上的挥发性有机酸的盐;4)对液态药物制剂的ph进行调节;以及5)使制剂适当含有添加剂。此处,药物动力学参数的改善可以通过测定成分1的tmax是否处于上述范围内(例如0.2~0.7(hr))来确认。4.管理品质的方法:本发明中,作为一个方式,提供一种方法,其是对含有成分1的皮下给药用液态药物制剂的品质进行管理的方法,其包括:对液态药物制剂中的成分1的α-螺旋含量和/或成分1中的形成α-螺旋的氨基酸残基数进行测定,将所得到的α-螺旋含量和/或成分1中的形成α-螺旋的氨基酸残基数的测定值与预先设定的基准值比较,在上述测定值为上述基准值以上的情况下,判断维持了液态药物制剂的品质。此处,预先设定的基准值为上述成分1的α-螺旋含量的特定范围下限(例如13.0%以上)。另外,也可以使与基准值进行比较的值为α-螺旋结构形成残基数,此时预先设定的基准值为上述成分1中的α-螺旋结构形成残基范围下限(例如4.5个以上)。或者,也可以使与基准值比较的值为基于圆偏振光二色性(cd)光谱测定(测定波长222nm)的平均残基摩尔椭圆率[θ],此时预先设定的基准值为上述圆偏振光二色性光谱测定平均残基摩尔椭圆率[θ]的范围上限(例如-6300以下)。此处,液态药物制剂的品质例如为液态药物制剂向皮下单次给药时所得到的药物动力学参数。作为药物动力学参数,可优选例示出成分1的绝对生物学利用率(%)、auclast、aucinf等。实施例以下举出实施例更详细地说明本发明。但本发明并不受以下实施例的束缚,可以在不脱离本发明的主旨的范围内以任意的方式实施。需要说明的是,在以下的实施例中,“配方”有时也被描述为与本发明的“液态药物制剂”相当的用语。实施例1(液态药物制剂的制备):(1)供于猴子药物动力学试验的液态药物制剂的制备:按照下述表1~2制备配方a~h。这些配方从其组成的方面出发分别与后述“(2)供于人药物动力学试验的液态药物制剂的制备”的配方a~h为大致相同的配方。按照下述表1制备配方a~d。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂溶液混合,用注射用水使其成为约46ml后,向该混合液中添加2.5ml的特立帕肽醋酸盐溶液(以特立帕肽计为2820μg/ml),制备约48.5ml的药液a。此处,各添加剂溶液和特立帕肽醋酸盐溶液各自的溶剂为注射用水。进而相对于该药液a添加盐酸,由此调整为表中“ph”栏中记载的ph,使用注射用水制备总量50ml的配方。对各配方进行过滤灭菌处理后,在塑料制小瓶中各填充1.5ml,由此制造填充有各配方的塑料制小瓶,供于猴子药物动力学试验。各配方的组成如表中的“最终含量”栏中记载。[表1]表1此外,按照下述表2制备配方e~h。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂与注射用水一起混合,使其总量为3000ml。其后,向该混合液1600ml中添加特立帕肽醋酸盐(以特立帕肽计为282mg),使其溶解,由此制备药液a。进而向该药液a中添加经稀释的盐酸,调整为表中“ph”栏中记载的ph,利用注射用水使其总量为2000ml,制备配方。对各配方进行过滤灭菌处理后,在塑料制小瓶中各填充1.5ml,由此制造填充有各配方的塑料制小瓶,供于猴子药物动力学试验。各配方的组成如表中的“最终含量”栏中记载。[表2]表2(2)供于人药物动力学试验的液态药物制剂的制备:按照下述表3制备配方a~h。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂(其中,l-蛋氨酸为预溶解而成的l-蛋氨酸溶液)与注射用水一起混合,加入特立帕肽醋酸盐(以特立帕肽计为1425.6mg),制备总量9.5kg的药液a。之后向药液a中添加经稀释的盐酸,由此调整为表中“ph”栏中记载的ph,之后使用注射用水制备总量10.10kg的配方。