用于检测突变的方法、电子设备和计算机存储介质与流程
本公开总体上涉及生物信息检测处理,并且具体地,涉及用于检测突变的方法、电子设备和计算机存储介质。
背景技术:
突变分为体细胞突变和生殖细胞突变。例如,体细胞突变是发生在正常机体细胞中的突变,比如发生在皮肤或器官中的突变。恶性肿瘤的散发形式可以通过体细胞突变引起。传统的检测体突变,特别是细胞突变的方案例如是:导入bam文件并读取igv图像,以及通过人工判断突变真假。
在上述传统的检测突变的方案中,导入bam文件并读取igv图像需要一定的时间。另外,人工判断真假突变的效率较为低下,而且会因为疲劳和经验局限等原因导致判断真假突变发生错误。因此,迫切需要能够有效提高检测突变的效率和准确性的方案。
技术实现要素:
本公开提供一种检测突变的方法、电子设备和计算机存储介质,能够有效提高检测突变的效率和准确率。
根据本公开的第一方面,提供了一种检测突变的方法。该方法包括:获取关于待测样本的序列与参考基因组序列的比对结果信息,关于待测样本包括待测血液样本和待测组织样本中的至少一种;基于比对结果信息,生成突变位点碱基信息,突变位点碱基信息包括碱基质量和比对质量中的至少一项;基于预测模型,提取突变位点碱基信息的特征值,预测模型经由对多个训练样本的机器学习而生成;以及基于所提取的特征值,预测突变信息。
根据本发明的第二方面,还提供了一种计算设备,该设备包括:存储器,被配置为存储一个或多个计算机程序;以及处理器,耦合至存储器并且被配置为执行一个或多个程序使装置执行本公开的第一方面的方法。
根据本公开的第三方面,还提供了一种非瞬态计算机可读存储介质。该非瞬态计算机可读存储介质上存储有机器可执行指令,该机器可执行指令在被执行时使机器执行本公开的第一方面的方法。
提供发明内容部分是为了以简化的形式来介绍对概念的选择,它们在下文的具体实施方式中将被进一步描述。发明内容部分无意标识本公开的关键特征或主要特征,也无意限制本公开的范围。
附图说明
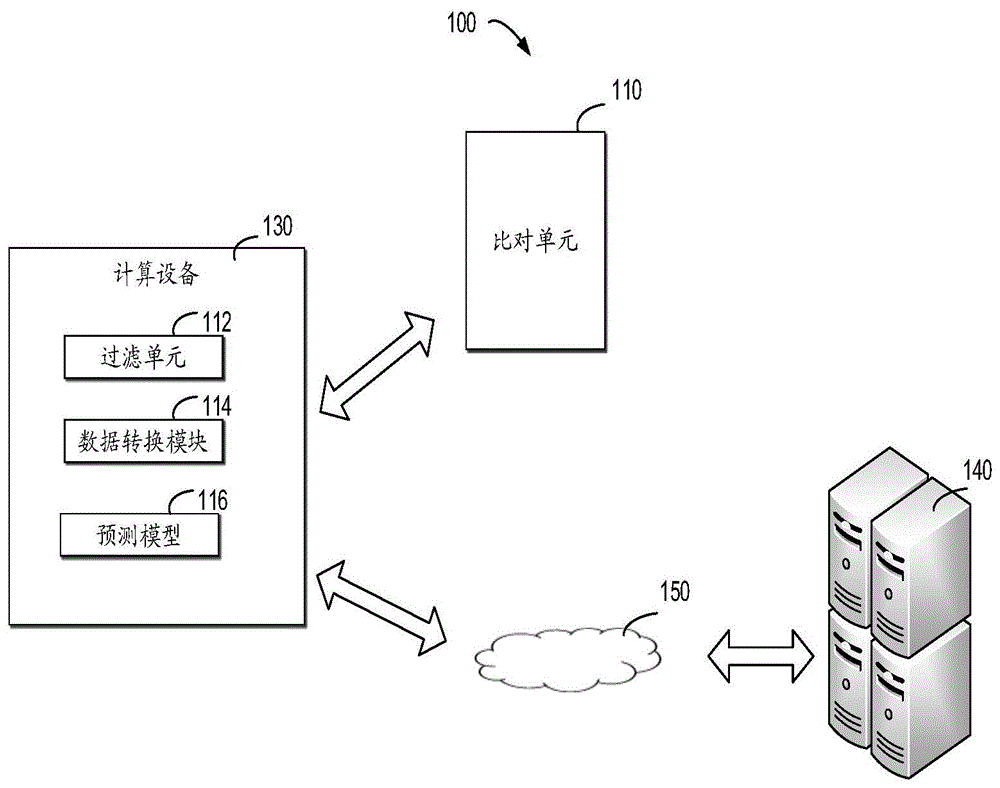
图1示出了根据本公开的实施例的用于检测突变的方法的系统100的示意图;
图2示出了根据本公开的实施例的用于指示单核苷酸多态性突变类型的比对信息200的示意图;
图3示出了根据本公开的实施例的用于指示碱基发生了删除或插入的突变类型的比对信息300的示意图;
图4示出了根据本公开的实施例的用于指示多个碱基变异为多个碱基delins的突变类型的比对信息400的示意图;
图5示出了根据本公开的实施例的用于检测突变的方法500的流程图;
图6示出了根据本公开的实施例的预测模型600的结构示意图;
图7示出了根据本公开的残差学习网络结构700的示意图;
图8示出了根据本公开的实施例的预测模型800的结构示意图;
图9示出了根据本公开的实施例的用于生成突变位点碱基信息的方法900的流程图;
图10示出了根据本公开的实施例的经由测序获得的比对结果信息1000的示意图;
图11示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1100的流程图;
图12示出了根据本公开的实施例的经由测序获得的比对结果信息1200的示意图;
图13示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1300的流程图;
图14示出了根据本公开的实施例的经由测序获得的比对结果信息1400的示意图;
图15示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1500的流程图;
图16示出了根据本公开的实施例的经由测序获得的比对结果信息1600的示意图;以及
图17示意性示出了适于用来实现本公开实施例的电子设备1700的框图。
在各个附图中,相同或对应的标号表示相同或对应的部分。
具体实施方式
下面将参照附图更详细地描述本公开的优选实施例。虽然附图中显示了本公开的优选实施例,然而应该理解,可以以各种形式实现本公开而不应被这里阐述的实施例所限制。相反,提供这些实施例是为了使本公开更加透彻和完整,并且能够将本公开的范围完整地传达给本领域的技术人员。
关于比对信息来源,以下实施例中,在进行之前,先对待测样本通过探针捕获得到的各个测序片段进行双末端测序得到双末端数据,该双末端数据包括一对对成对的读长;再将获得的双末端数据比对到参考基因组上得到比对信息。
在本文中使用的术语“包括”及其变形表示开放性包括,即“包括但不限于”。除非特别申明,术语“或”表示“和/或”。术语“基于”表示“至少部分地基于”。术语“一个示例实施例”和“一个实施例”表示“至少一个示例实施例”。术语“另一实施例”表示“至少一个另外的实施例”。术语“第一”、“第二”等等可以指代不同的或相同的对象。
经研究发现,在上述传统的用于检测突变的方案中,导入bam文件并读取igv图像需要一定的时间。另外,人工判断真假突变的效率较为低下,而且会因为疲劳和经验局限等原因导致判断真假突变发生错误。另外,由于二代测序一般会通过将样本序列打断为150bp片段进行测序,癌症组织一般取样时候会混合正常组织与肿瘤组织的混合抽样过程,加之,肿瘤组织本身具有异质性,因此随机抽样的待测样本的测序序列是否具有突变存在一定的概率,这是测序方面的难点。另外,在测序之后,有些150bp测序序列可能存在问题,例如测序序列有很多位置没有比对上,或者由于实验或者流程等原因导致一些测序序列的比对质量、深度比较低。上述测序比对结果的不完善导致关于支持突变的测序序列的检测结果存在一定的不准确性。
