一种显性表示蛋白口袋表层原子与配体相互作用满足程度的方法与流程
本发明涉及计算机辅助药物设计领域,尤其涉及一种显性表示蛋白口袋表层原子与配体相互作用满足程度的方法。
背景技术:
在药物设计的整个过程中,对于活性小分子的合理修改以提高其与蛋白靶点的生物活性是主要的工作之一,其准确有效的修改有利于提高药物开发的成本和时间周期。一般情况下,药物化学研究人员总是依靠着对于小分子化合物结构的观察并结合个人的经验来对小分子化合物的某些片段或者官能团进行修饰或者加以改变。亦或者,有计算条件并且掌握对接软件界面使用方法的研究人员,可以依据手动的方式观测对接后小分子与蛋白靶点的相对位置和接触关系,比如也可以依靠薛定谔或者autodock等提供的界面软件,而这样的方式极大地依赖于研究人员的经验,效率较低。
蛋白质与配体小分子的结合亲和力往往由多种物理化学相互作用项所综合组成,如氢键相互作用,范德华相互作用,电荷相互作用和疏水相互作用等。而对于可形成氢键的原子或者带正电或者负电的原子,其关键相互作用是否被满足对小分子的活性有着较大的影响。传统的经验打分函数一般都是由线性拟合而成,部分打分函数的最后分数体现在由蛋白口袋氨基酸原子与配体小分子原子通过经验方程计算后乘以拟合系数相加而成,故而原子与原子间某项相互作用的大小也可以一定程度的用经验方程结合与拟合系数的相乘来近似表示。
因此,对蛋白口袋表层的氨基酸残基原子相互作用的近似计算,并且以显性的不同颜色来表示所提供的相互作用满足情况图像和以客观的相对满足度大小来表示所提供的满足度列表,有利于药物化学研究人员更直观和目的性的判断出需要修改的结构部分和更为高效的进行分子修饰。
技术实现要素:
本发明的目的在于提供一种显性表示蛋白口袋表层原子与配体相互作用满足程度的方法,以帮助药物化学研究人员进行药物设计和修改。
实现本发明目的的具体技术方案是:
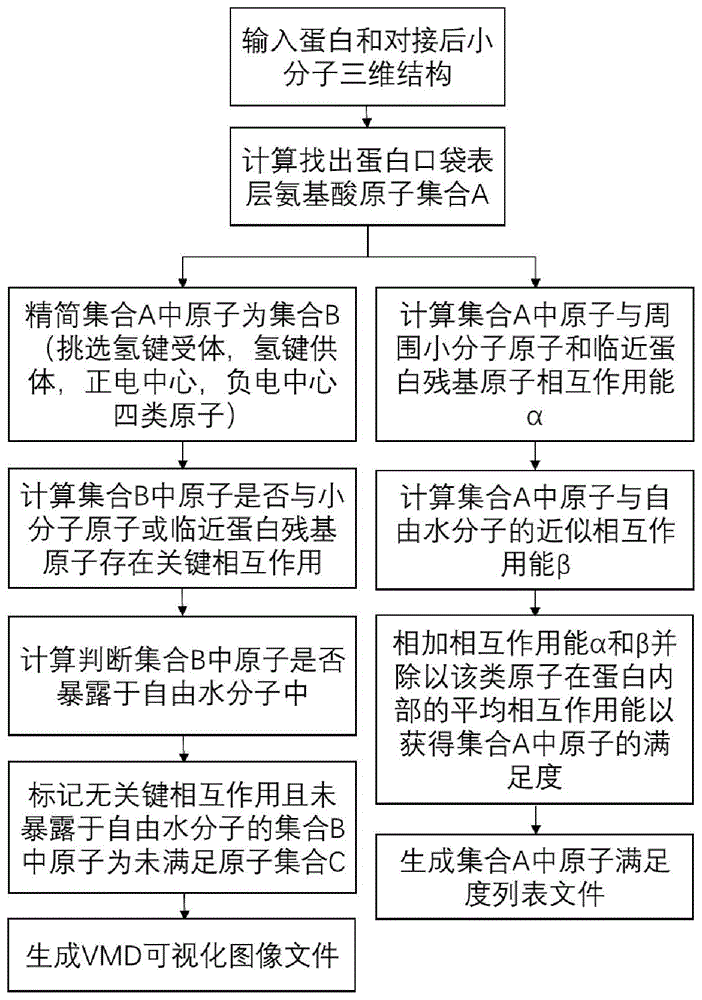
一种显性表示蛋白口袋表层原子与配体相互作用满足程度的方法,该方法包括以下具体步骤:
步骤1:从rcsb-pdb数据库中获取蛋白pdb三维结构,并用小分子画图软件画出小分子结构以生成小分子文件;
步骤2:分子对接,利用分子对接软件进行分子对接,小分子保留打分最好的构象作为对接后小分子结构;
步骤3:计算找出蛋白口袋表层氨基酸原子为原子集合a,利用naccess软件计算蛋白pdb三维结构中所有蛋白重原子的相对暴露表面积,并定义距离对接后小分子
步骤4:依据原子性质筛选集合a中原子为原子集合b,只保留可作为氢键受体,氢键供体,正电中心,负电中心四类原子作为集合b中的原子;
步骤5:计算判断集合b中各原子是否与小分子原子或临近蛋白残基原子存在关键相互作用,关键相互作用具体指可作为氢键受体的原子与在
步骤6:计算判断集合b中各原子是否暴露于自由水分子中,暴露于自由水分子中的判断标准为蛋白小分子复合物pdb三维结构中原子的相对暴露表面积大于0;
步骤7:标记按步骤5计算未存在关键相互作用且按步骤6计算未暴露于自由水分子中的原子为原子集合c,对于集合c中的所有原子都标记为-1,对于属于集合b而不属于集合c的原子标记为1;
步骤8:编写vmd软件可视化图像文件,包括:重写蛋白和小分子pdb文件并将步骤7中的标记信息记录于pdb文件的occupancy这一栏;编写vmd命令行文件,主要体现为以occupancy形式展示原子颜色,标记为1的显示为蓝色,标记为-1的显示为红色,未标记的显示为白色,以cpk形式展现步骤7中被标记的原子形状,其中occupancy形式和cpk形式都为vmd软件中的参数;
