用于治疗化学治疗诱导的医源性疼痛的C5aR抑制剂的制作方法
本发明涉及c5a受体(c5ar)抑制剂,其用于预防和治疗化学治疗诱导的医源性疼痛(chemotherapy-inducediatrogenicpain,ciip),尤其是与ciip相关的痛觉超敏(allodynia)。
背景技术:
不同类型的神经性并发症可能与抗肿瘤药物治疗有关,所述神经性并发症包括中枢神经毒性病症,其范围从轻微的认知缺陷到伴随痴呆甚至昏迷的脑病;以及周围神经毒性。医源性疼痛是癌症治疗的最常见神经性并发症,其代表了从轻微和暂时的症状到严重和永久性的多神经病形式的一组症状。
由于这种神经毒性由抗癌药物的施用引起,因此其通常被认为是化学治疗诱导的医源性疼痛(ciip)(m.cascella等,currentmedicalresearchandopinion,2017;42:1-3)。
“化学治疗诱导的医源性疼痛(ciip)”指示化学治疗对周围神经的剂量限制性神经毒性作用。许多不同的症状都与ciip有关:痛觉过敏、痛觉超敏和自发性感觉,例如灼烧、疼痛、麻木、痉挛和痒。特别地,尽管由化学治疗剂的神经毒性诱导的一些症状因患者而异,但导致疼痛性感觉异常的常见感觉障碍对于所有受影响的患者而言均是常见的。
ciip在约60%的癌症患者中发生(windebank等,jperiphernervsyst2008;13:27-46)并且可导致剂量限制或甚至治疗中止,从而最终影响患者的存活(mielke等,eurjcancer2006;42:24-30)。
特别地,通常与周围疼痛发作最有关的化学治疗剂包括:铂基药物,例如顺铂、卡铂和奥沙利铂;紫杉烷类,例如紫杉醇、卡巴他赛和多西他赛;埃博霉素类,例如伊沙匹隆;植物生物碱类,例如长春花碱、长春新碱、长春瑞滨和依托泊苷;沙利度胺、来那度胺和波马度胺;卡非佐米和硼替佐米;艾日布林(brewer等,gynecologiconcology2016;140:176-83)。
尽管在实验研究和临床试验中均已研究了多种神经保护性方法,但目前尚无可用的ciip预防策略或有效治疗,这也因为ciip的病因尚未完全阐明。
已经提出了多种机制来构成疼痛的发展和维持的基础。
一些证据表明炎性细胞因子/趋化因子,尤其是tnf-α、il-1β、il-6和ccl2可在ciip的化学治疗剂诱导的疼痛症状中发挥作用(wang等,cytokine2012;59(1):3-9)。然而,有力的证据也表明化学治疗药物对触觉神经元有直接作用(argyriou等,critrevonolhematol2012;82(1):51-77,boyette-davis等,pain,2011;152:308-13;pachman等,clinpharmacolther2011;90:377-387)。特别地,已经确定大多数化学治疗药物可容易地渗透血-神经屏障(blood-nerve-barrier,bnb)并与背根神经节(dorsalrootganglia,drg)和周围轴突结合(wang等,参见上文)。还存在这样的证据:这些药物随后直接破坏drg细胞和周围神经的结构,因此表皮层的触觉纤维崩溃并且小神经纤维损失(argyriou等,cancermanagres.2014;6:135-147)。
在细胞水平,神经毒性化学治疗剂破坏微管,干扰基于微管的轴突运输(lapointe等,neurotoxicology2013;37:231-9),通过诱导α-微管蛋白乙酰化来影响微管动力学,中断线粒体功能或直接靶向dna。来自用紫杉醇、奥沙利铂或长春新碱处理的实验动物和患者的神经活检示出了相同的形态学变化,表明潜在的常见致病机制。
补体的c5a肽片段由于其趋化活性和炎性活性,已被限定为“完全”促炎介质。实际上,另外的炎性介质,例如所选的趋化因子(例如il-8、mcp-1和rantes)对自吸引细胞具有高度选择性,而另一些炎性介质,例如组胺和缓激肽仅是弱趋化剂。有力的证据支持c5a在体内参与数种病理状况,包括缺血/再灌注、自身免疫性皮炎、膜增生性特发性肾小球肾炎、气道无反应性和慢性炎性疾病、ards和codp、阿尔茨海默病、幼年型类风湿关节炎(n.p.gerard,ann.rev.immunol.,12,755,1994)。还研究了c5a及其选择受体c5ar在与抗体依赖性ii型自身免疫有关的疾病的发展中,特别是在自身免疫性溶血性贫血(autoimmunehaemolyticanaemia,aiha)的发生(insurgence)中的病理学意义,aiha是一种特征在于产生针对引起溶血的自身红细胞(redbloodcell,rbc)的抗体的疾病。aiha是一种相当罕见的病症,估计发病率为1-3例/100,000/年。已经在实验动物模型中确定了c5a在igg依赖性aiha中的关键作用,该作用独立于该过敏毒素(anaphylotoxin)的趋化功能(v.kumar,j.clin.invest.,116(2),512,2006)。实际上,已经观察到缺乏c5ar的小鼠对该igg自身抗体诱导的疾病模型具有部分抗性,并且通过如下观察结果已经确定了c5ar与活化fcγ受体的串扰,特别是在肝巨噬细胞上:在施用抗红细胞抗体之后,在c5ar缺陷型小鼠中不存在库普弗(kupfer)细胞上的活化fcγr的上调;类似地,在fcγr缺陷型小鼠中,c5和c5a的产生被消除。这是先前未确定的fcγr介导的c5a生成途径的首个证据,表明c5a在抗体依赖性自身免疫病的发展中的作用以及c5a和/或c5ar阻断在与ii型自身免疫损伤相关的aiha中的潜在治疗益处。
wo2007/060215公开了(r)-芳基烷基氨基衍生物及其在治疗涉及c5a诱导的人pmn趋化性的疾病中以及在预防和治疗由缺血和再灌注引起的损伤中的用途,所述涉及c5a诱导的人pmn趋化性的疾病例如脓毒症、银屑病、大疱性类天疱疮、类风湿关节炎、溃疡性结肠炎、急性呼吸窘迫综合征、特发性纤维化、囊性纤维化、慢性阻塞性肺疾病、肾小球肾炎。