为了至少部分地解决上述问题以及其他潜在问题中的一个或者多个,本公开的示例实施例提出了一种用于检测突变的方案。该方案包括:获取关于待测样本的序列与参考基因组序列的比对结果信息,关于待测样本包括待测血液样本和待测组织样本中的至少一种;基于比对结果信息,生成突变位点碱基信息,突变位点碱基信息包括碱基质量和比对质量中的至少一项;基于预测模型,提取突变位点碱基信息的特征值,预测模型经由对多个训练样本的机器学习而生成;以及基于所提取的特征值,预测突变信息。
在上述方案中,通过基于序列与参考基因组序列的比对结果信息来生成突变位点碱基信息,以及基于经训练的预测模型预测突变信息;本公开无需耗费导入bam文件并读取igv图像的时间,对突变的识别无需受人力和经验局限,能够显著提高预测突变的效率和准确率。
图1示出了根据本公开的实施例的用于实施检测突变的方法的系统100的示意图。如图1所示,系统100包括:过滤单元112、数据转换模块114、预测模型116。在一些实施例中,系统100还包括:比对单元110、生信服务器140和网络150。
在一些实施例中,过滤单元112、数据转换模块114、预测模型116可以配置在一个或者多个计算设备130上;而比对单元110可以独立于计算设备130之外。计算设备130可以通过有线或者无线的方式与比对单元110进行数据交互。
计算设备130用于基于所获取关于待测样本的序列与参考基因组序列的比对结果信息预测突变信息。在一些实施例中,计算设备130可以具有一个或多个处理单元,包括诸如gpu、fpga和asic等的专用处理单元以及诸如cpu的通用处理单元。另外,在每个计算设备上也可以运行着一个或多个虚拟机。
在一些实施例中,计算设备130可以直接经由网络150获取来自生信服务器140的关于待测样本的序列与参考基因组序列的比对结果信息。在一些实施例中,计算设备130可以配置有一般的生信处理流程,并且经由该生信处理流程生成待测样本的序列与参考基因组序列的比对结果信息。
过滤单元112用于针对突变数据(例如关于支持突变的read的检测结果)进行过滤,并将经过滤的突变数据发送至数据转换模块114。例如,计算设备130在获取关于待测样本的测序序列与参考基因组序列的比对结果信息之后,由过滤单元112针对比对结果信息进行初步过滤。例如,如果过滤单元112确定具有一致性突变的reads数量大于或者等于预定值,则留下该突变数据,如果过滤单元112确定具有一致性突变的reads数量小于预定值该reads序列,则过滤掉该突变数据。通过采用上述手段,提高突变数据的可靠性。
数据转换模块114用于将比对结果信息转换为突变位点碱基信息。在一些实施例中,计算设备130可以经由已知分析流程seqan(https://github.com/seqan/seqan)获取经由待测样本的测序序列与参考基因组序列的比对后所确定的候选突变碱基序列,然后将候选突变碱基序列经由过滤单元112过滤后导入数据转换模块114,以便生成关于突变位点碱基信息的预测模型的输入矩阵。预测模型116用于基于所输入的突变位点碱基信息预测突变信息。预测模型116是经由对多个训练样本的机器学习而生成。预测模型116可以基于深度卷积神经网络(cnn)模型所构建,也可以基于时间卷积网络(temporalconvolutionalnetwork,tcn)模型所构建。预测模型116可以导入经由训练模型训练好的模型文件ckpt文件以便预测突变信息。该训练好的模型文件ckpt文件是经由训练模型针对多个训练样本的机器学习(例如深度网络学习)而生成的。预测模型116的输入是数据转换模块114输出的突变位点碱基信息。预测模型116的输出是所预测的突变信息。
训练模型的输入为多个训练样本。该训练样本例如是经由人工或者标注软件针对数据转换模块114所生成的突变位点碱基信息进行标注所形成的多个训练样本。在一些实施例中,可以将多个训练样本经过训练模型所获得训练输出矩阵与对应人工标注的数据进行对比,然后反馈给训练模型,以便通过深度网络学习提高准确率,最终获得最佳模型。通过多轮训练,训练模型生成优化模型文件ckpt文件参数。在一些实施例中,计算设备130还可以根据样本的信息,例如,癌症类型、癌症的污染指数、msi,样本突变中gc偏移等信息进行机器学习训练,以便进一步提升预测模型对于不同特征样本的碱基预测的准确率。
关于预测模型所输出的突变信息,下文将结合表一具体说明。在一些实施例中,突变信息例如包括突变类型。突变类型主要分为四种:单核苷酸多态性、单核苷酸位点变异、某一个位置或者多个位置的碱基发生了删除或插入、多个碱基变异为多个碱基delins中的至少一个。
关于单核苷酸缺失,其通常包括两种:单核苷酸多态性(singlenucleotidepolymorphism,snp)和单核苷酸位点变异(singlenucleotidevariants,snv)。单核苷酸多态性通常是在人群中有的突变,单核苷酸位点变异通常是在个体中发生的突变。图2示出了根据本公开的实施例的用于指示单核苷酸多态性突变类型的比对信息200的示意图。例如,如图2所示的比对信息200,其中,方框210所指示的位点的野生型的碱基t突变成了碱基a,则在方框210所指示的位点的突变类型为单核苷酸多态性snv。
