一种基于预测蛋白质亲和力的分子表征及其应用的制作方法
本发明涉及计算机辅助药物设计领域,特别是涉及一种基于预测蛋白质亲和力的分子表征及其应用。
背景技术:
在现有药物设计过程中,为缩短药物研究周期,控制药物研发成本,计算机药物辅助分子设计成为药物研发过程中必不可少的工具。计算机辅助分子设计可分为基于配体和基于受体两类方法。基于配体的方法多数是基于相似结构化合物具有相似生物活性这个假设,其中的关键点就是如何表征分子与生物活性之间的联系。
现有解决方案以及缺点:现有的分子表征可分为基于化学结构和基于生物活性两种。传统的化学结构分子表征针对分子的结构进行解剖,比如子结构碎片,拓扑结构,药效团特征以及物理化学性质等等。比较常用的有moe2d、cats、atompair、maccs、ecfp系列、fp系列等。这种分子表征虽然一定程度上涵盖了化合物与生物活性之间的联系,但是这种联系是不明确的,无法深度解读生物活性。随着技术的发展,基于各种生物试验的数据不断积累。研究者们尝试着使用生物试验数据表征化合物与生物活性之间的联系。例如,lamb等人[1]利用化合物扰动过后的基因信号的变动捕获到了化合物与疾病之间的关系。还有药物的副作用,分子与蛋白质靶点之间的试验结合亲和力等生物信息都被用来构建生物活性分子表征。然而,基于试验的分子表征仅仅适用于同样具有试验信息的分子中,这极大限制了生物活性分子表征的应用。为了扩大应用范围,研究者们通常会将不同小组得到的试验数据汇集形成一个大的数据集,这不可避免地产生了误差。即使如此,基于试验的生物活性数据分子表征也并不完全,并不是所有分子都具有广阔的试验谱,药企或研究组织会根据自身的药物研发进程而进行试验,这就导致了有的分子仅仅有几个试验结果或者具有完整试验谱的分子寥寥无几。这种不完整的数据矩阵的缺点通过表征传递给了分子,使得分子与生物活性之间的联系也并不完整。有的研究会默认缺失值为0,也就是分子与该试验的蛋白质没有结合作用力或者是分子没有该类生物活性,这种处理方式导致假阴性的产生。因此,我们需要开发基于计算机的生物活性分子表征冲破现有基于试验的生物活性分子表征的禁锢,并且使用尽可能简单的条件获取高准确度的结果。
技术实现要素:
为解决上述问题,本发明的目的在于,针对现有技术的不足,构建一种能够简单直接表征化合物和生物活性之间关系的分子表征及其应用。
为了实现上述目的,本发明提供了一种基于预测蛋白质亲和力的分子表征(即计算靶标谱),所述计算靶标谱采用以下方法构建得到:
(1)数据收集:收集蛋白质靶标及其信息;
(2)分子表征形成:通过计算机方法计算每个蛋白质靶标与化合物的结合亲和力值。
上述计算靶标谱,进一步的,所述s2中,所述计算机方法包括sar模型和对接。
上述计算靶标谱,进一步的,所述计算靶标谱采用以下方法构建得到:
(1)数据收集:收集蛋白质靶标及其活性数据;
(2)参数确定:计算每个蛋白质靶标的多种不同的描述符,并选择多种不同的机器学习算法;
(3)构建模型;将所述计算的描述符与所述机器学习算法两两组合,形成多个不同的单一模型;
(4)构建共识模型:将所述单一模型,计算平均概率值,作为分子与蛋白质靶标的结合强弱值,形成共识模型;
(5)形成计算靶标谱:在所述共识模型中,输入待测分子,输出计算靶标谱。
上述计算靶标谱,进一步的,所述s1中,所述蛋白质靶标包括酶、受体、转运体、离子通道;所述活性数据包括ic50、ec50、ki值。