wo2009/050258公开了(r)-4-(杂芳基)苯乙基化合物及其在治疗涉及c5a诱导的人pmn趋化性的疾病中以及在预防和治疗由缺血和再灌注引起的损伤中的用途,所述涉及c5a诱导的人pmn趋化性的疾病例如自身免疫性溶血性贫血(aiha)、银屑病、大疱性类天疱疮、类风湿关节炎、溃疡性结肠炎、急性呼吸窘迫综合征、特发性纤维化、肾小球肾炎。
已发表的论文a.moriconi等,pnas,111(47),16937-16942,2014公开了c5a过敏毒素受体(c5aanaphylatoxinreceptor,c5ar)的新强效别构抑制剂在数种急性和慢性炎性疼痛模型中以及在sni诱导的小鼠神经性疼痛模型(其使用胫侧和腓侧轴索切断(axotomy)在受伤的后爪上诱导慢性痛觉超敏)中降低机械性(mechanical)痛觉过敏的作用。诱导神经病和特定模型的特定特征所遵循的方法对于理解潜在机制和制定有效的管理治疗是至关重要的。特别是在化学治疗诱导的医源性疼痛的背景下,治疗方法的效力似乎与特定靶标的确定严格相关。为证实这一假设,科学证据不支持在治疗化学治疗诱导的医源性疼痛的管理中使用有效治疗一般性疼痛的药物(shindess等,supportcarecancer,24(2):547-553,2016)。
与在发表的论文a.moriconi等,pnas,111(47),16937-16942,2014中最初描述的创伤性损伤不同,当前数据涉及由化学治疗特异性机制诱导的神经毒性的验证模型,特别是涉及紫杉烷类(polomanorc等,pain,94,293,2001)。紫杉烷类,特别是紫杉醇,干扰微管网络的动态生理重组,微管网络对于细胞的重要功能是必要的,并且诱导氧化应激。c5a及其细胞膜受体c5ar与急性、炎性和神经性疼痛状态有关;但是,尚未研究c5a/c5ar在化学治疗诱导的医源性疼痛中的作用。
技术实现要素:
本发明人已令人惊讶地发现,c5ar的抑制能够降低或预防与全身性抗癌化学治疗的毒性相关的症状的发生,所述全身性抗癌化学治疗导致化学治疗诱导的医源性疼痛(ciip)。
此外,可用于预防和/或治疗化学治疗诱导的医源性疼痛的c5ar抑制剂不以任何方式干扰化学治疗剂的活性。
因此,本发明的第一目的是c5ar抑制剂,优选c5ar非竞争性别构抑制剂,其用于预防和/或治疗化学治疗诱导的医源性疼痛。
本发明的第二目的是c5ar抑制剂用于制备用于治疗和/或预防化学治疗诱导的医源性疼痛的药物的用途。
本发明的第三目的是用于预防和/或治疗ciip的方法,该方法包括向有此需要的对象施用治疗有效量的所述c5ar抑制剂的步骤。
本发明的第四目的是用于预防和/或治疗ciip的药物组合物,其包含根据本发明的c5ar抑制剂和可药用赋形剂。
附图说明
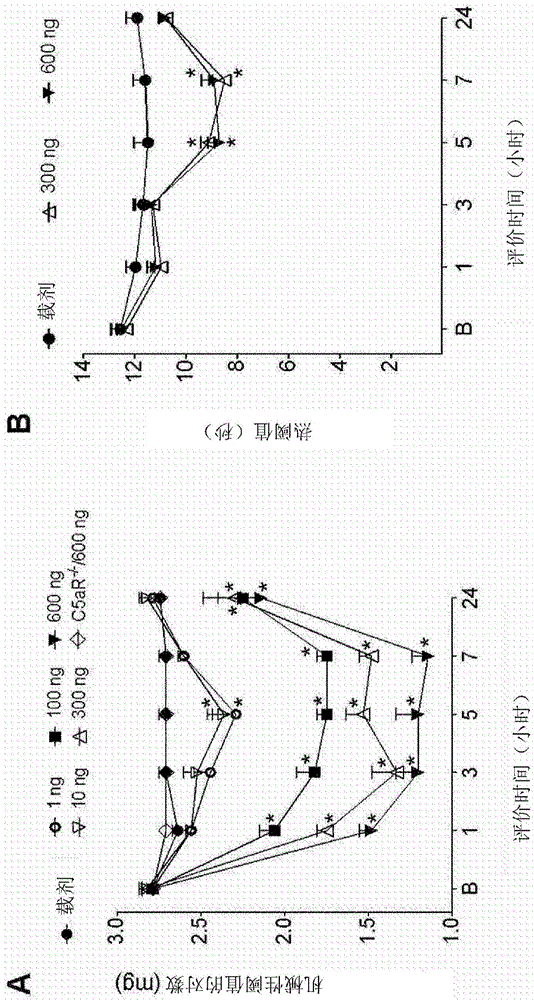
图1.在雄性balb/c小鼠中鞘内注射重组c5a(1、10、100、300和600ng/5μl)的剂量反应曲线(dose-responsecurve)。a,机械性阈值-vonfrey丝试验(vonfreyhairtest)-和b,热阈值-hargreaves试验。b,基线。数据由平均±sem表示。n=5。通过双因素anova、事后bonferroni’s检验进行统计分析。与载剂相比*p<0.05。
图2.a,机械性阈值-vonfrey丝试验-和b,热阈值-hargreaves试验-在雄性balb/c小鼠中在化学治疗诱导的医源性疼痛(ciip)发展期间。b,基线;pcx,紫杉醇;wt,野生型。数据由平均±sem表示。n=5。通过双因素anova、事后bonferroni’s检验进行统计分析。与wtpcx组相比*p<0.05。
图3a,3b.在首次注射pcx紫杉醇之后第8天(a和b)或第14天(c和d),在用1mg/kgdf2593a(箭头)经口处理之后,雄性balb/c小鼠的机械性阈值(a和c)和冷响应(b和d)。b,基线。数据由平均±sem表示。n=5。通过双因素anova、事后bonferroni’s检验进行统计分析。与pcx组相比*p<0.05。
图4.在balb/c雄性小鼠中,在化学治疗诱导的医源性疼痛(ciip)发展期间,a,机械性阈值-vonfrey丝试验和b,冷响应-丙酮试验。b,基线。12/12小时地用1mg/kgdf2593a经口处理动物,持续7天。在第1、3、5和7天,df2593a的施用是紫杉醇(pcx)之前1小时。在pcx注射之后4小时进行测量。数据由平均±sem表示。n=5。通过双因素anova、事后bonferroni’s检验进行统计分析。与pcx组相比*p<0.05。
图5a,5b.在首次注射紫杉醇(pcx)之后第8天(a和b)和第14天(c和d),在用不同剂量的df2593a鞘内处理之后,雄性balb/c小鼠的机械性阈值(a和c)或冷响应(b和d)。b,基线。数据由平均±sem表示。n=5。通过双因素anova、事后bonferroni’s检验进行统计分析。与pcx组相比*p<0.05。
图6.在df3966a处理之后3小时(紫杉醇之后4小时),评价了雄性balb/c小鼠的机械性(a)和冷(b)痛觉超敏。组之间差异的显著性通过双因素方差分析(anova)接着是bonferroni事后检验进行多重比较确定。相对于ctr的显著性水平设置为*p<0.05。
图7.用单独或与df3966y组合的c5a挑战下的α-微管蛋白水平。数据是3个不同实验的平均±sd。相对于未经处理的细胞ut,**p<0.01;相对于c5a#p<0.05。
图8.在基础条件下维持的drg细胞中电生理记录的代表性痕迹;处理a):在用紫杉醇挑战的drg细胞中电生理记录的代表性痕迹;处理b):在用紫杉醇+df3966y挑战的drg细胞中电生理记录的代表性痕迹。
图9.处理c):在用c5a挑战的drg细胞中电生理记录的代表性痕迹;处理d):在用c5a+df3966y挑战的drg细胞中电生理记录的代表性痕迹。