关于某一个位置或者多个位置的碱基发生了删除或插入(即indel),其是指某一个位点或者多个位点的碱基发生了插入(insertion),或者某一个位点或者多个位点的碱基发生了删除(deletion)。图3示出了根据本公开的实施例的用于指示碱基发生了删除或插入的突变类型的比对信息300的示意图。例如,如图3所示的比对信息300,左侧的方框310所指示的位点的碱基t发生了删除,右侧的方框320所指示的位点的碱基a发生了插入。
图4示出了根据本公开的实施例的用于指示多个碱基变异为多个碱基delins的突变类型的比对信息400的示意图。关于多个碱基变异为多个碱基delins中的至少一个,例如,如图4所示的比对信息400,方框410所指示的位点的多个碱基ggc变异为多个碱基atcgacg。
以下将结合图5描述根据本公开的实施例的用于检测突变的方法。图5示出了根据本公开的实施例的用于检测突变的方法500的流程图。应当理解,方法500例如可以在图17所描述的电子设备1700处执行。也可以在图1所描述的计算设备130处执行。应当理解,方法500还可以包括未示出的附加动作和/或可以省略所示出的动作,本公开的范围在此方面不受限制。
在框502处,计算设备130获取关于待测样本的测序序列与参考基因组测序序列的比对结果信息,待测样本包括待测血液样本和待测组织样本中的至少一种。
例如,采集待测个体的组织和/或血液样本之后,可以将所获取的采样样本的dna数据,然后随机抽样,以便例如通过二代测序技术生成关于待测样本的测序序列。然后,计算设备130将待测样本的测序序列与参考基因组序列(例如是人类hg19标准样本的基因序列)进行比对,以便生成比对结果信息。
关于待测样本,在一些实施例中,其可以是待测个体的肿瘤组织的采样样本或血液的采样样本。在一些实施例中,其可以是待测个体的肿瘤组织的采样样本和血液的采样样本。应该理解,血液样本中包含了生殖细胞突变信息。待测个体的肿瘤组织的采样样本中例如包括了体细胞突变信息。
关于比对结果信息,在一些实施例中,其例如是通过二代测序获得的片段化数据bam文件。在一些实施例中,bam文件指示待测个体的肿瘤组织的样本的测序序列(和/或血液样本的测序序列)与人类参考基因组(例如hg19)的测序序列的比对结果信息。该比对结果信息例如包括150bp的一条条的reads序列及其对应的位置。在一些实施例中,计算设备130可以将待测个体的肿瘤组织的样本的测序序列与参考基因组的测序序列进行比对的比对结果信息用于体细胞突变的预测。计算设备130也可以将待测个体的血液样本的测序序列与参考基因组的测序序列进行比对的比对结果信息用于生殖细胞突变的预测。计算设备130还可以汇总关于待测个体的肿瘤组织的样本和血液样本的测序序列的比对结果信息用于突变的预测,以便使得本公开的预测模型可以综合考虑生殖细胞突变信息和体细胞突变信息之间的关联,进而提高关于突变的预测结果的可靠性。
在一些实施例中,计算设备130在获取关于待测样本的测序序列与参考基因组序列的比对结果信息之后,还可以针对比对结果信息进行初步过滤。例如,计算设备130可以确定具有一致性突变的reads数量是否大于或者等于预定值,如果具有一致性突变的reads数量小于预定值,该reads序列及其对应的位置有可能存在测序的判读失误。通过采用上述手段,本公开可以通过初步过滤比对结果数据中因偶然性因素导致的突变数据错误,进而提高预测结果的可靠性。
在框504处,计算设备130基于比对结果信息,生成突变位点碱基信息,突变位点碱基信息至少包括碱基质量和比对质量中的至少一项。例如,计算设备130可以经由已知分析流程seqan(https://github.com/seqan/seqan)获取经由待测样本的测序序列与参考基因组序列的比对后所确定的候选突变碱基序列,然后将候选突变碱基序列导入数据转换模块114,以便生成关于突变位点碱基信息的预测模型的输入矩阵。
例如,计算设备130经由所配置的数据转换模块114匹配比对结果信息(例如bam文件),扫描比对的reads,寻找待测样本的序列可能不同于参考基因组序列的位点。提取关于突变位点碱基的多维特征,以便转为关于候选变异位点信息的多维特征输入矩阵。该多维特征中至少包括计算设备130所确定的关于待测样本血液样本和待测组织样本中的至少一种的碱基质量特征和新增比对(mapping)质量特征中的至少一种。下文将结合图9至图16具体说明数据转换模块114提取关于突变位点碱基的多维特征的处理过程,在此,不再赘述。
在一些实施例中,计算设备130可以基于关于待测血液样本和待测组织样本中的至少一种的碱基质量和比对质量,以及关于一致读长的特征、关于不一致读长的特征、关于正向链碱基一致读长数量特征、关于反向链碱基一致读长数量特征、关于正向链的突变型碱基不一致数量特征、关于反向链的突变型碱基不一致读长数量特征、关于参考基因组不一样且与突变型碱基不一致的多个特征、a碱基特征、c碱基特征、t碱基特征、g碱基特征、具突变位点的读长数量特征和参考基因组不一致读长数量特征中的多个特征,生成用于输入预测模型的多维特征矩阵。
