基于复杂网络与机器学习方法的肿瘤驱动基因预测系统与流程
本发明属于数据分析领域,具体是肿瘤数据研究领域,涉及一种基于复杂网络与机器学习方法的肿瘤驱动基因预测系统。
背景技术:
国家癌症中心发布的2017年最新《中国肿瘤现状和趋势》表明,恶性肿瘤(癌症)已经成为我国居民死亡主要原因之一,是威胁我国居民生命健康的最大杀手。肺癌和乳腺癌分别居我国男性和女性的肿瘤发病首位,癌症发病随年龄增加而逐渐上升,四十岁之后增长较快。与世界相比,中国癌症发病率约占全球22%,发病人数全球第一,死亡率高于全球平均水平。
目前,恶性肿瘤大数据网络建设初见成效,依托大数据构建医疗防线,搭建大数据中心环境和平台体系,开展基于肿瘤大数据中心和大数据平台的大数据应用研究,必将推动中国癌症事业向前发展。
驱动基因,是与癌症发生发展相关的重要基因,基于驱动基因的精准医学是治疗癌症的重要方向。我国肿瘤防控工作研究难点有:预防难、发现晚、不好治、不规范。其中,肿瘤治疗效果差、复发转移率高且肿瘤治疗副作用大、精准性差等是导致肿瘤治疗难度大的原因。本发明的目的是通过数据挖掘的方法,对潜在肿瘤驱动基因进行预测,在一定程度上加深对癌症的认识,进而推动癌症治疗的发展。
基因网络能够很好的反应基因-基因之间的复杂关系,为驱动基因识别提供了新的思路。基因网络中基因之间的作用关系存在正调控作用(促进基因表达)和反调控作用(抑制基因表达)。因此,在构建网络上,节点之间的边权表示是存在正负之分的。在处理关于肿瘤基因网络时,这种复杂的边关系要求更高的算法匹配性,以揭示肿瘤网络中基因间相互作用机制。带符号随机漫步重启算法(signedrandomwalkwithrestart,srwr)是一种用于符号网络的个性化排序算法。传统的基于随机漫步(randomwalk)的方法,如pagerank算法和带重启随机漫步算法(randomwalkwithrestart,rwr),只适用于假设为正边的网络,而不能在有符号的网络中对节点进行有效的排名,并且缺乏考虑复杂边关系的能力,而带符号随机漫步重启弥补了这一缺点。
肿瘤基因网络中节点的特征表达以及预测模型的训练是驱动基因挖掘算法中比较核心的内容,算法模型避开了人工筛选的盲目性,极大地节约了时间成本及资金成本,并且通过整合基因信息提高预测的准确性,保证算法的高效灵活和可扩展等。然而,目前还没有专门针对生物信息数据分析而设计的整合复杂网络和机器学习方法的肿瘤驱动基因预测的研究和应用。
技术实现要素:
本发明的目的在于消除临床实验的随机性,提供一种基于复杂网络与机器学习的肿瘤驱动基因预测方法,整合基因网络数据、基因突变数据和基因差异表达数据,通过引入节点的网络结构性指标、评价网络节点影响力的k-shell(k-壳)分解、提供个性化排序的带符号随机漫步重启算法,以及机器学习预测算法,发现候选驱动基因。
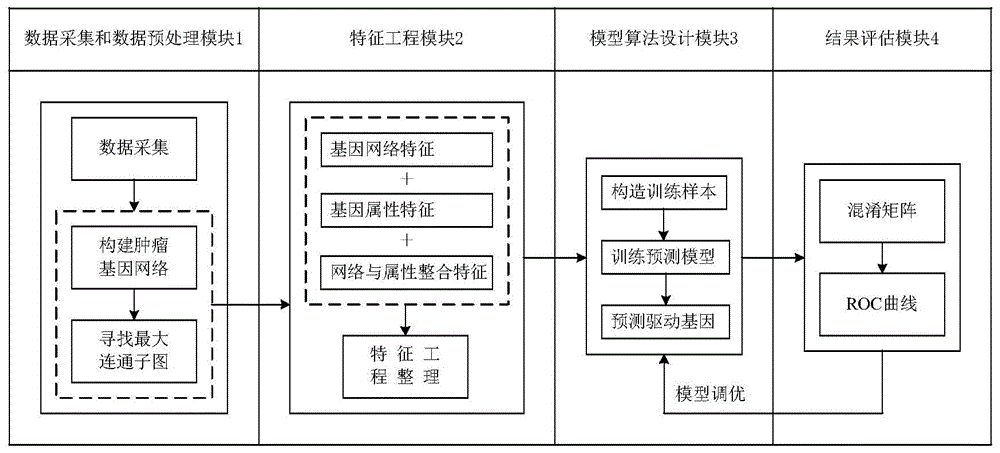
本发明包括数据采集和数据预处理模块、特征工程模块、模型算法设计模块、结果评估模块,具体如下:
(1).数据采集和数据预处理模块:
该模块包括数据采集、构建肿瘤基因网络,以及最大连通子图的筛选,为驱动基因预测提供数据基础,其中:
(1-1).数据采集:肿瘤基因相互作用关系数据、基因差异表达数据、患者基因突变数据。