对各配方进行过滤灭菌处理后,在安瓿中各填充2ml,由此制造填充有各配方的安瓿(配方制剂),供于人药物动力学试验。配方制剂为下述的制剂:其配方容量为0.2ml,填充有作为每一次的给药量含有以特立帕肽换算计为28.2μg的特立帕肽醋酸盐的配方。各配方的组成如表中的“最终含量”栏中记载。[表3]表3(3)对照液态药物制剂的制备:(3-1)对照配方1的制备:向市售的特立帕肽冻干制剂(“teribone皮下注射用56.5μg”旭化成制药公司制造;非专利文献1)中加入日本药典生理盐水0.45ml将其溶解,将所得到的药液利用注射器吸取0.2ml,制备对照配方1,将填充有对照配方1的注射器用作对照配方1制剂。需要说明的是,对照配方1为下述的配方:其容量为0.2ml,特立帕肽醋酸盐浓度以特立帕肽换算计为141μg/ml,作为每一次的给药量含有以特立帕肽换算计为28.2μg的特立帕肽醋酸盐。(3-2)对照配方2的制备:制备向市售的特立帕肽冻干制剂(“teribone皮下注射用56.5μg”旭化成制药公司制造;非专利文献1)中加入日本药典生理盐水1.0ml将其溶解而得到的对照配方2,将填充有对照配方2的注射器用作对照配方2制剂。需要说明的是,对照配方2为下述配方:其容量为0.89ml,特立帕肽醋酸盐浓度以特立帕肽换算计为63.5μg/ml,作为每一次的给药量含有以特立帕肽换算计为56.5μg的特立帕肽醋酸盐。(3-3)对照配方3的制备:按照下述表4制备对照配方3。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂与注射用水一起混合,制备总量3000g的溶液a。在2480g的溶液a中溶解特立帕肽醋酸盐(以特立帕肽计为352.5mg),使用溶液a使总量为2500ml,制备对照配方3。将对照配方3过滤灭菌后,向塑料制注射器中各填充0.2ml,将填充有对照配方3的注射器用作对照配方3制剂。需要说明的是,对照配方3为下述的配方:其容量为0.2ml,特立帕肽醋酸盐浓度以特立帕肽换算计为141μg/ml,作为每一次的给药量含有以特立帕肽换算计为28.2μg的特立帕肽醋酸盐。[表4]表4(4)供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备:按照下述表5制备配方a~h。这些配方从其组成的方面出发分别与上述“(2)供于人药物动力学试验的液态药物制剂的制备”的配方a~h为大致相同的配方。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂与注射用水一起混合,制备总量3000ml的溶液a。在1600ml的溶液a中溶解特立帕肽醋酸盐(以特立帕肽计为282mg),制备药液a。其后通过向药液a中添加经稀释的盐酸而调整为表中的“ph”栏中记载的ph,之后使用注射用水制备总量2000ml的配方。对各配方进行过滤灭菌处理后,在2ml的安瓿中各填充2ml,制造填充有各配方的安瓿(配方安瓿制剂),供于填充容器相关的稳定性试验。另外,对各配方进行过滤灭菌处理后,在塑料制注射器中各填充0.2ml,制造填充有各配方的塑料制注射器(配方注射器制剂),供于填充容器相关的稳定性试验。各配方的组成如表中的“最终含量”栏中记载。[表5]表5(5)供于稳定性试验的液态药物制剂的制备:按照下述表6制备配方a、b、e、f以及h。各配方的具体制备方法如下。首先,将表中的“添加剂”栏中记载的各添加剂与注射用水一起混合,制备总量3000ml的溶液a。在1600ml的溶液a中溶解特立帕肽醋酸盐(以特立帕肽计为282mg),制备药液a。其后向药液a中添加经稀释的盐酸,由此调整为表中“ph”栏中记载的ph,之后使用上述的溶液a制备总量2000ml的配方。对各配方进行过滤灭菌处理后,在2ml的安瓿中各填充2ml,制造填充有各配方的安瓿(配方安瓿制剂),供于稳定性试验。另外,对各配方进行过滤灭菌处理后,在塑料制注射器中各填充0.2ml,制造填充有各配方的塑料制注射器(配方注射器制剂),供于稳定性试验。