具体实施方式
如将在实验部分中详细公开的,本发明人已发现,充当c5ar活性抑制剂的分子在由紫杉醇诱导的医源性疼痛的动物模型中具有治疗效力。此外,本发明人还已发现,c5ar抑制能够抵消化学治疗剂对有助于其神经毒性作用的细胞骨架成分和组织的活性。
因此,本发明的第一目的是c5ar抑制剂,其用于治疗和/或预防化学治疗诱导的医源性疼痛(ciip)。
根据一个优选的实施方案,所述c5ar抑制剂用于预防和/或治疗与化学治疗诱导的医源性疼痛(ciip)有关的痛觉超敏。
根据本申请的术语“c5ar抑制剂”是指能够部分或完全抑制c5a和/或c5ar的生物学活性的任何化合物。这样的化合物可通过降低c5a的表达或活性或者通过抑制由c5a受体激活的胞内信号传导的触发而起作用。优选地,所述c5a抑制剂能够以等于或低于500nm,优选低于100nm的浓度抑制pmn中c5a诱导的趋化性的至少50%,优选至少60%。
本发明的第二目的是c5ar抑制剂用于制备用于治疗和/或预防化学治疗诱导的医源性疼痛(ciip)的药物的用途。
根据本发明的一个优选实施方案,所述药物用于治疗和/或预防与化学治疗诱导的医源性疼痛有关的痛觉超敏。
本发明的第三目的是用于治疗和/或预防化学治疗诱导的医源性疼痛的方法,该方法包括向有此需要的对象施用治疗有效量的如上文所限定的c5ar抑制剂的步骤。
根据本发明的一个优选实施方案,所述方法用于治疗和/或预防与化学治疗诱导的医源性疼痛有关的痛觉超敏。
本文中使用的“治疗有效量”是指足以实现疾病的治疗或预防的量。基于实现所期望的效果的有效量的确定在本领域技术人员的能力之内。有效量将取决于包括但不限于以下的因素:对象的体重和/或对象所遭受的疾病或不期望的病症的程度。本文中使用的术语“治疗”和“预防”是指所治疗的病症或与其相关的一种或更多种症状的各自的发作的根除/改善或预防/延迟,尽管患者可能仍然患有潜在的病症。
本发明的第四目的是包含与药学上合适的赋形剂联合的c5ar抑制剂的药物组合物,其用于治疗和/或预防化学治疗诱导的医源性疼痛(ciip)。
根据本发明的一个优选实施方案,所述药物组合物用于治疗和/或预防与化学治疗诱导的医源性疼痛有关的痛觉超敏。
根据一个优选的实施方案,本发明所有目的的c5ar抑制剂是c5a受体的非竞争性别构抑制剂。
根据本发明的“c5a受体的非竞争性别构抑制剂”意指显示出与位于tm区域的别构位点中的c5a受体相互作用的化合物,其抑制由激动剂结合激活的胞内信号转导事件,而不影响内源配体c5a在其受体上的任何结合。
根据本发明的优选的c5ar抑制剂选自(r)-芳基烷基氨基衍生物、(r)-4-(杂芳基)苯乙基化合物、及其可药用盐。
在以上化合物中,所述(r)-芳基烷基氨基衍生物优选为式(i)化合物或其可药用盐:
其中
r选自:
-2-噻唑基或2-
-c(ra)=n-w,其中w是直链或支链c1-c4烷基,
-cora、sora、so2ra、pora、po2ra,
其中
ra选自
-c1-c5-烷基、c3-c6-环烷基、c2-c5-烯基、未被取代或被选自以下的基团取代的苯基:卤素、c1-c4-烷基、c1-c4-烷氧基、卤代-c1-c4-烷氧基、羟基、c1-c4-酰氧基、苯氧基、氰基、硝基、氨基;
-杂芳基,其选自吡啶、嘧啶、吡咯、噻吩、呋喃、吲哚、噻唑、
-由直链或支链c1-c6-烷基、c3-c6-环烷基、c2-c6-烯基、c1-c6-苯基烷基组成的α或β羧基烷基残基,其任选地被另外的羧基(cooh)取代;
-式ii的ω-氨基烷基氨基:
其中
x表示:
-直链或支链c1-c6亚烷基、c4-c6亚烯基、c4-c6亚炔基,其任选地被co2r4基团或conhr5基团取代,其中r4表示氢或直链或支链c1-c6烷基或者直链或支链c2-c6烯基,其中r5表示氢、直链或支链c2-c6烷基或or4基团,r4如上所限定;
-(ch2)m-b-(ch2)n基团,其任选地被如上限定的co2r4或conhr5基团取代,其中b是氧或硫原子或氮原子,其任选地被c1-c4烷基取代,m为0或2至3的整数,并且n为2至3的整数,或者b为co、so或conh基团,m为1至3的整数,并且n为2至3的整数;
-或者x和与其结合的氮原子一起以及与r2基团一起形成含氮的3至7元杂环、单环或多环,并且r3表示氢、c1-c4烷基、c1-c4酰基、未被取代或被选自以下的基团取代的苯基:卤素、c1-c4-烷基、c1-c4-烷氧基、羟基、c1-c4-酰氧基、苯氧基、氰基、硝基、氨基;
r2和r3独立地是:
氢、任选地被氧或硫原子间断的直链或支链c1-c6烷基,c3-c7环烷基、c3-c6烯基、c3-c6-炔基、芳基-c1-c3-烷基、羟基-c2-c3-烷基;
或者r2和r3和与它们结合的n原子一起形成式(iii)的3至7元氮杂环
其中
y表示:
-单键、ch2、o、s或n-r6基团,其中r6表示氢、c1-c4烷基、c1-c4酰基、未被取代或被选自以下的基团取代的苯基:卤素、c1-c4-烷基、c1-c4-烷氧基、羟基、c1-c4-酰氧基、苯氧基、氰基、硝基、氨基,
并且p表示0至3的整数;
-式so2r7的残基,其中r7是c1-c6-烷基、c3-c6-环烷基、c2-c6-烯基、芳基和杂芳基;
r1是直链或支链c1-c5烷基、c3-c5环烷基;
ar是未被取代或被独立地选自以下的一个或更多个基团取代的苯基:卤素、c1-c4-烷基、c1-c4-烷氧基、羟基、c1-c4-酰氧基、苯氧基、氰基、硝基、氨基、c1-c4-酰基氨基、卤代-c1-c3-烷基、卤代-c1-c3-烷氧基、苯甲酰基、杂芳基羰基、杂芳基、直链或支链c1-c8-链烷磺酸盐/酯、直链或支链c1-c8-链烷磺酰胺、直链或支链c1-c8烷基磺酰基甲基;
或者ar是选自吡啶、吡咯、噻吩、呋喃、吲哚的杂芳基环。
在以上化合物中,特别优选的是所述式(i)化合物或其可药用盐,其中:r选自:
-2-噻唑基或2-