上述计算靶标谱,进一步的,所述s2中,所述描述符为moe2d、cats、cats2、gpidaph3、tgt、tgd、maccs、atompair、estate、fp系列、ecfp系列、ecfc系列描述符中的一种或多种;所述机器学习算法为rf、xgboosting、dl、ada、gbm、knn、svm算法中的一种或多种。
上述计算靶标谱,进一步的,所述计算靶标谱采用以下方法构建得到:
(1)数据收集:收集蛋白质靶标及其活性数据;
(2)参数确定:分别计算每个蛋白质靶标的三种不同的描述符:moe2d、cats和maccs,并选择三种不同的机器学习算法:rf、ada和gbm;
(3)构建模型;将三个不同的描述符和三个不同的机器学习算法两两组合,形成九个不同的单一模型;
(4)共识模型:将九个不同的单一模型,计算平均概率值,作为分子与蛋白质靶标的结合强弱值,形成共识模型;
(5)形成计算靶标谱:在所述共识模型中,输入待测分子,输出计算靶标谱。
基于一个总的技术构思,本发明提供了一种上述计算靶标谱在骨架跃迁中的应用,应用的方法为:
s1-1、分别将活性分子数据库和筛选数据库中的分子输入到共识模型中,输出计算靶标谱,,获得分子表征,计算基于化学结构的描述符,包括moe2d、maccs、cats、ecfp4、atompair、fp2、gpidaph3、tgt和tgd;
s1-2、依次选取活性分子数据库的不同靶标中的活性分子作为查询分子,其他活性分子和筛选数据库中的分子组合形成查询数据库;
s1-3、使用tanimoto系数计算查询分子与查询数据库中分子的相似性值;
s1-4、根据相似性值的大小排序,计算前100、200、500、1000、2000、5000化合物中活性分子骨架数量与所有活性分子骨架数量的比值,得到与查询分子不同骨架的活性分子的富集情况。
基于一个总的技术构思,本发明提供了一种上述计算靶标谱在预测蛋白质靶标中的应用,应用的方法为:
s2-1、将活性分子输入到所述共识模型中,输出活性分子的计算靶标谱,获得分子表征;
s2-1、根据活性分子表征预测的结合亲和力值对靶标进行排名预测活性分子的靶标。
基于一个总的技术构思,本发明提供了一种上述计算靶标谱在qsar模型预测肝毒性的应用,应用的方法为:
s3-1、从现有数据库中收集人体肝毒性数据和非肝毒性数据;
s3-2、采用不同的数据劈分策略将数据划分为训练集和测试集;
s3-3、将肝毒性和非肝毒性数据输入到所述共识模型中,输出计算靶标谱,获得cades分子表征;计算基于化学结构的moe2d、cats和maccs描述符;
s3-4、将所述cades分子表征和xgboosting算法构建模型;将基于化学结构的moe2d、cats和maccs描述符分别和xgboosting算法构建模型;
s3-5、组合所述计算靶标谱的模型和所述化学结构描述符的模型,构建共识模型;
s3-6、将待测分子输入所述共识模型中获得肝毒性数据。
上述的应用,进一步的,所述不同的劈分策略包括:随机劈分、bemis-murcko骨架劈分和碳骨架劈分。
基于一个总的技术构思,本发明提供了一种上述计算靶标谱在预测药物与药物相互作用中的应用,应用的方法为:
s4-1、将从文献收集的药物输入到共识模型中,输出计算靶标谱,获得分子表征;
s4-2、将所述药物分为正样本和负样本,所述正样本为所有真实的药物-药物相互作用对,负样本为药物之间的随机非相互作用对;
s4-3、将正样本中药物与药物相互作用对之间两个药物的分子表征结合作为药物对的分子表征,结合rf算法构建模型;
s4-4、将待测分子输入所述s4-3构建的模型中,预测药物与药物之间是否具有相互作用。
基于一个总的技术构思,本发明提供了一种上述计算靶标谱在确定药物作用模式网络中的应用,应用的方法为:
s5-1、将文献中获得的具有化学结构的药物输入到共识模型中,输出计算靶标谱,获得分子表征;
s5-2、从所述药物中,计算任意两个药物之间的pearson相关系数,根据解剖化学分类法,将药物网络可视化,其中每个节点对应一个药物,如果相应的相似性系数大于预定义的阈值则两个节点通过一条边连接;边缘的宽度与边缘连接的药物之间的相似度成正比。
与现有技术相比,本发明具有以下有益效果:
(1)本发明提供了能够简单直接表征化合物和生物活性之间结合关系的计算靶标谱。它从生物空间的角度阐述分子表征,将化合物和靶标之间的结合亲和力整合到分子表征中,通过机体的整体性预测得到新的活性或者生物信息;利用计算机快速得出结果,相较于基于试验的生物活性分子表征更加快速高效;仅需要化学结构信息,不局限于现有具备生物试验信息的分子中;适用范围广泛,作为分子表征时,可以应用到qsar模型中预测生物活性或毒性,也可以在虚拟筛选中富集活性分子及活性骨架。
(2)本发明提供了一种计算靶标谱在骨架跃迁中的应用。与传统用于骨架跃迁的分子表征相比,计算靶标谱引入了分子与蛋白质靶标之间的结合强弱关系表征分子,削弱了与化学结构的关联,因此计算靶标谱具有优越的骨架跃迁性能。
(3)本发明提供了一种计算靶标谱在预测蛋白质靶标中的应用。该方法要求简单,高效迅速,能较准确地计算出化合物的潜在靶标。
(4)本发明提供了一种计算靶标谱在qsar模型预测肝毒性的应用。计算靶标谱表征的是化合物与生物活性之间的关系,与化学化结构描述符的信息能相互补充,构建稳健准确的肝毒性模型。
(5)本发明提供了一种计算靶标谱在预测药物药物相互作用中的应用。计算靶标谱以生物活性的角度重新表征药物,能够准确地捕获药物与药物之间的相互作用。
(6)本发明提供了一种计算靶标谱在确定药物作用模式网络中的应用,计算靶标谱通过表征化合物与蛋白质靶标的结合亲和力关系,阐述药物在生物机体中的作用机制,将这种机制关系可视化,能够给研究者带来新的研究思路。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
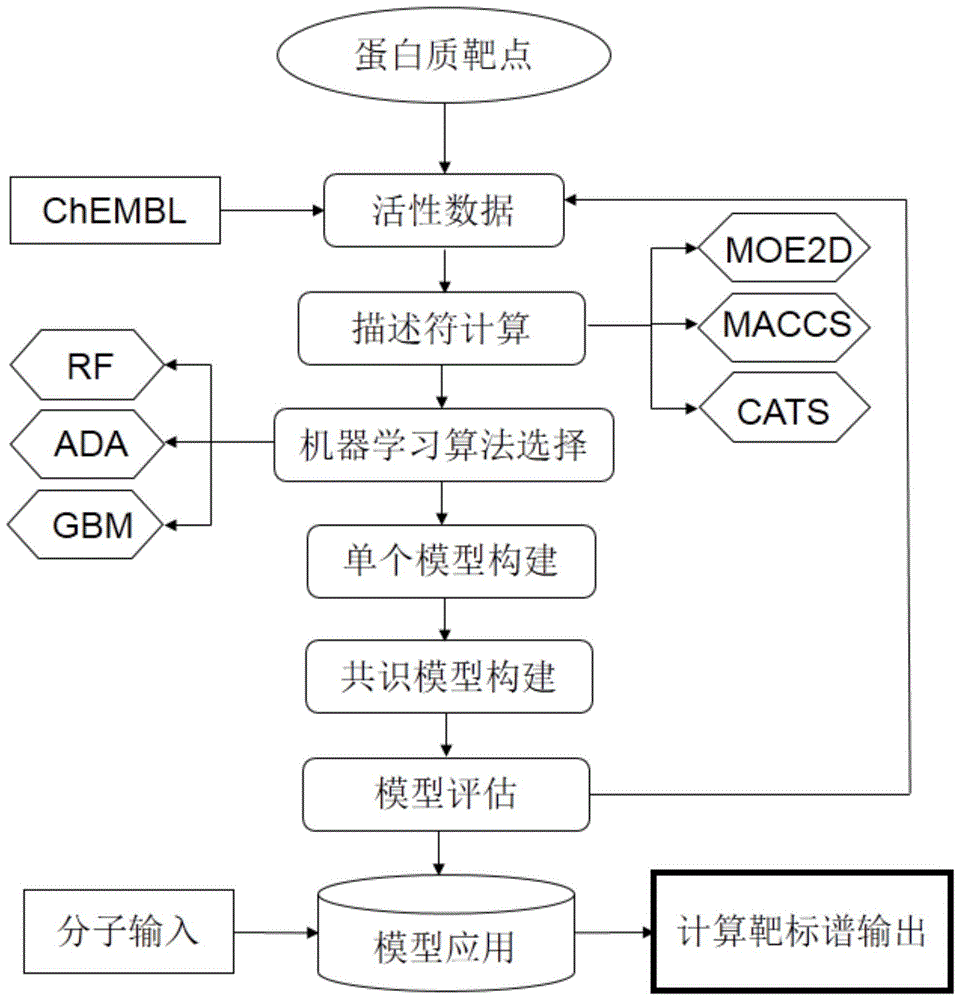
图1为本发明实施例1中计算靶标谱的构建流程图。
图2为本发明实施例2中计算靶标谱应用于骨架跃迁的相似性搜索框架。
图3为本发明实施例2中计算靶标谱应用于骨架跃迁的结果。
图4为本发明实施例6中计算靶标谱应用于药物作用模式的网络图。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。
若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段;若未特别指明,实施例中所用试剂均为市售。
本发明涉及到的百分号“%”,若未特别说明,是指质量百分比;但溶液的百分比,除另有规定外,是指100ml溶液中含有溶质的克数。
本发明所述重量份可以是μg、mg、g、kg等本领域公知的重量单位,也可以是其倍数,如1/10、1/100、10倍、100倍等。
实施例1:
一种计算靶标谱,其构建流程参见图1,具体构建步骤为:
(1)数据收集:收集靶标蛋白质及其活性数据。
从数据库和文献中收集多个蛋白质靶标,然后从chembl数据库中下载每个蛋白质靶标的活性数据,活性数据包括:ic50(halfmaximalinhibitoryconcentration,半抑制浓度),指对指定的生物过程抑制一半时所需的药物或抑制剂浓度),ec50(halfmaximaleffectiveconcentration,半数最大效应浓度),指在特定暴露时间后能达到50%最大生物效应对应的药物、或抑制剂的浓度),抑制常数(ki值,药物的半数解离浓度)中任意一种活性值的数据。本实施例的最终活性数据值为上述活性数据的均值。