各配方的组成如表中的“最终含量”栏中记载。[表6]表6实施例2(猴子药物动力学试验):(1)试验方法:分别使用上述实施例1的“(1)供于猴子药物动力学试验的液态药物制剂的制备”中制备的配方a~h以及上述实施例1的“(3)对照液态药物制剂的制备”中制备的对照配方1以及对照配方3来实施猴子药物动力学试验。将配方a~h、对照配方1和对照制剂3皮下给药至4~6岁的雌性食蟹猴,在给药后5、15、30、60、120以及180分钟的时间点从大腿静脉采血。pk试验分成2个试验(试验1和2)实施。各试验为交叉设计,在各期之间间隔有适当设定的休药期间。在各试验中使用6只动物。从通过这些的采血得到的血液中通过离心分离收集血浆,通过elisa法(高灵敏度人pth(1-34)elisa试剂盒、immutopicsinc.)测定血浆中的特立帕肽浓度。基于通过测定得到的血浆中特立帕肽浓度计算出血浆中浓度-时间曲线下面积(auc)。(2)试验结果:将试验结果记载于下述表7和8中。[表7]表7:试验1药物动力学的参数[表8]表8:试验2药物动力学的参数如上述表7和8所示显示出,将配方a、b、e、f以及h进行皮下给药时的auc比将对照配方3进行给药时的auc增加。另外显示出,将配方b进行给药时的auc比对照配方1增加。由以上的结果确认到,配方a、b、e、f以及h比对照配方3在食蟹猴中显示出了更好的体内动态。实施例3(人药物动力学试验):(1)试验(1)方法:使用上述实施例1的“(3)对照液态药物制剂的制备”中制备的对照配方2、3制剂,实施人药物动力学试验(1)。具体地说,在非盲检条件下在12例健康闭经后女性中实施药物动力学试验,将对照配方3向腹部、大腿部或上臂部进行单次皮下给药,将其中向腹部给药时的药物动力学参数与将对照配方2向上臂部皮下给药时的药物动力学参数进行比较。血浆中特立帕肽醋酸盐浓度利用在配方给药前、给药5、15、30、45分钟后、1、1.5、2、3、4以及6小时后采集的血液试样进行测定。由血浆中特立帕肽醋酸盐浓度通过不基于模型的方法根据下述式针对每一受试对象计算出药物动力学参数auclast、aucinf和cmax。auclast=基于线性梯形法(lineartrapezoidalrule)的截至最终观测时间为止的血浆中浓度-时间曲线下面积(人药物动力学试验试验(2)中的auclast也为相同定义)aucinf=基于线性梯形法(lineartrapezoidalrule)的截至无限大时间的血浆中浓度-时间曲线下面积(人药物动力学试验试验(2)中的aucinf也为相同定义)cmax=最高血浆中浓度(人药物动力学试验试验(2)中的cmax也为相同定义)对于所计算出的auclast、aucinf和cmax,按以下方法计算出对照配方3相对于对照配方2之比和95%信赖区间。对于对数转换后的auclast、aucinf和cmax,将受试对象(顺序组内)作为变量效果,将顺序组、制剂(对照配方2~3制剂)设为固定效果,使用基于混合效果模型的分散分析法进行分析。将所推定的制剂之差和95%信赖区间进行指数转换,以各配方之比和信赖区间的形式示出。另外,作为安全性评价项目,设置有害现象、临床检査(血液检査、生物化学检査、尿检査、免疫检査)、生命体征(腋窝体温、收缩/舒张压、脉搏数)、12导联心电图、体重,实施基于对照配方2、3制剂给药的安全性评价。将12例受试对象随机地分割成4组每组3例,经历4期按照下述表9的计划实施试验。[表9]表9组第1期第2期第3期第4期1对照配方2对照配方3(*)对照配方3(**)对照配方3(***)2对照配方3(**)对照配方2对照配方3(***)对照配方3(*)3对照配方3(*)对照配方3(***)对照配方2对照配方3(**)4对照配方3(***)对照配方3(**)对照配方3(*)对照配方2(*)腹部给药、(**)大腿部给药、(***)上臂部给药(2)试验(1)结果:将试验结果示于下述表10和11。