-c(ra)=n-w,其中w是直链或支链c1-c4烷基,
-cora、sora或so2ra,
其中ra如上所限定;
ar选自:
3’-苯甲酰基苯基、3’-(4-氯-苯甲酰基)-苯基、3’-(4-甲基-苯甲酰基)-苯基、3’-乙酰基-苯基、3’-丙酰基-苯基、3’-异丁酰基-苯基、4’-异丁基-苯基、4’-三氟甲磺酰基氧基-苯基、4’-苯磺酰基氧基-苯基、4’-三氟甲磺酰基氨基-苯基、4’-苯磺酰基氨基-苯基、4’-苯磺酰基甲基-苯基、4’-乙酰氧基苯基、4’-丙酰基氧基-苯基、4’-苯甲酰基氧基-苯基、4’-乙酰基氨基-苯基、4’-丙酰基氨基-苯基、4’-苯甲酰基氨基-苯基、3’-(呋喃-2-羰基)-苯基、3’-(苯并呋喃-2-羰基)-苯基、3’-(噻吩-2-羰基)-苯基、3’-(吡啶-2-羰基)-苯基、3’-(噻唑-2-羰基)-苯基、3’-(
在以上化合物中,还特别优选的是所述式(i)化合物或其可药用盐,其中:
r是
-2-噻唑基,其未被取代或被选自甲基或三氟甲基的基团取代;
-cora、so2ra、sora;
其中
ra选自:
-c1-c5-烷基、c3-c5-环烷基;
-苯基、2-吡啶基、2-噻唑基、2-呋喃基、2-吡咯基、2-噻吩基、2-吲哚基;
-由直链或支链c1-c6-烷基、c1-c6-苯基烷基组成的羧基烷基;
-式ii的ω-烷基氨基,
(ii)
其中
x表示:
直链或支链c1-c6亚烷基、c4-c6亚烯基、c4-c6亚炔基;
或者x和与其结合的氮原子一起以及与r2基团一起形成含氮的3至7元杂环单环,并且r3表示氢或c1-c4烷基;
r2和r3独立地是氢、直链或支链c1-c6烷基、c3-c7环烷基、c3-c6烯基、c3-c6-炔基;
或者r2和r3和与它们结合的n原子一起形成式(iii)的4至6元含氮杂环
其中y表示ch2、o、s或n-r6基团,其中r6表示氢、c1-c4烷基、c1-c4酰基,并且p表示0至2的整数;
r1是甲基;
ar选自:
3’-苯甲酰基苯基、3’-(4-氯-苯甲酰基)-苯基、3’-(4-甲基-苯甲酰基)-苯基、3’-乙酰基-苯基、3’-丙酰基-苯基、3’-异丁酰基-苯基、4’-异丁基-苯基、4’-三氟甲磺酰基氧基-苯基、4’-苯磺酰基氧基-苯基、4’-三氟甲磺酰基氨基-苯基、4’-苯磺酰基氨基-苯基、4’-苯磺酰基甲基-苯基、4’-乙酰氧基苯基、4’-丙酰基氧基-苯基、4’-苯甲酰基氧基-苯基、4’-乙酰基氨基-苯基、4’-丙酰基氨基-苯基、4’-苯甲酰基氨基-苯基、3’-(呋喃-2-羰基)-苯基、3’-(苯并呋喃-2-羰基)-苯基、3’-(噻吩-2-羰基)-苯基、3’-(吡啶-2-羰基)-苯基、3’-(噻唑-2-羰基)-苯基、3’-(
在以上化合物中,还特别优选的是所述式(i)化合物或其可药用盐,其中
其中r是
-2-噻唑基,其未被取代或被选自甲基或三氟甲基的基团取代;
-cora、so2ra
其中
ra选自:
-c1-c5-烷基、c3-c5-环烷基;
-苯基、2-吡啶基、2-呋喃基、2-噻吩基;
-式ii的基团
其中
x表示:
直链或支链c1-c6亚烷基,
r2和r3和与它们结合的n原子一起形成式(iii)的4至6元含氮杂环
其中y表示ch2,并且p表示0至2的整数;
r1是甲基;
ar选自:
3’-苯甲酰基苯基、3’-(4-氯-苯甲酰基)-苯基、3’-(4-甲基-苯甲酰基)-苯基、4’-三氟甲磺酰基氧基-苯基、4’-苯磺酰基氧基-苯基、3’-(呋喃-2-羰基)-苯基。
根据本发明特别优选的是式(i)化合物,其选自:
4-{(1r)-1-[(苯基磺酰基)氨基]乙基}苯基三氟甲磺酸盐/酯
n-[(1r)-1-(3-苯甲酰基苯基)乙基]苯磺酰胺
4-{(1r)-1-[(吡啶-3-基磺酰基)氨基]乙基}苯基三氟甲磺酸盐/酯
n-[(1r)-1-(3-苯甲酰基苯基)乙基]甲磺酰胺
n-{(1r)-1-[3-(2-糠酰基)苯基]乙基}噻吩-2-磺酰胺
n-{(1r)-1-[3-(2-糠酰基)苯基]乙基}甲磺酰胺
4-{(1r)-1-[(噻吩-2-基磺酰基)氨基]乙基}苯基三氟甲磺酸盐/酯
n-[(1r)-1-(3-苯甲酰基苯基)乙基]噻吩-2-磺酰胺
n-[(1r)-1-(3-苯甲酰基苯基)乙基]-3-吡咯烷-1-基丙烷-1-磺酰胺
5-({[(1r)-1-(3-苯甲酰基苯基)乙基]氨基}磺酰基)-2-糠酸甲酯
5-({[(1r)-1-(3-苯甲酰基苯基)乙基]氨基}磺酰基)-2-糠酸
4-{(1r)-2-甲基-1-[(甲基磺酰基)氨基]丙基}苯基三氟甲磺酸盐/酯
n-((1r)-1-{4-[1-甲基-1-(苯基磺酰基)乙基]苯基}乙基)甲磺酰胺
4-[(1r)-1-(异丁酰基氨基)乙基]苯基三氟甲磺酸盐/酯
4-{[(1r)-1-(吡啶-3-基羰基)氨基]乙基]}苯基三氟甲磺酸盐/酯
n-[(1r)-1-(3-苯甲酰基苯基)乙基]苯甲酰胺
n-[(1r)-1-(3-苯甲酰基苯基)乙基]-2-糠酰胺
n-[(1r)-1-(3-苯甲酰基苯基)乙基]环丁烷酰胺
n-[(1r)-1-(4-三氟甲磺酰基氧基)苯乙基]-4-哌啶-1-基丁酰胺
4-{(1r)-1-[(4-吡咯烷-1-基丁酰基)氨基]乙基]}苯基三氟甲磺酸盐/酯
3-{(1r)-1-[4-(4-三氟甲基-1,3-噻唑-2-基)氨基]乙基}苯基)(苯基)甲酮。
根据本发明特别优选的是选自n-[(1r)-1-(4-三氟甲磺酰基氧基)苯乙基]-4-哌啶-1-基丁酰胺(在本文中也表示为df2593y)及其可药用盐,优选其氯化物盐(在本文中也表示为df2593a)的式(i)化合物。
wo2007/060215中公开了式(i)化合物,其还公开了式(i)化合物的合成方法、作为c5ar抑制剂的活性以及在治疗涉及c5a诱导的人pmn趋化性的疾病中以及在预防和治疗由缺血和再灌注引起的损伤中的用途,所述涉及c5a诱导的人pmn趋化性的疾病例如脓毒症、银屑病、大疱性类天疱疮、类风湿关节炎、溃疡性结肠炎、急性呼吸窘迫综合征、特发性纤维化、囊性纤维化、慢性阻塞性肺疾病、肾小球肾炎。