在一些实施例中,计算设备130可以基于待测个体的血液样本的测序序列与参考基因组的测序序列的比对结果信息来生成关于突变位点碱基的多维特征输入矩阵(例如16维特征输入矩阵);和/或基于待测个体的肿瘤组织的样本的测序序列与参考基因组的测序序列的比对结果信息来生成关于突变位点碱基的多维特征输入矩阵(例如16维特征输入矩阵)。在一些实施例中,计算设备130可以基于血液样本的测序序列与参考基因组的测序序列进行比对的比对结果信息,生成关于突变位点碱基的第一多维矩阵(例如16维特征输入矩阵);基于肿瘤组织样本的测序序列与参考基因组的测序序列进行比对的比对结果信息,生成关于突变位点碱基的第二多维矩阵(例如16维特征输入矩阵);汇总第一多维矩阵和第二多维矩阵,以便生成用于预测模型的输入矩阵(例如32维特征输入矩阵),预测模型是基于深度卷积神经网络模型所构建。通过采用上述手段,本公开的所预测的突变信息能够综合考虑生殖细胞突变信息和体细胞突变信息之间的关联与影响,因此具有更高的可靠性。
在框506处,计算设备130基于预测模型,提取突变位点碱基信息的特征值,预测模型经由对多个训练样本的机器学习而生成。
在一些实施例中,预测模型是基于深度卷积神经网络(cnn)模型所构建。该cnn模型主要包括由输入层、卷积层、激活函数层、池化层、全连接层和输出层。
关于输入层,可以将基于比对结果信息而转化生成关于突变位点碱基信息的多维特征输入矩阵作为cnn模型的输入。
关于卷积层,其用于提取多维特征输入的不同特征。每层卷积层由若干卷积单元组成,每个卷积单元的参数都是通过反向传播算法最佳化得到的。
关于激活函数层,其例如使用线性整流(rectifiedlinearunits,relu),以用于增强判定函数和整个神经网络的非线性特性。
卷积层还包括多个block残差学习单元。
关于池化层,其用于通过池化处理缩小碱基信号的特征尺寸,以便有效的去除由上一层的结果作为输入而带来的计算量较大的问题。
关于全连接层,该层是cnn模型中消耗参数最多的层,该层判别结果可以作为输出结果。该cnn模型的输出特征向量被输入至softmax分类器。下文将结合图6至8具体说明本公开cnn模型的网络结构,在此,不再赘述。
在一些实施例中,预测模型例如是基于时间卷积网络(temporalconvolutionalnetwork,tcn)模型所构建。该tcn模型采用一维的卷积网络,以及使用空洞卷积核(dilatedconvolutions)来获取整个序列的全局信息,并且也设置了残差结构(residualblock)。通过采用基于tcn模型构建的预测模型,可以获得更快的计算速度。
关于预测模型的输入,即突变位点碱基信息,在一些实施例中,其为k*n的输入矩阵,例如是32*193。其中,k代表特征维度,例如而不限于是16或32。n代表每一维度特征所包括的数据个数,该数据个数与突变位点的前后预定数量的碱基位点数量相关联,例如,前文提及的193对应于:突变位点以及该突变位点的前后各96个的碱基位点的总数。
相较而言,本公开的cnn模型不同于传统的cnn模型。传统的cnn模型通常用于处理图像输入,其输入通常是三维输入矩阵(例如,输入特征被编码成红-绿-蓝(rgb)堆积图像),传统的cnn模型的输出也是三维输出矩阵。而本公开的预测模型的输入(即突变位点碱基信息)为一维k*n的输入矩阵,有利于显著地避免预测模型的输入数据过于冗余的问题,能够显著减少预测突变信息的计算量,以及提高预测速度。
在框508处,基于所提取的特征值,预测突变信息。
在一些实施例中,所预测的突变信息例如至少可以指示:染色体、是否发生了突变,以及如果发生突变,突变型信息,对应参考基因的信息、突变频率、其他信息等等。在一些实施例中,突变型信息例如包括:snp、snv、indel等。突变信息所指示的上述信息例如而不限于以数据表的形式输出,例如是vcf文件。以下结合表一具体说明用于指示所预测的部分突变信息的vcf文件。
表一
以上述表一前几行为例,“chr1”指示染色体,“2493264”指示突变位置,“rs560742705”指示基因编号,“c”指示对照组,“t”指示变异组。其他信息例如包括:关于蛋白质及氨基酸的注释等等。
在上述方案中,通过采用基于比对结果信息生成突变位点碱基信息,再将突变位点碱基信息输入至预测模型来预测突变信息,而非将比对结果信息直接输入预测模型进行预测,本公开能够显著地避免预测模型的输入数据过于冗余的问题,减少预测突变信息的计算量,进而提高预测速度,而且,相对于传统的检测突变的方法(“tpu,tensorprocessingunit”层面的硬件配置要求),明显降低了对硬件配置的要求(例如仅需少量的gpu,例如1个gpu即可支持预测运算)。另外,在上述方案中,通过使得输入预测模型的突变位点碱基信息中至少包括碱基质量和比对质量中的至少一项,本公开的预测模型所提取的特征中与碱基质量和比对质量有关联,因此能够显著提高检测突变的准确性。
在一些实施例中,方法500还包括:计算设备130可以获取多个预测模型所分别预测的多个体细胞突变信息;以及基于多个体细胞突变信息,确定关于待测个体的体细胞突变的预测结果。例如,多个预测模型的输出特征向量被输入至softmax分类器。