将对照配方3进行皮下给药时cmax为将对照配方2进行皮下给药时的约1/2,auclast和aucinf为约1/4(表11)。[表10]表10:药物动力学的参数(***)上臂部给药[表11]表11:各药物动力学的参数相关的对照配方3相对于对照配方2之比和其95%的信赖区间(***)上臂部给药(3)试验(2)方法:分别使用上述实施例1的“(2)供于人药物动力学试验的液态药物制剂的制备”中制备的配方a~h制剂以及上述实施例1的“(3)对照液态药物制剂的制备”中制备的对照配方2制剂实施人药物动力学试验(2)。受试对象为24名健康闭经后女性。试验通过在非盲检条件下对于将配方a~h向腹部单次皮下给药得到的药物动力学参数与将对照配方2向上臂部进行皮下给药得到的药物动力学参数进行比较来实施。本试验以2个队列进行,将i、ii、iii以及iv组作为队列1,将v、vi、vii以及viii组作为队列2。对于每一队列,将12例随机地分成4组、每组各3例。按照下述表12所示的给药计划,将配方a~h和对照配方2对受试对象进行给药。表12中的“-”是指配方a~h和对照配方2均未给药。在各期进行1次给药,各期的天数按照本试验的目的适当设定。[表12]表12组第1期第2期第3期第4期第5期第6期i对照配方2配方a配方b配方c配方d-ii对照配方2配方c配方a配方d配方b-iii对照配方2配方e配方f配方g配方h-iv对照配方2配方g配方e配方h配方f-v-配方b配方d配方a配方c对照配方2vi-配方d配方c配方b配方a对照配方2vii-配方f配方h配方e配方g对照配方2viii-配方h配方g配方f配方e对照配方2血浆中特立帕肽醋酸盐浓度利用在配方给药前、给药15、30、45分钟后、1、1.5、2、3、4以及6小时后采集的血液试样进行测定。由血浆中特立帕肽醋酸盐浓度通过不基于模型的方法针对每一受试对象计算出药物动力学参数auclast、aucinf和cmax。对于所计算出的auclast、aucinf和cmax,按照以下方法计算出配方a~h相对于对照配方2之比和95%信赖区间。首先对计算出的auclast、aucinf和cmax进行对数转换,接着,对于对数转换后的auclast、aucinf和cmax,将受试对象(顺序组内)作为变量效果,将顺序组、配方设为固定效果,使用基于混合效果模型的分散分析法进行分析。将所推定的各配方之差和95%信赖区间进行指数转换,以各配方之比和信赖区间的形式示出。此外,使用与配方a~h不同的特立帕肽醋酸盐制剂实施其他人的药物动力学试验,使用所得到的aucinf(11.4ng·min/ml)(非专利文献24;2.7.1.2.2生物利用度)以及由上述配方a~h和对照配方3计算出的aucinf,按照下述式计算出血浆中特立帕肽的绝对生物学利用率(%)。[数4]成分1的绝对生物学利用率(%)=[{(基于皮下给药而得到的成分1的aucinf)×(基于静脉给药的成分1的给药量)}]/[{(基于静脉给药而得到的成分1的aucinf)×(基于皮下给药的成分1的给药量)}]×100(%)需要说明的是,上述的其他药物动力学试验是以下述步骤为方法的临床药理试验:对于健康的30岁年龄段和60岁年龄段的男性各5例,将以特立帕肽计含有14.1μg的特立帕肽醋酸盐制剂进行3分钟静脉内持续给药。另外对于进行了配方a~h给药的受试对象(各配方为12例病例)和进行了对照配方2给药的受试对象(计24例病例)中的副作用表现进行了观察。作为副作用表现率(%),设其为将表现出各副作用的人数除以给药人数并乘以100而得到的值。进一步对于进行了配方a~h和对照配方2给药的受试对象中的血清钙值上升进行了观察。血清钙值上升为给药后经过6小时后的血清钙值与给药前的血清钙值之差(平均值)。关于tmax,也以各被给药者的血浆中特立帕肽醋酸盐浓度达到最高的时间的平均值的形式计算出。(4)试验(2)结果:(4-1)试验:将试验结果示于下述表13~19。