步骤9:分别计算步骤3中集合a中各原子与周围小分子原子和临近蛋白残基原子相互作用能α,两原子相互作用能计算方式为:两原子之间的范德华相互作用能乘以范德华系数、疏水相互作用能乘以疏水系数、氢键相互作用能乘以氢键系数、电荷相互作用能乘以电荷系数之和;其中,范德华系数、疏水系数、氢键系数、电荷系数由线性拟合得到,范德华相互作用能、疏水相互作用能、氢键相互作用能、电荷相互作用能定义如下:
1>范德华相互作用能为:
其中,vdw指两原子间的范德华相互作用能,d0指两原子的半径之和,dis指两原子之间的距离;
2>疏水相互作用能为:
其中,hc指两原子间的疏水相互作用能,d0指两原子的半径之和,dis指两原子之间的距离;
3>氢键相互作用能为:
要求两原子分别为氢键供体和氢键受体,且两原子间的距离小于
其中,hb指两原子间的氢键相互作用能,dis指两原子之间的距离;
4>电荷相互作用能为:
其中,ele指两原子间的电荷相互作用能,dis指两原子之间的距离;
步骤10:分别计算步骤3中集合a中各原子与自由水分子的近似相互作用能β,原子与自由水分子的近似相互作用能计算公式定义如下:
其中,hohi表示原子ai与自由水分子的近似相互作用能,r表示气体摩尔常数,t表示热力学温度,这里表示室温下温度等于298k,acci表示原子ai在蛋白小分子复合物结构中的相对暴露表面积,
步骤11:相加相互作用能α和β并除以该类原子在蛋白内部的平均相互作用能以获得集合a中原子的满足度,同类氨基酸原子在蛋白内部的平均相互作用能由蛋白内部与该原子同类的氨基酸原子的相互作用能统计平均而得,具体如下:每一个与原子ai同类的氨基酸原子mij的相互作用能为该原子与临近蛋白残基原子的相互作用能之和;其中,原子mij是指与原子ai同类的氨基酸原子中的第j个原子,临近蛋白残基原子具体指去除与原子mij所在氨基酸残基直接相连的氨基酸残基后所有与原子mij在
步骤12:生成集合a中原子满足度列表文件。
本发明通过识别蛋白口袋表层原子与结合小分子和周围其它蛋白原子的相互作用关系,标记无关键相互作用的蛋白口袋表层原子和小分子原子生成可视化文件,并计算蛋白口袋表层原子的相互作用满足程度生成满足度列表。构建了一种更加直观的观测蛋白口袋表层氨基酸残基原子的相互作用情况的方法。
本发明的基本功能是直观的观测蛋白口袋表层氨基酸残基原子的相互作用满足情况。相比于直接观测,本发明的有益效果为:第一,以图像的形式显性的表示蛋白口袋表层氨基酸残基原子和小分子上的原子是否满足关键相互作用,其中满足的原子以蓝色表示,不满足的以红色表示;第二,以相对满足度大小来直观的体现蛋白口袋表层氨基酸残基原子与周围环境原子的相互作用满足程度,对满足程度普遍较低的系列原子所在的子口袋所对应位置的分子片段可考虑进行修饰,有利于有方向性的进行分子结构优化修改。
附图说明
图1为本发明流程图;
图2为vmd可视化图像;
图3为满足度列表。
具体实施方式
本发明可以通过输入蛋白pdb三维结构和小分子文件,然后识别蛋白口袋表层原子与结合小分子和周围其它蛋白原子的相互作用关系,标记无关键相互作用的蛋白口袋表层原子和小分子原子生成可视化文件,并计算蛋白口袋表层原子的相互作用满足程度生成满足度列表,从而直观显性的体现蛋白口袋表层氨基酸残基原子的相互作用情况。
本发明的具体步骤:
步骤1:从rcsb-pdb数据库中获取蛋白pdb三维结构,并用小分子画图软件画出小分子结构以生成小分子文件;
步骤2:分子对接,利用分子对接软件进行分子对接,小分子保留打分最好的构象作为对接后小分子结构;
步骤3:计算找出蛋白口袋表层氨基酸原子为原子集合a,利用naccess软件计算蛋白pdb三维结构中所有蛋白重原子的相对暴露表面积,并定义距离对接后小分子
步骤4:依据原子性质筛选集合a中原子为原子集合b,只保留可作为氢键受体,氢键供体,正电中心,负电中心四类原子作为集合b中的原子;
步骤5:计算判断集合b中各原子是否与小分子原子或临近蛋白残基原子存在关键相互作用,关键相互作用具体指可作为氢键受体的原子与在