为了取得更准确稳定的模型,我们去除活性数据数量小于200的蛋白质靶标,留下832个蛋白质靶标,包括239个激酶、270个其他酶、192个受体、46个离子通道、30个转运体和55个其他类别。具体蛋白质靶标如表1所示。
表1:实施例1中计算靶标谱中应用的蛋白质靶标
(2)数据预处理及劈分:对活性数据进行预处理,根据活性值划分为正集和负集。
为了去除建模中数据导致的噪音,活性数据用molecularoperatingenvironment(moe,版本2018)进行预处理,通过wash功能去除金属离子,除盐,加氢,对强酸强碱质子化。在每个蛋白质靶标中,将活性值低于10μm的数据作为正集,活性值高于10μm的数据作为负集。若负集数据不足,从其他蛋白质靶标中抽取部分数据作为负集,以减少数据不平衡带来的偏差。
(3)模型构建:选择描述符和机器学习算法,构建模型。
3.1、选择表征分子物理化学性质、药效团和结构碎片的描述符:moe2d、cats和maccs。
3.2、选择基于树、连接和核的机器学习算法:rf、ada和gbm。
3.3、将三种不同的描述符和三种不同的机器学习算法两两组合,每个蛋白质靶标构建9个不同的单一模型,分别是:moe2d&rf模型、moe2d&ada模型、moe2d&gbm模型、cats&rf模型、cats&ada模型、cats&gbm模型、maccs&rf模型、maccs&ada模型、maccs&gbm模型。一共具有832*9个模型。为了取得更准确的结果,将832*9个模型整合单一模型的输出结果,计算平均概率值,作为分子与蛋白质靶标的结合强弱值,形成共识模型。
(4)模型评估:用五折交叉验证方法来评估构建的模型。
五折交叉验证是指,每个蛋白质靶点中的建模数据被分为同等大小的五个数据集,其中使用四个数据集训练模型,使用剩下的一个数据集测试构建的模型。这个过程重复五次,每次测试模型都使用不同的数据集,一个模型将得到五个结果,总共具有832*9*5个结果。使用acc(accuracy,准确度,表示分类正确的样本数所占比例),auc(theareaunderthereceiveroperatingcharacteristiccurve,受试者操作曲线下面积,是判断二分类预测模型整体性能的指标)这两个指标来评估构建模型的性能。
结果表明,模型的acc值和auc值分别在70~99.8%和75.9~100%的范围内。这个评估结果表明模型可有效预测新分子。
(5)形成计算靶标谱:在共识模型中,输入待测分子,输出计算靶标谱。
实施例2:
一种实施例1的计算靶标谱在骨架跃迁中的应用,其应用流程参见图2,具体构建步骤为:
(1)活性分子数据库:采用来自文献:vogt,m.;stumpfe,d.;geppert,h.;bajorath,j.scaffoldhoppingusingtwo-dimensionalfingerprints:truepotential,blackmagic,orahopelessendeavor?guidelinesforvirtualscreening.jmedchem2010,53,(15),5707-5715.构建的数据库,包括17个靶标的活性分子,只选择ki或ic50小于1μm,重原子个数在10-50内以及侧链集团中的非氢原子个数小于骨架中非氢原子个数的分子。每个靶标中具有至少10个不同bemis-murcko骨架(由bemis和murcko提出,定义为一个分子去除r-基团但保留环系统之间的连接基所提取出的支架),每个骨架有5个代表分子。