关于相对于对照配方2之比的95%信赖区间的上限值大于0.5的配方,在auclast中为配方a、b、e、f和h,在aucinf中为配方a、e、f和h,在cmax中为全部的配方a~h。未确认到配方a~h和对照配方2间的顺序效果(表14)。[表13]表13:药物动力学的参数(auc,cmax)[表14]表14:药物动力学的参数(auc,cmax)相关的配方a~h相对于对照配方2之比和其95%的信赖区间(95%ci表示95%信赖区间)[表15]表15:药物动力学的参数(绝对生物学利用率)由以上结果可知,从药物动力学的观点出发,优选配方a、b、e、f以及h。[表16]表16:药物动力学的参数(tmax)[表17]表17:平均值推移中的血浆中特立帕肽醋酸盐浓度为100pg/ml(或者250pg/ml)以上的状态的经过时间(hr)[表18]表18:副作用(呕吐/恶心/注射部位红斑)表现率(%)在进行了配方a~h的给药的受试对象中确认到了头痛、腹部膨胀、腹泻、恶心、呕吐和注射部位红斑的有害现象,完全未确认到其他有害现象。另外,这些有害现象之中,头痛、恶心、呕吐和注射部位红斑被确认为副作用。呕吐、恶心和注射部位红斑各自的副作用表现率如上述表18所述。[表19]表19:给药后经过6小时后的血清钙浓度增加值(相对于给药前血浆中钙浓度;平均值)根据上述结果,从伴随单次给药的安全性(特别是消化道副作用)的观点出发,tmax小或者血浆中特立帕肽醋酸盐浓度为阈值以上的经过时间短的特立帕肽醋酸盐液态制剂总体上可以说是优异的。实施例4(圆偏振光二色性(cd)光谱测定试验):(1)试验方法:使用圆二色性分散计(日本分光株式会社销售;j-720),将上述实施例1的“(4)供于圆偏振光二色性(cd)光谱测定试验的液态药物制剂的制备”中制备的配方a~h制剂以及上述实施例1的“(3)对照液态药物制剂的制备”中制备的对照配方1,3分别加入到1mm的皿中,在20℃通过8次积分实施圆偏振光二色性(cd)光谱测定。另外,作为空白溶液,使用各配方的安慰剂溶液。试验分2次实施,测定1的测定对象为对照配方1、对照配方3、配方b、配方d(合计4个配方),测定2的测定对象为配方a~h(合计8个配方),进行各配方的圆偏振光二色性光谱测定。(2)试验结果:将试验结果记载于下述表20~22中。[表20]表20:测定1的测定结果[表21]表21:测定2的测定结果(1)[表22]表22:测定2的测定结果(2)此处,表中“平均残基摩尔椭圆率[θ]”是将222nm测定波长下的测定值([mdeg])换算成残基摩尔椭圆率([deg.cm2/dmol])而得到的数值,“α-螺旋含有比例”是使用下述数学式基于平均残基摩尔椭圆率[θ]推算出的α-螺旋含有比例。[数5]α螺旋含有比例=-(平均残基摩尔椭圆率[θ]+2340)/30300(非专利文献10)将测定2的测定结果(测定波长190~260nm)中的配方a~h的平均残基摩尔椭圆率[θ]分别示于图1a~h中。此外,将测定2的测定结果(测定波长210~230nm部分)中的配方a~h的平均残基摩尔椭圆率[θ]示于图1i中。另外,在测定2的测定结果中,将平均残基摩尔椭圆率[θ]222与auclastratio(auclast相关的各配方相对于对照配方2之比)的关系示于图2,将α-螺旋含有比例与auclastratio的关系示于图3。上述“供于人药物动力学试验的液态药物制剂的制备”中制备的配方a~h制剂分别与上述“供于猴子药物动力学试验的液态药物制剂的制备”中制备的配方a~h制剂为大致相同的制剂。因此,在即使将前者配方a~h制剂与后者配方a~h制剂进行交换,猴子药物动力学试验的结果也几乎没有变化的前提下,对于本发明中的液态药物组合物所含有的特立帕肽或其盐所具有的α-螺旋的含量和平均残基摩尔椭圆率[θ]222与将该组合物进行猴子皮下给药时的特立帕肽或其盐的药物动力学参数的关系进行了研究。将其结果示于图4~5。