在以上c5ar抑制剂中,所述(r)-4-(杂芳基)苯乙基化合物优选为式(ii)化合物或其可药用盐:
其中
x是选自以下的杂原子
-s、o和n
y是h或选自以下的残基:
-卤素、直链或支链c1-c4-烷基、c2-c4-烯基、c1-c4-烷氧基、羟基、-cooh、c1-c4-酰氧基、苯氧基、氰基、硝基、-nh2、c1-c4-酰基氨基、卤代-c1-c3-烷基、苯甲酰基、直链或支链c1-c8-链烷磺酸盐/酯、直链或支链c1-c8-链烷磺酰胺、直链或支链c1-c8-烷基磺酰基甲基;
z是选自以下的杂芳基环:
未被取代的四唑和
被一个羟基取代并且任选地另外地被一个或更多个选自以下的基团取代的三唑、吡唑、
在以上化合物中,优选的是式(ii)化合物或其可药用盐,其中:
x是选自以下的杂原子
-s和o
y是h或选自以下的残基:
-卤素、直链或支链c1-c4-烷基和卤代-c1-c3-烷基;优选选自三氟甲基、氯、甲基和叔丁基;
z是选自以下的杂芳基环:
未被取代的四唑和被一个羟基取代并且任选地另外地被一个或更多个选自以下的基团取代的三唑、吡唑、异
根据本发明的特别优选的式(ii)化合物选自:
n-{4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-4-(三氟甲基)-1,3-噻唑-2-胺;
4-甲基-n-{4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-1,3-噻唑-2-胺;
4-叔丁基-n-{4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-1,3-噻唑-2-胺;
n-{4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-1,3-噻唑-2-胺;
n-{4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-4-(三氟甲基)-1,3-
4-甲基-n-{4-[(1r)-1-(1h四唑-5-基)乙基]苯基}-1,3-
5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1h吡唑-1-醇;
4-甲基-5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1h-吡唑-1-醇;
5-[(1r)-1-(4-([4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1h-1,2,3-三唑-1-醇;
5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]异
4-甲基-5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]异
5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]异噻唑-3-醇;
4-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1,2,5-
4-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1,2,5-噻二唑-3-醇;
5-[(1r)-1-(4-{[4-(三氟甲基)-1,3-噻唑-2-基]氨基}苯基)乙基]-1h1、2、4-三唑-1-醇。
根据本发明的特别优选的式(ii)化合物选自1-n-[4-[(1r)-1-(1h-四唑-5-基)乙基]苯基}-4-(三氟甲基)-11,3-噻唑-2-胺(在本文中也表示为df3966y)及其可药用盐,优选其钠盐(在本文中也表示为df3966a)。
wo2009/050258中公开了式(ii)的c5ar抑制剂,其还公开了该抑制剂的合成方法、作为c5ar抑制剂的活性以及在治疗涉及c5a诱导的人pmn趋化性的疾病中以及在预防和治疗由缺血和再灌注引起的损伤中的用途,所述涉及c5a诱导的人pmn趋化性的疾病例如自身免疫性溶血性贫血(aiha)、银屑病、大疱性类天疱疮、类风湿关节炎、溃疡性结肠炎、急性呼吸窘迫综合征、特发性纤维化、肾小球肾炎。
根据本发明的化学治疗诱导的医源性疼痛可以是由具有神经毒性副作用的任何化学治疗剂诱导的那些。优选地,所述化学治疗剂选自:铂基药物、紫杉烷类、埃博霉素类、植物生物碱类、沙利度胺、来那度胺和泊马度胺、卡非佐米、硼替佐米和艾日布林。更优选地,所述化学治疗剂选自:顺铂、卡铂、奥沙利铂、紫杉醇、卡巴他赛、多西他赛、伊沙匹隆、长春花碱、长春新碱、长春瑞滨、依托泊苷、沙利度胺、来那度胺、泊马度胺、卡非佐米、硼替佐米和艾日布林。根据一个优选的实施方案,化学治疗诱导的医源性疼痛是由紫杉烷类,更优选由紫杉醇引起的。
实施例
方法和材料
药物和试剂
以下材料获自指定的来源:重组小鼠c5a购自r&d,批次mjj0714041,在bsa0.1%中稀释并保持在-70℃下。紫杉醇(
生化试剂盒和试剂
celltag700stainicw试剂盒(li-cor#目录926-41091)
gro/kc,peprotech#目录400-10
重组大鼠cxcl1/groα/kc/cinc-1蛋白-r&dsystems,目录#515-cn,批次44s0211121
紫杉醇(taxol)-tocris-r&dsystems#1097批次7a/177205
c5a成分-重组小鼠补体-r&dsystems#2150-c5批次mjj0715041
hoechst33342-thermofischersc.#h3570
动物