(1-2).构建肿瘤基因网络:筛选肿瘤样本中,相互作用关系显著的基因对构建肿瘤基因网络g=(v,e),v表示节点集,e表示边集;节点代表基因,边代表两个基因所编码蛋白质之间的相互作用关系,且边的权值有正负之分,即基因间促进表达的边权为正数,基因间抑制表达的边权为负数。
(1-3).筛选最大连通子图:剔除个别孤立节点,筛选出最大连通子图,即网络g′。
(2).特征工程模块:
在机器学习相关问题的研究中,数据和特征决定了预测结果的上限,而模型和算法只是逼近这个上限而已。本发明的特征工程模块就是解决前半句的特征选择问题,即为了模型算法更好的学习基因特征,达到最准确的预测结果。该模块是基于网络g′的特征工程,包括特征工程提取和特征工程整理。
(2-1).特征工程提取:包括三类特征提取,分别是基因网络特征、基因属性特征、网络与属性整合特征;
所述的基因网络特征包括网络结构属性和k-shell值;
所述的基因属性特征包括基因突变频次和基因差异表达量;
所述的网络与属性整合特征包括以基因突变频次和基因差异表达量分别为初值进行带符号随机漫步重启srwr得到的结果。
(2-1-1).基因网络结构属性包含四个指标:度中心性、接近中心性、介数中心性、特征向量中心性。
计算方法分别是:
a.度中心性:节点度越大意味着节点的度中心性越高,在网络中就越重要;
cd(i)表示节点i的度中心性,∑j∈na(i,j)表示节点i和节点j直接相连的数量,n表示节点个数;如节点i和节点j直接相连,a(i,j)=1,如节点i和节点j不直接相连,a(i,j)=0。
b.接近中心性:反映在网络中某一节点与其他节点之间的接近程度;
ccl(i)表示节点i的接近中心性,d(i,j)表示节点i与节点j的距离。
c.介数中心性:以经过某节点的最短路径数目来刻画节点的结构属性;
cb(i)表示节点i的介数中心性,σst表示节点s与节点t之间最短路径总数,σst(i)表示节点s与节点t之间通过节点i的最短路径数量。
d.特征向量中心性:一个节点的结构属性既取决于其邻居节点的数量,也取决于其邻居节点的结构属性;
m(i)表示节点i的邻居集;如节点i和节点t直接相连,ai,t=1,如节点i和节点t不直接相连,ai,t=0;
ax=λx;
a表示邻接矩阵,a=(ai,t);x是矩阵a的特征值λ对应的特征向量;
给定初值x(0),然后采用迭代算法计算向量x,直到归一化的x′(t)=x′(t-1),迭代算法为:
(2-1-2).k-shell值:k-shell分解是计算网络中节点影响力的方法,基于k-shell算法挖掘肿瘤基因网络中基因节点的结构属性,递归地剥离网络中度数小于k的节点,计算每个基因的k-shell值,作为每个基因的特征。
(2-1-3).基因突变频次:根据肿瘤患者的突变基因数据,统计突变基因在不同患者中出现的次数,得到基因突变频次作为基因属性的突变特征。
(2-1-4).基因差异表达量:筛选基因的差异表达数据,即基因在正常细胞和肿瘤细胞中的表达量,是否具有显著差异,作为基因属性的表达特征。
(2-1-5).网络与属性整合特征:采用带符号随机漫步重启算法,揭示肿瘤基因网络中基因间的作用机制,并以基因的突变频次和差异表达量分别作为初值,进行带符号随机漫步重启srwr得到整合特征,即计算每个基因的srwr得分向量,步骤如下:
①计算节点的抑制表达得分r-和促进表达得分r+:
②计算节点srwr得分r=r+-r-。
(2-2).特征工程整理:
是对以上特征工程提取的所有特征进行整理,整理成预测模型能够识别的数据格式,并对缺失值进行处理:
(2-2-1).以网络g′中的基因为准,整合所有特征;
(2-2-2).处理基因差异表达特征和基因突变特征的缺失数据,对基因差异表达特征的数据缺失做均值填充,对基因突变特征的数据缺失直接补零。
在特征工程模块,基于基因相关特征的提取与整理,得到了可提供给模型学习和训练的结构化数据。
(3).模型算法设计模块:包括构造训练样本、预测模型设计。
(3-1).构造训练样本:肿瘤基因包括已知的驱动基因和非已知驱动基因组成的普通基因,且普通基因都有成为驱动基因的概率,无非是概率大小的问题,本发明的目的就是从普通基因中预测出可能是驱动基因概率较大的基因,所以,可以假设普通基因是驱动基因的概率肯定不大于已知驱动基因是驱动基因的概率,基于这个假设采用pairwise模型构造训练样本,即从驱动基因中抽取一个正样本的同时也从普通基因中抽取一个负样本构造训练样本,得到数量相同的成对正负训练样本,将数据随机划分成90%和10%,其中90%的数据用于后续模型训练,寻找最优模型参数,剩下10%的数据用于测试模型的预测效果。
(3-2).预测模型设计:采用随机森林,以决策树作为基学习器构建随机森林,具体如下:
(3-2-1).从划分好的用于训练的样本中随机采样m个样本,每个样本的特征属性均来自特征工程模块提取的特征,构成一个训练集,并训练一个决策树模型,其中训练过程中的节点划分是以信息增益为准则选取特征属性划分决策树的,并且这里是从所有特征中随机抽取部分特征寻找最优解;信息增益:
(3-2-2).重复(3-2-1),进行n次采样,并训练得到n个决策树;
(3-2-3).将生成的n个决策树组成随机森林,然后将划分好的用于测试的样本放入训练好的随机森林模型中进行预测,并根据决策树分类结果投票决定最终的预测结果;投票规则如下:
h(x)表示基因x的最终预测类别,0表示普通基因,1表示驱动基因;hi(x)表示基因x在决策树hi下的预测类别:当x在hi下的预测类别为驱动基因,则
(4).结果评估模块:采用混淆矩阵和roc曲线验证模型的预测效果;
所述的混淆矩阵是评判模型结果的指标,属于模型评估的一部分,用n行n列的矩阵形式表示,把预测结果的精度显示在一个混淆矩阵里面,每一列代表预测类别,每一列的总数表示预测为该类别的数据的数目,每一行表示数据的真实归属类别,每一行的总数表示该类别的数据实例的数目。
所述的roc曲线是从医疗分析领域引入的一种新的分类模型性能评价方法,适用于二分类的研究问题,roc空间将假阳性率fpr定义为x轴,真阳性率tpr定义为y轴,roc曲线所覆盖的面积定义为auc,且auc值越大,即越接近于1,表示模型的预测效果越好。
roc曲线的真阳性率tpr和假阳性率fpr通过混淆矩阵计算如下:
tp表示将测试集中的驱动基因预测为驱动基因的个数;fp表示将测试集中的普通基因预测为驱动基因的个数;fn表示将测试集中的驱动基因预测为普通基因的个数;tn表示将测试集中的普通基因预测为普通基因的个数;
根据模型算法设计模块(3-2-3)的预测结果,绘制roc曲线并计算auc,并寻找最优预测结果下的模型参数,所述的auc为roc曲线所覆盖的面积;在最优预测模型下,计算所有普通基因是驱动基因的概率,筛选出可能性大的基因作为候选驱动基因集。