由这些结果与上述实施例3的结果的对比可知,在药物动力学与α-螺旋的含量和形成α-螺旋的氨基酸残基数之间明显存在相关关系。即,上述实施例3中,显示为良好的药物动力学的配方(配方a、b、e、f以及h)的α-螺旋含量和形成α-螺旋的氨基酸残基数均示出了比这些以外的配方(配方c、d、g以及对照配方3)更大的值。发明人认为,通过利用本发明,能够比以往更有效、经济、安全地获得具有特别显著的药物动力学性质的含有特立帕肽或其盐的人皮下给药用液态药物组合物。实施例5(稳定性试验):(1)试验方法:使用上述的“供于稳定性试验的液态药物制剂”中制备的配方a、b、e、f和h安瓿制剂、以及上述的“供于稳定性试验的液态药物制剂”中制备的配方a、b、e、f和h注射器制剂等,实施稳定性试验。具体地说,将各配方制剂在25℃/60%rh的稳定性试验器中保存后,第3个月进行采样,通过高效液相色谱测定稳定性。(2)试验结果:将试验结果记载于下述表23和24中。表中的“相对于开始时的含量”表示将保存前的特立帕肽量设为100时在第3个月所残留的特立帕肽量的比例(%)。表中的“类似物总量”表示将第3个月所存在的(特立帕肽量和类似物总量)设为100时在第3个月所存在的类似物总量的比例(%)。[表23]表23:玻璃安瓿制剂的稳定性配方相对于开始时含量类似物总量配方a94.07.2配方b92.27.4配方e93.37.0配方f93.77.0配方h93.97.1[表24]表24:塑料注射器制剂的稳定性配方相对于开始时含量类似物总量配方a92.18.0配方b90.08.3配方e91.87.5配方f90.68.0配方h90.08.1实施例6(药物动力学相关的模拟试验1):分别假定:某一理论的含有成分1的制剂,其是将该制剂向人皮下单次给药时所得到的吸收速度常数ka为0.48(1/hr)的制剂(制剂a);以及其他理论的含有成分1的制剂,其是将该制剂向人皮下给药时所得到的吸收速度常数ka为2(1/hr)的制剂(制剂b),通过利用本身公知的药物动力学模型的模拟法确认吸收速度的变化对成分1的血浆中浓度推移所带来的影响。药物动力学模型使用分析用软件phenixwinnonlin7.0software(certara公司:原pharsightcorporation公司)的伴有1次吸收和1次消失过程的1-房室模型。将本实施例和实施例7中使用的1-房室模型的概要示意性示于图7中。使制剂a和制剂b各自的清除率和分布容积为适当的相同值,使制剂a和制剂b分别包含的成分1量均为28.2μg。将模拟结果的概要示于下述表25中。需要说明的是,此处,在1-房室模型中,应用下述式(a)。[数6]c(t)=d*ka/(v/f)/(ka-ke)*{exp(-ke*t))-exp(-ka*t)}…式(a)(其中,式中,t表示时间(time),ka表示吸收速度常数,ke表示消失速度常数,v/f表示表观分布容积(apparentvolumeofdistribution),c表示浓度,d表示给药量(dose)。)[表25]表25制剂a制剂bauc(hr*pg/ml)499.1499.1tmax(hr)0.5380.315超过100pg/ml的时间(hr)约2.4约1.5ka(l/hr)0.482实施例7(药物动力学相关的模拟试验2):(1)试验(1)方法:基于将下述表26记载的no.1~12各制剂供于人药物动力学试验而得到的结果,使用与实施例6同样的1-房室模型,分别计算出v/f、ka以及cl/f,对制剂中的成分1浓度与计算出的ka的关系性进行研究。具体地说,对于制剂中的成分1浓度(x)与计算出的ka(y)进行单回归分析,计算出其斜率、截距以及决定系数。其中,此处,ka表示吸收速度常数,v/f表示分布容积,cl/f表示清除率,1-房室模型为与基于上述式(a)的模型等同的模型。[表26]表26(其中,此处,制剂a-2~4是分别将上述配方a~e制剂中的任一制剂的配方填充在不同的医疗用容器中而成的制剂。)