(2)筛选数据库:准备chembl(85644)、specs(212863)、naturalproducts(120686)、chemblock(198643)和asinex(567106)5个不同设计和大小的筛选数据库,降低数据库对结果的干扰。数据库中的分子的预处理过程与活性分子数据库分子相同。
(3)将活性分子数据库和筛选数据库中的分子输入到实施例1的共识模型中,输出每个新分子与共识模型中832个蛋白质靶标的结合亲和力强弱值。每个新分子将会得到832*1的数据矩阵,作为该分子的计算靶标谱进行应用。
为了更方便进行相似性搜索,根据0.5的阈值将最终结果转化为指纹形式。然后分别计算活性分子数据库和筛选数据库化合物的分子表征,包括moe2d、maccs、cats、ecfp4、atompair、fp2、gpidaph3、tgt和tgd,用作对比。
(4)依次选择活性分子数据库中每一个靶点的活性数据分别作为查询分子,将剩下的活性化合物和筛选数据库组成查询数据库。
(5)使用tanimoto系数计算查询分子与查询数据库中分子的相似性值。
(6)根据相似性值的大小排序,计算前100、200、500、1000、2000、5000化合物中活性骨架回收率(活性分子骨架数量与所有活性分子骨架数量的比值),得到与查询分子不同骨架的活性分子的富集情况。
(7)结果参见图3:所有分子表征均显现出一定的骨架跃迁能力。所有分子表征总体富集能力的顺序为:计算靶标谱>moe2d>ecfp4,gpidaph3>其他。计算靶标谱在相似性值排名前100个化合物中,平均能够回收chembl中38.0%(np:43.7%,chemblock:24.4%,specs:23.3%,asinex:40.2%)的活性骨架。计算靶标谱在五个数据库中的平均活性骨架回收率为33.9%,表明在前100种化合物中发现了近三分之一的活性支架类型。以本研究中的17个靶标中最低的10种骨架类型为标准,计算靶标谱在每一个靶标中最少能富集到3.3个活性骨架。在五个筛选数据库中,对于前5000个化合物,计算靶标谱的活性骨架回收率分别为69.5%、65.3%、52.1%、50.6%和59.7%。总体活性骨架回收率大于50%,甚至达到69.5%,这已经证实计算靶标谱从筛选数据库中回收活性骨架的强大能力。最重要的是,我们发现计算靶标谱的活性骨架回收率在五个数据库中是最高的或略低于最高。它表明,计算靶标谱总体上具有比本研究中所用的其他分子表征更好的骨架跃迁性能。
实施例3
一种实施例1的计算靶标谱预测蛋白质靶标的应用,其构建过程如下所述:
(1)将药物溴隐亭输入到共识模型,输出溴隐亭的计算靶标谱,形成分子表征。
(2)我们将计算靶标谱中的预测概率值作为化合物与蛋白质靶标之间的结合亲和力值,根据该结合亲和力值对靶标进行排名。
(3)在计算靶标谱中显示,溴隐亭可能与新的蛋白质d(1a)多巴胺受体(uniprotid:q95136)相互作用。甲磺酸溴隐亭是具有强多巴胺能活性的半合成麦角生物碱衍生物,且适用于治疗帕金森综合症。通过查阅相关文献,发现溴隐亭与d(1a)多巴胺受体之间的结合亲和力(ki=1.444μm)。并且溴隐亭的大多数已批准靶标排名在预测前30,这表明计算靶标谱可以在一定程度上预测药物的潜在靶标。
实施例4
一种实施例1的计算靶标谱在qsar模型预测肝毒性的应用,具体构建步骤为:
(1)从sider、livertox和drugbank数据库和文献收集基于人体的肝毒性数据和非肝毒性数据,使用moe中的wash功能对数据进行预处理。
(2)采用不同的数据劈分策略将预处理后的数据划分为训练集和测试集,具体为:随机劈分(比例为80%﹕20%),bemis-murcko骨架劈分(根据bemis-murcko骨架类别划分训练集和测试集,其中训练集和测试集的骨架不同,比例为75%-85%﹕15%-25%)和碳骨架劈分(根据碳骨架类别划分训练集和测试集,其中训练集和测试集的骨架不同,比例为75%-85%﹕15%-25%)。设置两种骨架劈分的目的是研究计算靶标谱在真实情况下的肝毒性预测表现。
(3)将肝毒性和非肝毒性数据输入到实施例1的共识模型,输出计算靶标谱,作为cades分子表征。计算基于化学结构的描述符,包括moe2d、cats和maccs。
(4)将步骤(3)中cades分子表征和xgboosting算法构建模型。基于化学结构的moe2d、cats和maccs描述符分别和xgboosting算法构建模型。然后使用acc、auc、spe(specificity,特异性,表示预测正确的负样本数与真实正确的负样本数的比例)和sen(sensitivity,敏感度,表示预测正确的正样本数与真实正确的正样本数的比例)评估模型。考察结果参见表2。
表2:计算靶标谱与其他分子表征应用于肝毒性预测的比较结果
从表2的结果可知:在随机劈分中,使用本申请的cades模型和maccs、moe2d训练的模型预测性能相近,acc接近70.0%,该结果表明cades模型具有与其他优秀常见化学结构描述符相当的肝毒性预测性能。在bemis-murcko骨架劈分中,训练集和测试集中的分子骨架完全不同,这导致评估参数低于随机劈分,平均准确度从69.4%降低到66.1%。但是,我们发现,基于cades模型表现最佳,具有67.0%的acc、76.2%的sen、53.0%的spe和71.9%的auc,这是因为cades模型擅于捕获生物信息且对化学结构依赖程度低。我们还根据碳支架定义划分了训练集和测试集,结果显示计算靶标谱的模型比其他模型表现更好。这些结果表明计算靶标谱在实际应用中更适合肝毒性预测。
(5)组合步骤(4)中计算靶标谱训练的模型和化学结构描述符训练的模型,构建共识模型。具体组合方式为:cats+cades、maccs+cades、moe2d+cades。
(6)将测试集数据输入到步骤(5)中的共识模型中,考察共识模型应用于肝毒性预测的结果。结果参见表3。
表3:计算靶标谱与其他分子表征组合构建共识模型应用于肝毒性预测的结果
从表3的结果可知:共识模型通常具有比单个描述符训练的模型更好的肝毒性预测性能,这是因为计算靶标谱和化学结构描述符信息的相互补充,使得单一模型无法区分的化合物被正确区分。但是,在共识模型中也有将单一模型预测正确的化合物预测错误的化合物。总而言之,共识模型利大于弊,整合计算靶标谱和化学结构描述符确实可以提升肝毒性的预测性能。
实施例5
一种实施例1的计算靶标谱预测药物与药物相互作用的应用,其构建过程如下所述:
(1)根据文献:yao,z.j.;dong,j.;che,y.j.;zhu,m.f.;wen,m.;wang,n.n.;wang,s.;cao,d.s.targetnet:awebserviceforpredictingpotentialdrug-targetinteractionprofilingviamulti-targetsarmodels.jcomputaidedmoldes.2016,30,413-24.收集的1125种药物和6743种药物-药物相互作用对,将这1125种药物输入到模型中,输出实施例1的计算靶标谱,形成分子表征。