本发明从数据挖掘的角度研究肿瘤驱动基因,通过不断的特征整合与处理,结合网络节点的结构特征、k-shell分解、带符号随机漫步重启、机器学习等方法实现驱动基因预测。因此,本发明能够有效分析肿瘤数据,并预测驱动基因,进而为医学实验验证提供科学指导,提高肿瘤驱动基因挖掘效率,在一定程度上促进肿瘤治疗工作的发展。
附图说明
图1是本发明流程图。
具体实施方式
下面结合技术方案和附图,详细说明本发明的具体实施。
现有乳腺癌的基因网络数据、差异表达信息,以及乳腺癌患者的突变基因等数据,包括1050个患者基因突变数据,14000个左右的基因。
如图1所示,一种基于复杂网络与机器学习方法的肿瘤驱动基因预测系统,包括数据采集和数据预处理模块1、特征工程模块2、模型算法设计模块3、结果评估模块4。
第一步:根据已有的肿瘤样本中的基因相互作用关系数据,筛选相互作用关系显著的基因对(通过设置阈值,一般选择p-value小于0.05的边),构建肿瘤基因网络g=(v,e),v表示节点集,e表示边集;节点代表基因,边代表两个基因所编码蛋白质之间的相互作用关系,且边的权值有正负之分,即基因间促进表达的边权为正数,基因间抑制表达的边权为负数。
第二步:剔除个别孤立节点,筛选出最大连通子图,即网络g′。
1)第三步:特征工程模块包括三类特征提取,分别计算了网络中节点的网络结构属性、k-shell值、肿瘤基因突变频次,以及基因的差异表达数据和srwr得分。
2)1、计算网络g′中节点的网络结构属性,包含四个指标:度中心性、接近中心性、介数中心性、特征向量中心性,作为基因网络的结构属性特征。
(1).度中心性:节点度越大意味着节点的度中心性越高,在网络中就越重要;
cd(i)表示节点i的度中心性,∑j∈na(i,j)表示节点i和节点j直接相连的数量,n表示节点个数;如节点i和节点j直接相连,a(i,j)=1,如节点i和节点j不直接相连,a(i,j)=0。
(2).接近中心性:反映在网络中某一节点与其他节点之间的接近程度;
ccl(i)表示节点i的接近中心性,d(i,j)表示节点i与节点j的距离。
(3).介数中心性:以经过某节点的最短路径数目来刻画节点的结构属性;
cb(i)表示节点i的介数中心性,σst表示节点s与节点t之间最短路径总数,σst(i)表示节点s与节点t之间通过节点i的最短路径数量。
(4).特征向量中心性:一个节点的结构属性既取决于其邻居节点的数量,也取决于其邻居节点的结构属性;
m(i)表示节点i的邻居集;如节点i和节点t直接相连,ai,t=1,如节点i和节点t不直接相连,ai,t=0;
ax=λx;
a表示邻接矩阵,a=(ai,t);x是矩阵a的特征值λ对应的特征向量;
给定初值x(0),然后采用迭代算法计算向量x,直到归一化的x′(t)=x′(t-1),迭代算法为:
2、k-shell值:k-shell分解是计算网络中节点影响力的方法,基于k-shell算法挖掘肿瘤基因网络中基因节点的结构属性,递归地剥离网络中度数小于k的节点,计算每个基因的k-shell值,作为每个基因的特征。
3、基因突变频次:根据乳腺癌患者的基因突变数据,统计突变基因在不同患者中出现的次数,得到基因突变频次作为基因属性的突变特征;