实验是在雄性balb/c野生型(wt)和c5ar缺陷型(c5ar-/-)小鼠(6至8周龄)中进行的。对于用df2593a进行的实验,将动物安置在圣保罗大学的ribeiraopreto医学院的动物护理设施中,使其在20℃±1℃的塑料笼中,自由饮水和进食,并控制明暗循环。当进行经口急性处理时,将动物禁食2至4小时。动物在实验之前至少1小时被带到测试室,并且仅使用一次。动物护理和处理程序符合国际疼痛研究协会指南,并由ribeiraopreto医学院动物伦理委员会根据协议120/2014控制。
对于用df3966a进行的实验,将动物安置在意大利那不勒斯大学药学部门的动物护理设施中,使其在具有受控的温度(22±1℃)、湿度(60±10%)和光照(每天12小时)的房间中;食物和水可随意获得。所有行为测试均在9:00am至5:00pm之间进行,并且动物仅使用一次。动物护理和操作根据国际和国家法律和政策(有关动物实验的欧盟指令2010/63/eu,arrive指南和巴塞尔宣言,包括3r概念)进行。在此报道的操作由那不勒斯费德里克二世大学的动物实验伦理学委员会(cvs)并由卫生部(ministerodellasalute)根据协议n.2014-00884607批准。
ciip(化学治疗诱导的医源性疼痛)实验方案
所使用的方案是根据polomano等,2001(适用于小鼠)进行的。动物腹膜内(i.p.)接受4mg/kg紫杉醇,进行间隔的四天(第1、3、5和7天)。在治疗期间,在紫杉醇注射之后4小时测量机械性和冷痛觉超敏。
急性后处理
在首次紫杉醇注射之后第8天和第14天以1mg/kg的剂量经口给予df2593a。在施用之后2、4、6和24小时评价药物的作用。
慢性预处理
在ciip的诱导阶段期间,以1mg/kg的剂量12/12小时地经口给予df2593a,持续7天。在第1、3、5和7天,df2593a的施用是在紫杉醇之前1小时。
从第1天到第14天,30mg/kg/os地施用df3966a(每天两次;8:00am和20.00pm)。在1-3-5-7天,df3966a在紫杉醇之后1小时施用。
鞘内处理
在首次注射紫杉醇(ciip)之后的第8天和第14天,以10、30或100μg/5μl的剂量注射df2593a。
通过鞘内的方式以5μl的体积给予重组小鼠c5a,并且其作用在注射之后持续1、3、5、7和24小时。
机械性伤害性爪试验
在df2593a实验中,使用vonfrey细丝测试了机械性痛觉过敏。在开始测试之前15至30分钟,将小鼠放置于安静的室中的具有线栅地板的丙烯酸类笼(12×10×17cm)中。在ciip小鼠的右爪上施加新月形的系列细丝。将能够引起退缩运动的较低细丝记录为机械性阈值。在处理之前和之后对动物进行测试,并且结果表示为机械性阈值的对数。
在df3966a实验中,使用动态足底触觉计(dynamicplantaraesthesiometer,dpa,ugobasile,italy)测量了对触觉刺激的敏感性。将动物置于具有网格金属地板的室中,地板上覆盖有塑料圆顶,这使得动物能够自由行走,但不能跳跃。然后将机械刺激传递到后爪的足底中部皮肤。截止值固定为50g。在紫杉醇首次施用之前,然后在紫杉醇首次施用之后的第3、5、7、10和14天对两只爪进行测试。
冷痛觉超敏-丙酮试验
在与vonfrey试验相同的装置中,并且在机械性试验之后近15分钟,进行丙酮试验。用1ml注射器释放一滴(50μl)的纯丙酮,使液体散布到两个爪的表面(背侧和足底)。用计时器记录爪的所有运动,例如舔、退缩或抬起,总测试时间为2分钟。
热潜伏期-hargreaves试验
如先前所述(hargreaves等,1988)进行hargreaves试验。使动物在受控的温度下的习惯于在玻璃表面上,这允许红外光在爪上均匀地递送。在激活光源之后,打开计时器。动物一旦在光线外作出退缩运动,新月温度就到达爪,并且计时器就关闭。记录的时间被认为是热潜伏期。固定了20秒的截止以避免爪的损伤。结果以秒表示。
背根神经节(drg)神经元的原代培养
遵循良好限定的方案,通过解离出生后(p2)spraguedawley大鼠神经节(envigo,bressoitaly)来获得背根神经节神经元(o’meara等,2011;owen等,2012)。为了使污染的细胞最小化地被引入到培养物中,从drg修剪掉任何过长的根。分离之后,如下酶解drg:
-在37℃下,1.5mg/ml木瓜蛋白酶暴露20分钟,
-在37℃下,4mg/ml胶原酶暴露30分钟。
随后,将细胞用火抛光的玻璃巴斯德(pasteur)移液管机械地解离,以获得单一细胞混悬液。
一旦完成解离,将细胞以选定的密度接种到包被有层黏连蛋白(10ug/ml,在室温(rt)下过夜)的96孔黑色多孔板上。在补充有10%fbs、1×n2、1×b27、100iu/ml青霉素、10mg/ml链霉素的dmem高葡萄糖培养基中孵育1周之后,根据实验需要处理细胞。
用于α-微管蛋白评价的细咆内western分析
使如先前所报道地分离的背根神经节神经元进行以下实验挑战方案:
紫杉醇、c5a和药物在细胞内western实验之前24小时施用。
在如先前所述用适当的刺激物处理24小时之后,将细胞用1×pbs洗涤,并用3.7%甲醛固定15分钟。除去固定溶液之后,将细胞用1×pbs洗涤,并在室温下用1×pbs-0.1%tritonx-100透化5分钟。然后将细胞在4℃下用在odyssey封闭缓冲液中以1∶500稀释的抗乙酰化α-微管蛋白抗体按照制造商的方案进行染色过夜。
第二天,将细胞用1×pbs-0.1%tween20洗涤,并用第二抗体(如方案所建议的,在odyssey封闭缓冲液中以1∶800稀释)和celltag700stain0.2m染色。为了降低背景,将最终浓度为0.2%的tween20添加到odyssey封闭缓冲液中。然后在odysseyclx成像系统上读取96孔板并获取图像。数据分析是通过使用imagestudio2.1软件进行的。
α微管蛋白乙酰化
将drg来源的神经元培养7天,然后用以下进行处理:
-在存在或不存在df3966y(0.1、1和10μm)或df2593a(0.1、1和10μm)下的紫杉醇(200nm、500nm、1μm和5μm、20μm)
-或者在存在或不存在df3966y(1μm)下的c5a(500ng/ml和1μg/ml)处理24小时之后,将细胞通过免疫荧光法进行染色。
免疫荧光法
将细胞用蔗糖-多聚甲醛固定,然后进行染色过夜。将第一抗体(抗iii微管蛋白和抗突触结合蛋白1)在1×pbs-4%bsa-2%正常山羊血清和0.3%tritonx-100中以1∶500稀释。
第二天,将细胞用1×pbs-4%bsa洗涤,并使用在1×pbs-4%bsa-2%正常山羊血清和0.3%tritonx-100中以1∶500稀释的第二样体。在孵育1小时之后,将细胞用1×pbs洗涤。hoechst用作复染剂。
图像分析
用arrayscanxtihcareader(thermofisherscientific)以40倍物镜对每孔采集16张图像。所有分析均用hcsstudio软件(thermofisherscientific)进行。
对于突触计数,已经设定了阈值,具体地,所有对突触结合蛋白染色呈阳性的0.365至2.457μm2的对象均被视为突触。
电生理治疗
将背根神经节(drg)来源的神经元培养7天,然后用以下组合盲法进行处理:10nm紫杉醇,10nm紫杉醇+1μmdf3966y,1μg/mlc5a,1μg/mlc5a+1μmdf3966y。
施用化合物1分钟30秒(短急性刺激)或5分钟(长慢性刺激)。施加化合物之后,在生理对照溶液中清洗等效时间(约10分钟)。
电生理记录
通过膜片钳技术在全细胞配置中进行电生理记录。浴式施加标准的胞外溶液并且其包含以下(mm):nacl135、kcl2、cacl22、mgcl22、hepes10、葡萄糖5,ph为7.4。标准移液管溶液包含以下(mm):天门冬氨酸钾130、nacl10、mgcl22、cacl21.3、egta10、hepes10,ph为7.3。在电流钳模式下,通过pclamp8.2软件和multiclamp700a放大器(axoninstruments)获取记录。
数据分析
对于体内实验,数据报告为平均±sem。图例中的字母n是指每个实验的每个实验组中使用的小鼠数目。实验组之间的差异通过anova(单因素)进行比较,并且个体的比较用bonferroni事后检验随后进行。当在刺激物注射之后的不同时间处测量高伤害性响应时,使用双向anova来比较各组。p<0.05被认为是显著的。
结果
重组c5a的鞘内注射降低机械性和热伤害性阈值
本结果表明,小鼠中重组c5a(100、300和600ng/5μl)的鞘内注射在注射之后长达24小时以剂量依赖性方式促进机械性阈值的降低。在c5ar-/-小鼠中c5a促伤害性作用被取消。较高剂量还导致注射之后5至7小时之间的热潜伏期降低(图1,a和b)。
c5a/c5ar的缺乏决定了医源性疼痛的较少发展
本发明人证明了ciip的发生和维持二者均在c5ar-/-动物中受到损害。实际上,在ciip的诱导阶段和维持阶段期间,c5ar-/-小鼠的机械性(图2,a)和热(图2,b)超敏感性均较少。
这些结果指出了c5a在ciip中的相关性,表明了c5a在不同疼痛途径和纤维的敏化中的作用并且包含了重要的临床症状。
实施例1
df2593a在ciip中的作用
在ciip的以下两个阶段期间,在雄性小鼠中1mg/kgdf2593a的经口施用均能够降低机械性超敏感性:在第8天,诱导结束(图3a,a)和在第14天,建立ciip(图3b,c)。除此之外,在df2593a处理之后,由ciip引起的冷响应(图3a,b和图3b,d)降低。从药物施用之后2小时到6小时,均观察到这些作用。
综上所述,这些数据表明,在建立的疼痛状态下用df2593a进行的经口治疗性治疗能够降低由物理或化学神经病灶引起的机械性和冷超敏感性,其在施用之后持续至少6个小时。
实施例2
在诱导ciip期间用df2593a进行的全身性治疗预防了医源性疼痛的发展
当在ciip的诱导阶段期间长期给予时,df2593a预防了机械性(图4,a)和冷(图4,b)伤害性响应的发展,尽管仅在治疗期间。从ciip模型的第一天到第7天,向没有食物限制的小鼠给予df2593a1mg/kgp.o.的每日两次的剂量(12/12小时)。该药物总是在紫杉醇的施用之前1小时给予。在基线处测量动物,然后从首次紫杉醇注射直至第8天(建立疼痛的时间点)每天测量动物。
该药物能够有效地预防在ciip诱导阶段期间机械性和冷响应的发展。不管怎样,在最后一次df2593a剂量之后24小时未观察到效果,这与先前的结果以及该药物的药代动力学谱一致。
类似的,在具有建立的ciip的雄性小鼠中(第8天和第14天)通过鞘内途径注射10、30或100μg/5μldf2593a能够降低伤害性行为并改善机械性和冷痛觉超敏。尽管10μg/5μl的效力也具有统计学意义,但最相关的效果在较高剂量:在第8天的30和100μg/5μl(图5a,a和b)下观察到。
实施例3
df3966a在紫杉醇诱导的机械性和冷痛觉超敏中的作用
在紫杉醇施用之后,与sham大鼠相比,载剂对照组(ctr)表现出明显的机械性和冷痛觉超敏。特别地,在dpa试验中,爪退缩阈值在第5、7、10和14天导致了显著降低的疼痛迹象(图6,a)。
当与载剂对照动物相比时,用df3966a处理的动物在第3、5、7、10和14天表现出机械性痛觉超敏的显著降低(图6,a)。在冷痛觉超敏实验中,在对照组中,爪退缩阈值数在第3、5、7、10和14天导致了显著降低的疼痛迹象(图6,b)。当与载剂对照动物相比时,用df3966a处理的动物在第3、5、7、10和14天表现出冷痛觉超敏的显著降低。获得的结果清楚地表明,在紫杉醇施用之后第5、7、10和14天,df3966a导致了机械性和冷痛觉超敏的显著降低。
实施例4
在紫杉醇治疗下,df2593a和df3966y对α-微管蛋白水平的影响