图6示出了根据本公开的实施例的预测模型600的结构示意图。应当理解,预测模型600还可以包括未示出的附加网络层和/或可以省略所示出的网络层,本公开的范围在此方面不受限制。
如图6所示,预测模型600例如包括卷积层602、激活函数层604、块(block)残差学习单元606、610、614和618、池化层608、612、616和620、全连接层620和输出结果622。
关于卷积层602,在一些实施例中,其例如被配置为具有32个通道、13个内核大小。
关于激活函数层604,其激活函数为relu。relu能够缓解梯度消失问题(vanishinggradient),以便提高网络中的低层训练速度。
预测模型600配置有四个块(block)残差学习单元606、610、614和618。block残差学习单元606、610、614和618具有类似于resnet结构的短路连接。每一个block残差学习单元由卷积核为3*3的卷积层和卷积核为5*5的卷积层组成。采用block残差学习单元的目的是:利用残差学习与针对原始特征的直接学习相比更为容易,既可以保留深层网络的深度,又可以有浅层网络的优势去避免退化问题。下文结合公式(1)和图7示意性说明block残差学习单元的原理。
图7示出了根据本公开的残差学习网络结构700的示意图。block残差学习单元的结构例如通过图7所示前向神经网络714附加短路连接712(shortcut)加以实现,其中短路连接712(shortcut)相当于简单执行了同等映射,不会产生额外的参数和计算复杂度。
h(x)=f(x)+x(1)
在上述公式(1)中,h(x)代表图7所示输入到求和710之后的网络映射。f(x)代表残差函数。当残差函数f(x)为0时,此时堆积层仅做了恒等映射h(x)=x,网络性能不会下降。实际的残差函数f(x)不会为0,引入残差的映射对输出的变化更加敏感,这也会使得堆积层在输入特征的基础上学习到新的特征,从而使得网络模型拥有更好的性能。
在一些实施例中,对于基于tcn模型所构建的预测模型,block残差学习单元的结构(未示出)例如包括:输入经由空洞卷积、权重归一化、激活函数、两轮的dropout处理,生成残差函数f(x);输入经由1*1卷积过滤(filters),生成短路连接(shortcut)的“x”,以用于与残差函数f(x)进行叠加,以便生成求和之后的网络映射。
与传统的均一化处理不同,在预测模型的block残差学习单元之间,使用批处理最大池层,即池化层608、612、616或620。最后的block残差学习单元618的输出被馈送至一个尺寸为240的全连接层620。全连接层620的输出结果(例如输出特征向量)被输入至一个softmax分类器(未示出)。
上述方案不同于传统的cnn模型。传统的cnn模型中的卷积操作和池化操作都是线性操作,而本公开的样本,在进行分类时并不是线性关系,因而需要在传统的cnn模型中引入非线性元素,使得本公开的cnn模型能解决非线性问题。另外,本公开通过池化处理缩小碱基信号特征尺寸,能有效的去除由上一层的结果作为输入而带来的计算量较大的问题。
图8示出了根据本公开的实施例的预测模型800的结构示意图。应当理解,预测模型800还可以包括未示出的附加网络层和/或可以省略所示出的网络层,本公开的范围在此方面不受限制。
在一些实施例中,计算设备130可以获取多个预测模型(例如,第一预测模型810、第二预测模型820至第n预测模型830)所分别预测的多个体细胞突变信息;以及基于多个体细胞突变信息,确定关于待测个体的体细胞突变的预测结果。例如,通过将多组预测模型输出结果进行汇总,最终通过打分模型得到多模型平均预测结果,以便进一步保证预测模型的泛化能力。
如图8所示,预测模型800包括多个预测模型,例如第一预测模型810、第二预测模型820至第n预测模型830。第一预测模型810、第二预测模型820至第n预测模型830的关于突变信息的输出结果被输入至一个softmax分类器840,以便计算多组模型平均预测判别结果。通过采用上述技术手段,能够降低单个模型的误差。
在一些实施例中,在训练模型时,例如可以将数据分为多组(例如而不限于100组),基于每一组数据经由预测模型会输出预测结果,然后将第二组数据导入该预测模型进行训练,然后输出预测结果,以此类推。经过每一轮训练,会有多个(例如100个)经训练的预测模型,然后针对多个经训练的预测模型进行选择,以用于实际预测。
通过采用上述手段,可以通过采用多个预测模型来降低单个预测模型所带来的输出结果的误差。
图9示出了根据本公开的实施例的用于生成突变位点碱基信息的方法900的流程图。图10示出了根据本公开的实施例的经由测序获得的比对结果信息1000的示意图。应当理解,方法900例如可以在图17所描述的电子设备1700处执行。也可以在图1所描述的计算设备130处执行。应当理解,方法900还可以包括未示出的附加动作和/或可以省略所示出的动作,本公开的范围在此方面不受限制。