4、基因差异表达量:筛选基因的差异表达数据,即基因在正常细胞和乳腺癌细胞中的表达量,是否具有显著差异,将此作为基因属性的表达特征。
5、网络与属性整合特征:采用带符号随机漫步重启算法,揭示肿瘤基因网络中基因间的作用机制,并以基因的突变频次和差异表达量分别作为初值,进行带符号随机漫步重启srwr得到整合特征,即计算每个基因的srwr得分向量,步骤如下:
①计算节点的抑制表达得分r-和促进表达得分r+:
②计算节点srwr得分r=r+-r-;
第四步:整理以上特征工程提取的所有的特征,并针对肿瘤基因的突变特征和差异表达特征进行缺失值处理。
以网络g′中的基因为准,整合所有特征;处理基因差异表达特征和基因突变特征的缺失数据,对基因差异表达特征的数据缺失做均值填充,对基因突变特征的数据缺失直接补零。
第五步:构造训练样本:肿瘤基因包括已知的驱动基因和非已知驱动基因组成的普通基因,且普通基因都有成为驱动基因的概率,无非是概率大小的问题,本发明的目的就是从普通基因中预测出可能是驱动基因概率较大的基因,所以,可以假设普通基因是驱动基因的概率肯定不大于已知驱动基因是驱动基因的概率,基于这个假设采用pairwise模型构造训练样本,即从驱动基因中抽取一个正样本的同时也从普通基因中抽取一个负样本构造训练样本,得到数量相同的成对正负训练样本,将数据随机划分成90%和10%,其中90%的数据用于后续模型训练,寻找最优模型参数,剩下10%的数据用于测试模型的预测效果。
第六步:预测模型算法选择被誉为“代表集成学习技术水平的方法”的随机森林,以决策树作为基学习器构建随机森林。具体实现过程如下:
1).从划分好的90%用于训练的样本中随机有放回地采样400个基因样本构成一个训练集,并训练一个决策树模型,其中训练过程中的节点划分是以信息增益为准则选取特征属性划分决策树的。“信息增益”定义为:
2).重复1),进行n次采样,并训练得到n个决策树;
3).将生成的n个决策树组成随机森林,然后将划分好的用于测试的样本放入训练好的随机森林模型中进行预测,并根据决策树分类结果投票决定最终的预测结果;投票规则如下:
h(x)表示基因x的最终预测类别,0表示普通基因,1表示驱动基因;hi(x)表示基因x在决策树hi下的预测类别:当x在hi下的预测类别为驱动基因,则
第七步:绘制roc曲线并计算auc,并寻找最优预测结果下的模型参数。在最优预测模型参数下,计算所有普通基因是驱动基因的概率,筛选出可能性比较大的基因作为候选驱动基因集。
roc曲线是从医疗分析领域引入的一种新的分类模型性能评价方法,适用于二分类的研究问题,roc空间将假阳性率fpr定义为x轴,真阳性率tpr定义为y轴,roc曲线所覆盖的面积定义为auc,且auc值越大,即越接近于1,表示模型的预测效果越好;
roc曲线的真阳性率tpr和假阳性率fpr通过混淆矩阵计算如下:
tp表示将测试集中的驱动基因预测为驱动基因的个数;fp表示将测试集中的普通基因预测为驱动基因的个数;fn表示将测试集中的驱动基因预测为普通基因的个数;tn表示将测试集中的普通基因预测为普通基因的个数。根据模型算法设计模块的预测结果,绘制roc曲线并计算auc,并寻找最优预测结果下的模型参数,所述的auc为roc曲线所覆盖的面积;在最优预测模型下,计算所有普通基因是驱动基因的概率,筛选出可能性大的基因作为候选驱动基因集。
- 还没有人留言评论。精彩留言会获得点赞!