(2)将分子表征中的数据分为正样本和负样本,正样本为所有真实的药物-药物相互作用对,负样本为1125种药物之间的随机非相互作用对。负样本随机取样,取样量为正样本的数量,以避免不平衡数据引起的偏差。
(3)将药物-药物相互作用对之间两个药物的计算靶标谱组合作为描述符,结合rf算法构建模型。
(4)使用三个不同级别的测试集评估模型:
级别1:训练集中包含两种药物的ddi关联。
级别2:训练集中仅包含一种药物的ddi关联。
级别3:训练集中不包括两种药物的ddi关联。
采用留一法交叉验证评估模型。
(5)结果参见表4:
表4:分子表征应用于药物-药物相互作用的结果
从表4的结果可知:级别1的模型获得了最佳的预测性能,其acc、sen和spe分别为91.8%、92.8%和88.8%。级别2的模型的acc、sen和spe分别为71.6%、74.5%和68.6%。级别3的模型的acc、sen和spe分别为68.9%、62.9%和75.1%。准确度排序为级别1>级别2>级别3,这是因为不同级别蕴含的信息依次减少,这个结果建议模型中应当包含尽可能丰富的信息。整体模型的高准确度表明计算靶标谱可以用作在临床试验和药物开发过程中评估药物药物相互作用的有效分子表征。
实施例6
一种实施例1的计算靶标谱确定药物作用模式网络的应用,其构建过程如下所述:
(1)将从drugbank收集的已批准且具有化学结构的909种药物输入到实施例1的模型中,输出计算靶标谱,获得分子表征。
(2)计算任意两个药物之间的pearson相关系数,根据解剖化学分类法(anatomicaltherapeuticchemicalclassificationsystem,atc)将药物网络可视化。其中每个节点对应一个药物,并且如果相应的相似性系数大于预定义的阈值(此网络的相似性阈值为0.97),则两个节点通过一条边连接。边缘的宽度与边缘连接的药物之间的相似度成正比。
(3)结果参见图4:在网络中,具有相似作用模式的药物被连接或位于同一社区中。首先,我们检查了两种连接药物的atc一级水平是否相同。图中可以清楚地看到835条边中的327条边表示相同的atc级别,这表明atc中的第一级可以在一定程度上反映药物作用模式。例如,flunisolide和fluocinonide具有相似的计算靶标谱,相似系数为0.987。我们发现两种药物作用的原理都是激活糖皮质激素受体。然而,氟尼松是一种皮质类固醇激素,通常用于治疗变应性鼻炎,而fluocinonide则是用于治疗湿疹的局部糖皮质激素。另外,medrysone和methylprednisolone也具有非常相似的计算靶标谱,相似系数为0.993。medrysone用于治疗过敏性结膜炎、春季结膜炎、巩膜炎和肾上腺素敏感性,而methylprednisolone用于类风湿关节炎的短期辅助治疗。根据文献,我们发现它们是通过诱导磷脂酶a2抑制蛋白(统称为脂皮质激素)发挥作用。据推测,这些蛋白质通过抑制它们共同的前体花生四烯酸的释放来控制炎症的有效介质如前列腺素和白三烯的生物合成。这些结果显示计算靶标谱对药物作用模式网络预测的准确性。通过计算靶标谱预测药物作用模式网络仅需要计算相似性度量,无需提前了解药物作用方式,这极大推进了药物作用模式在药物研究中的发展和应用。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!