在框902处,计算设备130基于突变位点的位置信息,获取突变位点及其前后预定数量的碱基位点的读长碱基信息。例如,计算设备130基于经由二代测序获得的比对结果信息(例如为bam文件),寻找可能不同于参考基因组(人类基因组hg19,如标记1010所示)的位点信息(例如是候选的突变位点1012)。bam文件中保存的是一条一条的reads序列,及所对应的位置,通过碱基位置很容易获得包含这个位置(例如虚线框1022所示碱基位点)所有reads序列1020。例如,计算设备130获取特定位置的碱基位点(例如虚线框1012指示的突变位点),以及该特定位置前后各预定数量(例如是前96个碱基位点,如标记1014所示,以及后96个碱基位点,如标记1016所示)的碱基位点,共193个碱基位点的所有reads信息。
在框904处,计算设备130将所获取的读长碱基信息与参考基因组数据进行比对。例如,将193个碱基位点的所有reads信息和对应参考基因组数据(例如人类基因组hg19)的数据进行对比。
在框906处,计算设备130基于所获取的读长碱基信息中与参考基因组数据一致的读长碱基信息,生成关于一致读长的特征。
在框908处,计算设备130基于所获取的读长碱基信息中与参考基因组数据不一致的读长碱基信息,生成关于不一致读长的特征。例如计算设备130针对一致的reads进行统计,以及针对不一致reads也进行统计,以及基于统计结果分别生成包括对应于193个碱基位点的193位特征值的特征维度为一维的、关于一致读长的特征向量,以及包括对应于193个碱基位点的193位特征值的特征维度为一维的、关于不一致读长的特征向量。通过采用上述手段,本公开可以通过基于突变位点的位置信息生成关于一致读长和不一致读长的两维特征向量,能够以较少维度的特征来体现突变碱基位置的reads的关键信息,利于简化预测突变信息的计算,提高预测速度。
图11示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1100的流程图。图12示出了根据本公开的实施例的经由测序获得的比对结果信息1200的示意图。应当理解,方法1100例如可以在图17所描述的电子设备1700处执行。也可以在图1所描述的计算设备130处执行。应当理解,方法1100还可以包括未示出的附加动作和/或可以省略所示出的动作,本公开的范围在此方面不受限制。
在框1102处,计算设备130将所获取的读长碱基信息分别与参考基因组的正向链碱基和反向链碱基进行比对。
在框1104处,计算设备130基于与正向链碱基或反向链碱基一致的读长数量,分别生成关于正向链碱基一致读长数量特征和关于反向链碱基一致读长数量特征。例如,计算设备130通过输入的点突变数据的位置信息,可以分别统计与人类参考基因组(例如hg19)的正向链碱基或反向链碱基一致的reads数。例如,如图12所示,人类基因组hg19(如标记1210所示)正向链在虚线框1212所示位点发生突变碱基a,计算设备130针对在该突变位点与碱基a一致的所有读长1220进行统计,生成关于碱基一致读长数量特征,例如是1*193的一维的特征向量ref(a)。该特征向量ref(a)例如包括预定数量(例如193位)的特征值,每一位的特征值例如用于指示与参考基因组对应碱基位点的碱基一致的读长数量或者一致的概率。人类参考基因组(例如hg19)的虚线框1212所示突变位点的碱基为a,例如,经统计,在与碱基a一致的所有读长1220中,该突变位点(例如虚线框1222所示位点)与碱基a一致的reads数量为100,则特征向量ref(a)中对应于突变位点的第97位的特征值例如为100。以此类推,经统计,与人类参考基因组虚线框1212所示突变位点的前一位的碱基(例如是碱基t)一致的reads数量为98,例如某两条reads对应位点的碱基为c,与碱基t不一致,则对应于突变位点的第96位的特征值例如为98。
在框1106处,计算设备130基于突变型碱基的形式与正向链突变型碱基或反向链突变型碱基不一致的读长数量,分别生成关于正向链的突变型碱基不一致数量特征和关于反向链的突变型碱基不一致读长数量特征。应当理解,人类参考基因组是双链结构(即一个位置具有正向链和反向链两条链)。例如,如图12所示,人类基因组hg19正向链和反向链如虚线框1232-1和1232-2所示突变位点的突变型碱基的形式为碱基t。计算设备130可以分别基于与人类基因组hg19与正向链突变型碱基或反向链突变型碱基不一致的读长数量1230,生成2*193的两维的特征向量alt(t)。
在框1108处,计算设备130基于与正向链碱基或反向链碱基不一致的读长数量并且与突变型碱基不一致的读长数量,分别生成关于参考基因组不一样且与突变型碱基不一致的多个特征。例如,计算设备130可以针对与参考基因组不一致并且与突变型碱基的形式不一致的reads进行统计。在计算突变型reads需要分别针对人类参考基因组正向链碱基与反向链碱基的情况进行分析与统计。例如,如图12所示,计算设备130基于与人类基因组hg19的正向链和反向链不一致和突变型碱基的形式(例如c,g缺失)不一致的读长1240,生成4*193的四特征维度的特征向量other(c,g缺失)。基于上述统计,计算设备130可以生成八特征维度的特征。该八特征维度的特征例如包括:关于正向链的一致读长数量特征、关于反向链的一致读长数量特征、关于正向链突变型碱基一致读长数量特征、关于反向链突变型碱基一致的读长的数量,以及关于参考基因组不一样且与突变型碱基不一致的四个特征。