(2)试验(1)结果:使用1-房室模型进行计算而得到的各制剂的ka如下述表27所示。使用这些ka进行单回归分析,结果如下述数学式所示,确认到制剂中的成分1的浓度(x)与ka(y)高相关。[数7]y(1/hr)=0.0047x(μg/ml)+0.3261(其中,r2=0.744)[表27]表27no.ka(1/hr)10.5720.5130.7041.0151.2261.0570.9981.0490.41100.89110.86120.84(3)试验(2)方法:进一步将使用1-房室模型进行计算而得到的各制剂(其中为成分1的浓度大于100μg/ml、且生物学利用率高的no.4~8和10~12)的ka和kel代入到下述式中,计算出各制剂的理论tmax。[数8]tmax=in(ka/kel)/(ka-kel)(其中,ka≠kel)(4)试验(2)结果:将计算出的结果汇总于下述表28中。其结果,各制剂的ka宽为0.84~1.22。需要说明的是,由于未确认到通过不基于模型的方法得到的no.4~8制剂的tmax(实施例3试验结果(2)表16中记载的配方a、b、e、f以及h的tmax)明显背离下表中记载的no.4~8制剂的理论tmax,因此可认为使用1-房室模型进行计算而得到的各制剂的各药物动力学参数(v/f、ka以及cl/f)为妥当的推算值。[表28]表28no.tmax(hr)ka(1/hr)kel(1/hr)40.391.015.2750.491.223.2160.471.053.8370.430.994.5480.431.044.43100.470.894.11110.470.864.30120.520.843.65此外,将上述表的最大和最小ka(0.84(1/hr)和1.22(1/hr))输入到上述单回归分析的数学式中时,制剂中的成分1的浓度为109~190(μg/ml)。将利用不基于模型的方法以中央值的形式得到的no.10~12制剂的tmax示于下述表29中。[表29]表29no.tmax(hr)100.5110.5120.625参考例(成分1的tmax为特定范围内的发明相关的参考例):在双重盲检条件下在30例健康闭经后女性中实施临床试验,对于将特立帕肽28.2μg或56.5μg进行单次皮下给药时的药物动力学、骨代谢标记物和安全性与安慰剂进行比较。特立帕肽28.2(或56.5)μg制剂为将含有特立帕肽醋酸盐的冻干制剂使用1ml的日本药典生理盐水在用时溶解而得到的注射剂。具体地说,特立帕肽28.2μg制剂为下述的制剂:其容量为1.0ml,作为每一次的给药量含有以特立帕肽换算计为28.2μg的特立帕肽醋酸盐;特立帕肽56.5μg制剂为下述的制剂:其容量为1.0ml,作为每一次的给药量含有以特立帕肽换算计为28.2μg的特立帕肽醋酸盐。副作用表现率(%)为将表现出各副作用的人数除以给药人数并乘以100而得到的值。进而对于被施用了特立帕肽28.2(或56.5)μg制剂的受试对象中的血清钙值上升进行了观察。血清钙值上升为给药后经过6小时后的血清钙值与给药前的血清钙值的差值(平均值)。关于tmax,也以各被给药者的血浆中特立帕肽醋酸盐浓度达到最高的时间的平均值的形式计算出。将试验结果记载于下述表30~33中。[表30]表30:药物动力学的参数(tmax)[表31]表31:平均值推移中的血浆中特立帕肽醋酸盐浓度为100pg/ml(或者250pg/ml)以上的状态的经过时间(hr)[表32]表32:副作用(呕吐、恶心、注射部位红斑)表现率(%)[表33]表33:给药后经过6小时后的血清钙浓度增加值(相对于给药前血浆中钙浓度;平均值)工业实用性本发明的液态药物制剂从药物动力学的观点出发是优异的。本发明的药物动力学参数的改善方法也是突破性的主药控制方法。因此,本发明在医药品产业中极为有用。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1