培养7天之后,以200nm的浓度将紫杉醇给予至drg神经元(scuteri等,2006)。
如表1中所报道的,相对于对照(ut)细胞,200nm紫杉醇表现出乙酰化α-微管蛋白的微小但显著的提高。
表1
数据为2个不同实验的平均±sd。相对于ut,*p<0.05;相对于紫杉醇,#p<0.05;相对于紫杉醇##p<0.01
如表1中所示,化合物df2593a似乎以剂量依赖性方式逆转了紫杉醇作用。特别地,当以0.1μm的浓度施加时,该药物不能逆转紫杉醇诱导的乙酰化α-微管蛋白的提高。当以1μm的浓度施加df2593a时,该药物逆转了紫杉醇诱导的挑战,并且所观察到的乙酰化α-微管蛋白的水平与对照(ut)细胞相似。在10μm时,该药物似乎对drg神经元产生显著的有益作用,其中相对于对照(ut)细胞乙酰化α-微管蛋白降低了约25%。
化合物df3966y(0.1、1、10μm)显著逆转了紫杉醇诱导的挑战。在最低测试浓度(0.1μm和1μm)下,该药物似乎对drg神经元产生有益作用。
实施例5
df3966y在c5a诱导的神经元毒性中的作用
为了确定所观察到的作用是否也可由c5a触发,在存在或不存在1μmdf3966y的情况下培养7天之后,向drg神经元给予1μg/ml(111.11nm)的重组小鼠c5a。
孵育24小时之后,如先前所述,使用细胞内western技术量化乙酰化α-微管蛋白。
图7显示出1μg/mlc5a能够提高乙酰化α-微管蛋白的水平,并且df3966y显著降低由c5a暴露诱导的挑战。
实施例6
df3966y对神经元功能的影响
在未经处理的细胞中以及在用单独或药物的存在下的紫杉醇和c5a刺激的细胞中,评价了细胞数目、突触数目和树突状分枝。
作为化学治疗诱导的毒性的标志物,drg神经元上突触的总数目被定量评价为相对于未经处理(ut)的对照的突触数目百分比。
树突状分枝直接影响突触的发生和引入。良好发展的分枝表示功能性突触网络。出于该原因,已经量化了树突状分枝所占的面积。
数据作为通过分析每个实验的每个孔的16个视野而获得的值的总和来获得。计算了相对于相对平均的未经处理组的值的百分比。下表2至7中描绘了来自4个独立实验的百分比平均。
表2-在单独或组合的pac(紫杉醇)和df3966y下的细胞数目。数据是3个不同实验的平均±sd。
向drg神经元施用紫杉醇诱导细胞数目的轻微降低。该作用可能是由于存在这样的增殖细胞(purkinje细胞)而非真正的神经元死亡:紫杉醇在该增殖细胞上发挥其抗增殖作用。因此,用df3966y进行的处理,当单独施用或与pac一起施用时,对细胞数目不产生任何显著影响。
表3-在单独或组合的c5a和df3966y下的细胞生存力。数据是2个不同实验的平均±sd。
获得的结果表明,当单独或与1μmdf3966y组合施用时,c5a不会诱导神经元死亡。
表4-在单独或组合的pac和df3966y下的突触数目。数据是3个不同实验的平均±sd。
紫杉醇诱导突触数目的明显降低。用1μmdf3966y进行的处理显著抵消紫杉醇诱导的作用。
这些数据清楚地表明了1μmdf3966y对由化学治疗诱导的神经元功能损害的积极作用。
表5-在单独或组合的c5a和df3966y下的突触数目。数据是3个不同实验的平均±sd。
数据显示,在这些实验条件下,c5a不会诱导神经元活性损伤。
表6-在单独或组合的紫杉醇和df3966y下的树突树面积。数据是3个不同实验的平均±sd。
由树突状分枝所占的面积的非显著性降低是由紫杉醇诱导的,并且用df3966y进行的处理看似可抵消这样的降低。
这些发现与通过量化突触数目获得的结果一致,表明紫杉醇对神经元功能的毒性作用和化合物的积极作用。
表7-在单独或组合的c5a和df3966y下的树突树面积。数据是3个不同实验的平均±sd。
数据表明,急性刺激不足以诱导细胞损伤。
所描述的结果清楚地表明了紫杉醇对神经元功能的有害作用。在另一方面,c5a在此时间点未显示出对神经元功能的任何显著影响,可能是因为其可能是化学治疗诱导的神经元毒性的晚期介质。
用df3966y进行的处理产生了有益的作用,抵消了紫杉醇诱导的神经元突触损失。
实施例7
df3966y和紫杉醇对drg电生理的影响
如图8(基础条件)中所示,生理对照溶液中drg神经元的动作电位放电率(firingrate)长时间的保持稳定,持续多达30至40分钟。
当施用紫杉醇时(图8-处理a),存在动作电位放电率的即时和显著的提高(相对于ctrl,+54.64%),这是去极化阶段的典型特征,在此之后,由于电压设门通道的失活,在更长的暴露时间下,电活性降低(相对于ctrl,-83.51%)。通过施用对照生理溶液,该作用完全恢复。
当在df3966y的存在下施用紫杉醇时(图8-处理b),无论是在第一即时阶段还是在更长的暴露时间下,均未观察到电活性的显著变化。因此,df3966y似乎能够完全恢复因紫杉醇暴露而改变的电活性,无论是在短的暴露时间还是较长的暴露时间下均如此。
实施例8
df3966y和c5a对drg电生理的影响
当向drg神经元施用c5a时(图9-处理c),观察到放电率的即时提高(+55.61%),在此之后,在更长的暴露时间下,电活性显著降低(降低约70%)。通过施用对照生理溶液,该作用完全恢复。
当在df3966y的存在下向drg神经元施用c5a时(图9-处理d),在短的时间点未观察到电活性的显著变化。此外,在较长的暴露时间(3至5分钟)下,df3966y抑制了约35%的电活性,从而改善了c5a诱导的作用。通过施用对照生理溶液,该作用完全恢复。
当单独施用时,两种挑战(紫杉醇和c5a)对drg神经元均产生有害作用,尽管其本质不同:紫杉醇是比c5a更强效的刺激物。
在急性刺激下观察到的即时作用是(在紫杉醇和c5a的情况下)放电率的提高,这是去极化阶段的典型特征,在此之后,由于电压设门通道的失活,在更长的暴露时间下,电活性降低。
这种作用可被df3966y抵消,特别是在紫杉醇刺激时。
- 还没有人留言评论。精彩留言会获得点赞!