图13示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1300的流程图。图14示出了根据本公开的实施例的经由测序获得的比对结果信息1400的示意图。应当理解,方法1300例如可以在图17所描述的电子设备1700处执行。也可以在图1所描述的计算设备130处执行。应当理解,方法1300还可以包括未示出的附加动作和/或可以省略所示出的动作,本公开的范围在此方面不受限制。
在框1302处,计算设备130基于突变位点的位置信息,获取关于突变位点的前后预定数量的碱基位点的a、c、t、g碱基的数量。例如,计算设备130通过输入的位点突变数据的位置信息,可以分别统计该突变位置前后各预定数量(例如是前96个碱基位点以及后96个碱基位点)的序列长度上a、c、t、g碱基的个数。
在框1304处,计算设备130基于a、c、t、g碱基的数量,分别生成用于分别指示a、c、t、g碱基分布情况的a碱基特征、c碱基特征、t碱基特征和g碱基特征。例如,计算设备130基于所分别统计该突变位置前后各96个碱基位点的序列长度上a、c、t、g碱基的个数,生成4*193的四特征维度的特征向量。例如,如图15所示,计算设备130可以基于突变位点(如虚线框1412所示)及其前后各96的碱基位点的a、c、t、g碱基的数量,生成4*193的四特征维度的特征向量actg,如标记1414所示。例如,标记1414所示的四特征维度的特征向量actg的中的第一行特征序列特征值分别指示193个碱基位点上对应分布碱基a的数量。
图15示出了根据本公开的实施例的用于生成突变位点碱基信息的方法1500的流程图。图16示出了根据本公开的实施例的经由测序获得的比对结果信息1600的示意图。应当理解,方法1500例如可以在图17所描述的电子设备1700处执行。也可以在图1所描述的计算设备130处执行。应当理解,方法1500还可以包括未示出的附加动作和/或可以省略所示出的动作,本公开的范围在此方面不受限制。
在框1502处,计算设备130确定具有突变位点的读长数量,以生成具突变位点的读长数量特征。例如,如图16所示,计算设备130统计具有参考基因组1614的突变位点(如虚线框1612所示)的reads(如虚线框1620所示)数量,以生成1*193的一特征维度的特征向量,如标记1630所示。
在框1504处,计算设备130确定具有突变位点的读长中与参考基因组不一致的读长数量,以生成参考基因组不一致读长数量特征。例如,如图16所示,计算设备130生成1*193的一特征维度的特征向量,如标记1640所示。
通过上述图9至图16所示的特征提取,计算设备130可以针对待测血液样本和待测组织样本中的至少一种获得14特征维度的特征向量。在一些实施例中,计算设备130还需进行深度计算,以便对上述多个特征维度的特征向量进行归一化处理。对于图9至图16所示的14特征维度的特征向量而言,其该深度计算例如是统计在突变位点比对上的reads的总数,以生成的深度值。对于关于碱基质量和比对质量的特征向量而言,其深度值为质量范围。然后将多个特征维度的特征向量的特征值除以深度值,以便进行均一化处理。
在一些实施例中,计算设备130可以将针对待测组织样本的16维度的特征向量与针对待测血液样本的16特征维度(包括碱基质量和比对质量)的特征向量进行汇总,以便形成拥有32特征维度的特征向量,以用于输入预测模型来预测突变信息。在一些实施例中,计算设备130可以基于针对待测肿瘤组织样本的16特征维度的特征向量和针对待测血液对照样本的16特征维度的特征向量,以用于输入预测模型来预测体细胞突变信息。应该理解,待测个体的待测血液对照样本中包含了生殖细胞突变信息。待测个体的待测肿瘤组织样本中包括了体细胞突变信息。基于上述针对待测肿瘤组织样本的16特征维度的特征向量和针对待测血液对照样本的16特征维度的特征向量能够过滤掉所预测的突变信息中有关生殖细胞突变的信息,进而获得准确的体细胞突变信息。
图17示意性示出了适于用来实现本公开实施例的电子设备1700的框图。设备1700可以是用于实现执行图5、9、11、13、15所示的方法500、900、1100、1300、1500的设备。如图7所示,设备1700包括中央处理单元(cpu)1701,其可以根据存储在只读存储器(rom)1702中的计算机程序指令或者从存储单元1708加载到随机访问存储器(ram)1703中的计算机程序指令,来执行各种适当的动作和处理。在ram1703中,还可存储设备1700操作所需的各种程序和数据。cpu1701、rom1702以及ram1703通过总线1704彼此相连。输入/输出(i/o)接口1705也连接至总线1704。
设备1700中的多个部件连接至i/o接口1705,包括:输入单元1706、输出单元1707、存储单元1708,处理单元1701执行上文所描述的各个方法和处理,例如执行方法200至700。例如,在一些实施例中,方法500、900、1100、1300、1500可被实现为计算机软件程序,其被存储于机器可读介质,例如存储单元1708。在一些实施例中,计算机程序的部分或者全部可以经由rom1702和/或通信单元1709而被载入和/或安装到设备1700上。当计算机程序加载到ram1703并由cpu1701执行时,可以执行上文描述的方法500、900、1100、1300、1500的一个或多个操作。备选地,在其他实施例中,cpu1701可以通过其他任何适当的方式(例如,借助于固件)而被配置为执行方法500、900、1100、1300、1500的一个或多个动作。
