一种稳定的头孢克洛颗粒药物组合物的制作方法
本发明涉及一种稳定的头孢克洛颗粒药物组合物,属于药物制剂技术领域。
背景技术:
头孢克洛由礼来实验室研发,为第二代头孢菌素,具有广谱抗菌的作用,其作用机理与其它头孢菌素相同,主要通过抑制细胞壁的合成达到杀菌作用。分别在美国、日本等国家上市,现已在包括中国在内的世界多个国家销售,商品名主要有ceclor、希刻劳®、distaclor、kefral、keflor、口福乐等。目前在我国有干混悬剂、胶囊剂、片剂和颗粒剂等多种剂型销售。
根据日本盐野义公司公开的头孢克洛细粒剂if文件和专利cn108743547a显示,头孢克洛对湿、热均不太稳定,在产品储存过程中会不同程度地出现含量明显下降、杂质升高的现象。药物含量是药物起效的关键指标,药物杂质是药物副作用和不良反应的重要来源,头孢克洛稳定性不佳的特点对药品的有效性和安全性带来了隐患。
cn103330685b和cn108743547a采用药物包合技术,利用包合物包裹主药的方式对主药头孢克洛进行处理,降低了药物降解的速度,但上述工艺采用药物包合技术需增加减压干燥等步骤,提高了工艺复杂度,增加了生产成本。
二氧化硅是药物制剂中常用辅料,根据辅料手册(r.c.罗,p.j.舍斯基,p.j.韦勒.药用辅料手册[m].北京:化学工业出版社,2005.)描述,在固体制剂中可作为助流剂和崩解剂使用,作为助流剂使用时推荐用量为0.1~0.5%。
cn201510905949.0提出了一种用较高比例(丁苯酞和二氧化硅的质量比为0.1-4:1,即二氧化硅在药物载药过程中最高占总重90.9%)、强吸附性的二氧化硅粉末作为药物载体吸收液体状的丁苯酞,解决了丁苯酞固体剂型成分复杂、载药量小、药物稳定性较差以及制备工艺繁琐复杂、耗时较长的问题,但是需选择高孔容的二氧化硅作为载体(如syloid®244fp或cab-o-sil®m-5p),成本较高,且因高孔容二氧化硅粒径非常细、用量大时易形成高粉尘环境,产品生产时需关注粉尘爆炸的风险,对设备和操作有较高的防爆需求,同时对生产人员具有潜在呼吸道刺激乃至肺纤维化的风险。
cn201810929288公开了特定比例微粉化的二氧化硅、纳米化的二氧化硅与麦芽糊精的补强体系,利用二氧化硅具有改善药物引湿性的作用,改善了中药组合物的吸湿率,从而提高了药物稳定性;cn107213127b在说明书中提及“二氧化硅还能在一定程度上缓解山梨醇的吸湿性提高固体分散体的稳定性”。
技术实现要素:
发明目的:针对现有技术的不足,提供一种稳定的头孢克洛颗粒组合物,为临床提供信得过药品。
在使用二氧化硅作为助流剂对头孢克洛颗粒处方研究时,意外地发现在明显超过推荐用量(0.1~0.5%)时,占组合物总重1%~5%的二氧化硅不但起助流作用,还对头孢克洛的稳定性有正面意义。
试验还发现,并非是通过二氧化硅的抗吸湿性特点来提高药物稳定性的。如试验例2所示:使用本专利实施例和对比例的裸露颗粒在高湿环境下放置,第47小时水分均接近4%;实施例产品含量和有关物质相对0月无明显变化,对比例产品含量呈下降趋势,有关物质相对0月呈上升趋势,表明实施例产品获得了意想不到的稳定性效果。