步骤6:计算判断集合b中各原子是否暴露于自由水分子中,暴露于自由水分子中的判断标准为蛋白小分子复合物pdb三维结构中原子的相对暴露表面积大于0;
步骤7:标记按步骤5计算未存在关键相互作用且按步骤6计算未暴露于自由水分子中的原子为原子集合c,对于集合c中的所有原子都标记为-1,对于属于集合b而不属于集合c的原子标记为1;
步骤8:编写vmd软件可视化图像文件,包括:重写蛋白和小分子pdb文件并将步骤7中的标记信息记录于pdb文件的occupancy这一栏;编写vmd命令行文件,主要体现为以occupancy形式展示原子颜色,标记为1的显示为蓝色,标记为-1的显示为红色,未标记的显示为白色,以cpk形式展现步骤7中被标记的原子形状,其中occupancy形式和cpk形式都为vmd软件中的参数;生成的图像如图2所示,其中,红色的圆球表示未存在关键相互作用也未暴露于自由水分子中的集合b中的原子,蓝色的圆球表示存在关键相互作用或者暴露于自由水分子中的集合b中的原子;
步骤9:分别计算步骤3中集合a中各原子与周围小分子原子和临近蛋白残基原子相互作用能α,两原子相互作用能计算方式为:两原子之间的范德华相互作用能乘以范德华系数、疏水相互作用能乘以疏水系数、氢键相互作用能乘以氢键系数、电荷相互作用能乘以电荷系数之和;其中,范德华系数、疏水系数、氢键系数、电荷系数由线性拟合得到,范德华相互作用能、疏水相互作用能、氢键相互作用能、电荷相互作用能定义如下:
1>范德华相互作用能为:
其中,vdw指两原子间的范德华相互作用能,d0指两原子的半径之和,dis指两原子之间的距离;
2>疏水相互作用能为:
其中,hc指两原子间的疏水相互作用能,d0指两原子的半径之和,dis指两原子之间的距离;
3>氢键相互作用能为:
要求两原子分别为氢键供体和氢键受体,且两原子间的距离小于
其中,hb指两原子间的氢键相互作用能,dis指两原子之间的距离;
4>电荷相互作用能为:
其中,ele指两原子间的电荷相互作用能,dis指两原子之间的距离;
步骤10:分别计算步骤3中集合a中各原子与自由水分子的近似相互作用能β,原子与自由水分子的近似相互作用能计算公式定义如下:
其中,hohi表示原子ai与自由水分子的近似相互作用能,r表示气体摩尔常数,t表示热力学温度,这里表示室温下温度等于298k,acci表示原子ai在蛋白小分子复合物结构中的相对暴露表面积,
步骤11:相加相互作用能α和β并除以该类原子在蛋白内部的平均相互作用能以获得集合a中原子的满足度,同类氨基酸原子在蛋白内部的平均相互作用能由蛋白内部与该原子同类的氨基酸原子的相互作用能统计平均而得,具体如下:每一个与原子ai同类的氨基酸原子mij的相互作用能为该原子与临近蛋白残基原子的相互作用能之和;其中,原子mij是指与原子ai同类的氨基酸原子中的第j个原子,临近蛋白残基原子具体指去除与原子mij所在氨基酸残基直接相连的氨基酸残基后所有与原子mij在
步骤12:生成集合a中原子满足度列表文件。生成的列表如图3所示,其中res_atom_name这一列表示蛋白中具体某个氨基酸残基原子,atom_num这一列表示蛋白原子原子序号,energy这一列表示蛋白原子与周围原子得相互作用能大小,atom_dehyde这一列表示蛋白原子与自由水分子得近似相互作用能,atom_satu这一列表示蛋白原子的相对满足度。拿图3列表中第一行数据举例,其蛋白原子属于tyr(络氨酸),氨基酸序号为6,原子名为ce1,原子序号为111,其与小分子原子和周围氨基酸残基原子的相互作用能为-0.761012,其与自由水分子的近似相互作用能为0.010770,其相对满足度为1.215402,表明该原子此时相对于该类原子在蛋白内部的被满足情况更好。
- 还没有人留言评论。精彩留言会获得点赞!