需要进一步说明的是,本公开可以是方法、装置、系统和/或计算机程序产品。计算机程序产品可以包括计算机可读存储介质,其上载有用于执行本公开的各个方面的计算机可读程序指令。
计算机可读存储介质可以是可以保持和存储由指令执行设备使用的指令的有形设备。计算机可读存储介质例如可以是但不限于电存储设备、磁存储设备、光存储设备、电磁存储设备、半导体存储设备或者上述的任意合适的组合。计算机可读存储介质的更具体的例子(非穷举的列表)包括:便携式计算机盘、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦式可编程只读存储器(eprom或闪存)、静态随机存取存储器(sram)、便携式压缩盘只读存储器(cd-rom)、数字多功能盘(dvd)、记忆棒、软盘、机械编码设备、例如其上存储有指令的打孔卡或凹槽内凸起结构、以及上述的任意合适的组合。这里所使用的计算机可读存储介质不被解释为瞬时信号本身,诸如无线电波或者其他自由传播的电磁波、通过波导或其他传输媒介传播的电磁波(例如,通过光纤电缆的光脉冲)、或者通过电线传输的电信号。
这里所描述的计算机可读程序指令可以从计算机可读存储介质下载到各个计算/处理设备,或者通过网络、例如因特网、局域网、广域网和/或无线网下载到外部计算机或外部存储设备。网络可以包括铜传输电缆、光纤传输、无线传输、路由器、防火墙、交换机、网关计算机和/或边缘服务器。每个计算/处理设备中的网络适配卡或者网络接口从网络接收计算机可读程序指令,并转发该计算机可读程序指令,以供存储在各个计算/处理设备中的计算机可读存储介质中。
用于执行本公开操作的计算机程序指令可以是汇编指令、指令集架构(isa)指令、机器指令、机器相关指令、微代码、固件指令、状态设置数据、或者以一种或多种编程语言的任意组合编写的源代码或目标代码,该编程语言包括面向对象的编程语言—诸如smalltalk、c++等,以及常规的过程式编程语言—诸如“c”语言或类似的编程语言。计算机可读程序指令可以完全地在用户计算机上执行、部分地在用户计算机上执行、作为一个独立的软件包执行、部分在用户计算机上部分在远程计算机上执行、或者完全在远程计算机或服务器上执行。在涉及远程计算机的情形中,远程计算机可以通过任意种类的网络—包括局域网(lan)或广域网(wan)—连接到用户计算机,或者,可以连接到外部计算机(例如利用因特网服务提供商来通过因特网连接)。在一些实施例中,通过利用计算机可读程序指令的状态信息来个性化定制电子电路,例如可编程逻辑电路、现场可编程门阵列(fpga)或可编程逻辑阵列(pla),该电子电路可以执行计算机可读程序指令,从而实现本公开的各个方面。
这里参照根据本公开实施例的方法、设备(系统)、和计算机程序产品的流程图和/或框图描述了本公开的各个方面。应当理解,流程图和/或框图的每个方框以及流程图和/或框图中各方框的组合,都可以由计算机可读程序指令实现。
这些计算机可读程序指令可以提供给语音交互装置中的处理器、通用计算机、专用计算机或其它可编程数据处理装置的处理单元,从而生产出一种机器,使得这些指令在通过计算机或其它可编程数据处理装置的处理单元执行时,产生了实现流程图和/或框图中的一个或多个方框中规定的功能/动作的装置。也可以把这些计算机可读程序指令存储在计算机可读存储介质中,这些指令使得计算机、可编程数据处理装置和/或其他设备以特定方式工作,从而,存储有指令的计算机可读介质则包括一个制造品,其包括实现流程图和/或框图中的一个或多个方框中规定的功能/动作的各个方面的指令。
也可以把计算机可读程序指令加载到计算机、其它可编程数据处理装置、或其它设备上,使得在计算机、其它可编程数据处理装置或其它设备上执行一系列操作步骤,以产生计算机实现的过程,从而使得在计算机、其它可编程数据处理装置、或其它设备上执行的指令实现流程图和/或框图中的一个或多个方框中规定的功能/动作。
附图中的流程图和框图显示了根据本公开的多个实施例的设备、方法和计算机程序产品的可能实现的体系架构、功能和操作。在这点上,流程图或框图中的每个方框可以代表一个模块、程序段或指令的一部分,该模块、程序段或指令的一部分包含一个或多个用于实现规定的逻辑功能的可执行指令。在有些作为替换的实现中,方框中所标注的功能也可以以不同于附图中所标注的顺序发生。例如,两个连续的方框实际上可以基本并行地执行,它们有时也可以按相反的顺序执行,这依所涉及的功能而定。也要注意的是,框图和/或流程图中的每个方框、以及框图和/或流程图中的方框的组合,可以用执行规定的功能或动作的专用的基于硬件的系统来实现,或者可以用专用硬件与计算机指令的组合来实现。
以上已经描述了本公开的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。
以上仅为本公开的可选实施例,并不用于限制本公开,对于本领域的技术人员来说,本公开可以有各种更改和变化。凡在本公开的精神和原则之内,所作的任何修改、等效替换、改进等,均应包含在本公开的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!