本发明技术的原理尚不清楚,推测本发明用量范围内的二氧化硅有助于提高头孢克洛的稳定性的原理可能是:药物的化学反应一般在溶媒(如水蒸气)的介入下反应速率较快,固-固表面发生化学反应的速率非常慢。由于二氧化硅表面积较大,容易与其它原辅料形成巨大的分子间作用力,从而使颗粒结构更紧实,减少了颗粒与外界水氧交换的强度;二氧化硅结构疏松并易在颗粒内部形成无数疏水的类似毛细管的结构,通过颗粒表层的水分子易通过毛细管作用运输至二氧化硅“管路”内部;二氧化硅具有较强的吸附性,使“管路”内部的水分子难以逃逸在“毛细管”附近形成了水分相对饱和的稳定的结构。二者共同作用下,降低了头孢克洛与氧分子或水分子等发生氧化、水解等反应的速率,药物稳定性得以提高。这种推测并非定论,仅是本发明“意外发现”的一种可能的理论解释。
本发明的技术方案是:
一种头孢克洛颗粒药物组合物,单位剂量的组合物中含有:头孢克洛(以c15h14cln3o4s计)6.5%~17.8%,二氧化硅1%~5%,填充剂65%~90%,粘合剂1%~5%,崩解剂1%~5%,所述百分比为质量百分比。
本发明所述头孢克洛颗粒药物组合物,单位剂量的药物组合物总重量不大于2.5g。
本发明所述头孢克洛颗粒药物组合物,优选的二氧化硅用量为2~4%。
填充剂选自甘露醇、蔗糖、玉米淀粉、糊精中的一种或几种。
粘合剂选自甲基纤维素、乙基纤维素、羟丙甲纤维素、羟丙纤维素、聚乙烯吡咯烷酮中的一种或几种;粘合剂以干粉形式加入,也可以全部或部分溶解于润湿剂后加入。
崩解剂选自交联羧甲基纤维素钠、羧甲基淀粉钠、交联聚维酮、低取代羟丙纤维素中的一种或几种。
本发明所述头孢克洛颗粒的药物组合物还可以含有适量的ph调节剂和/或着色剂。
ph调节剂选自为苹果酸、富马酸、醋酸、酒石酸、盐酸、磷酸、乳酸中的一种或几种,ph调节剂占组合物总重的0.1%~0.5%;着色剂组成为氧化铁、胡萝卜素、胭脂虫红、柠檬黄、日落黄中的一种或几种,着色剂占组合物总重的0.2%~0.5%。加入ph调节剂的目的是为了使药物组合物满足ph=3.0~5.0的药典要求。
本发明所述头孢克洛颗粒的药物组合物,还可以添加矫味剂,所述矫味剂选自单糖浆、山梨醇、阿司帕坦、三氯蔗糖、甜菊糖苷、葡萄糖、木糖醇、麦芽糖醇、香精中的一种或几种;矫味剂占组合物总重的0.2%~2%。
本发明所涉及的头孢克洛颗粒的药物组合物,所涉及的润湿剂为水或乙醇水溶液,并在干燥过程中去除。
本发明所述头孢克洛颗粒组合物的制备方法,包括以下步骤:
步骤1.配制外加辅料(例如润湿剂/粘合剂)
步骤2.将处方量的头孢克洛、填充剂、崩解剂、二氧化硅、部分或全部粘合剂加入制粒机中混合均匀;
步骤3.搅拌下向存有步骤2物料的制粒机中加入外加辅料,如润湿剂或配制好的部分/全部粘合剂,制软材;
步骤4.将步骤3制备的软材使用摇摆颗粒机或快速制粒机制湿颗粒;
步骤5.湿颗粒转移至沸腾干燥机或烘箱中干燥,干燥时控制物料温度不超过60℃;
步骤6.使用筛分机整粒筛除不符合要求颗粒(要求不能通过10目标准筛和能通过80目标准筛的颗粒之和不超过15%)
步骤7.将步骤6所得颗粒总混均匀后,根据需求进行颗粒分装得到成品。
本发明所述制备方法,矫味剂、ph调节剂和/或着色剂可以在步骤2环节加入,也可以溶解于润湿剂后在步骤3环节加入。
本发明采用的二氧化硅用量占药物组合物总重的1~5%,优选占2~4%,明显超过推荐用量(0.1~0.5%),但最大总用量符合fda公布的iig和日本添加物事典用量要求,患者服用安全性可以得到保障。
经过加速条件和中间条件的稳定性考察,如试验例1所示:
加速条件稳定性试验表明:经6个月稳定性考察,采用本专利的实施例2较未采用本专利的对比例3稳定性期间含量降低更小、杂质增长明显更慢;使用本专利制备的药物组合物(实施例1~7),相对于剂型最为接近的参比制剂希克劳®(头孢克洛干混悬剂)杂质增幅更小、含量降低更少,表现出了显著的质量优势;
中间条件稳定性试验表明:经12个月稳定性考察,实施例2优于实施例1和实施例4,显示出二氧化硅用量占总重量的2%~4%可实现更好的应用效果,并且实施例均优于对比例,说明本发明技术获得了意想不到的效果。
对比例1和2不能得到理想实施效果,表明高于本专利要求的二氧化硅浓度不利于工艺的顺利进行(对比例1),低于比例本专利要求的二氧化硅浓度不能满足稳定性要求(对比例2)。
附图说明:
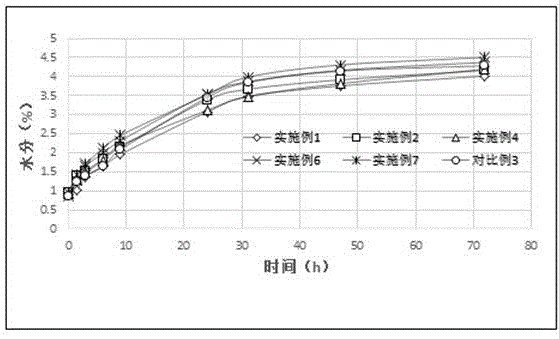
图1:头孢克洛颗粒高湿挑战试验吸湿增重速率图。
有益效果:
综上,与现有技术相比,本发明所得药物组合物有效提高了药物稳定性,有利于提高药物有效性和安全性;本方法生产成本低、操作简便,二氧化硅占总重比例远小于cn201510905949.0,避免了二氧化硅干粉潜在的形成高粉尘环境和对劳动者的伤害的风险。
具体实施方式
下述实施例仅为本发明的优选实施例,并非对本发明保护范围的限制,但凡采用本发明的设计原理,以及在此基础上进行非创造性劳动而作出的变化,均应属于本发明的保护范围之内。
实施例1:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2.内加原辅料预混:将处方量头孢克洛、二氧化硅、羟丙甲纤维素、低取代羟丙纤维素、甘露醇、玉米淀粉、阿司帕坦和天然等同草莓粉末香精加入湿法制粒机中,混合均匀。
步骤3.湿法制软材:搅拌下,向步骤2所得混合物中加入步骤1所得的外加辅料制备适宜软材。
步骤4.制湿颗粒:将步骤3制备的软材使用摇摆颗粒机制湿颗粒。
步骤5.干燥:湿颗粒转移至沸腾干燥机中,设置物料温度55℃干燥,干燥至颗粒水分≤1.5%时停机出料。
步骤6.整粒:使用标准筛筛分颗粒,要求不能通过10目标准筛和能通过80目标准筛的颗粒之和不超过15%。
步骤7.分装:步骤6所得颗粒混合均匀后,根据需求进行分装得成品。
实施例2:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤7同实施例1。
实施例3:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量20%乙醇中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤7同实施例1。
实施例4:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤7同实施例1。
实施例5:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量磷酸和聚乙烯吡咯烷酮(k30),搅拌至溶液均匀澄清透明即得。
步骤2至步骤7同实施例1。
实施例6:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量柠檬黄和磷酸,搅拌至溶液均匀澄清透明即得。
步骤2至步骤7同实施例1。
实施例7:
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量柠檬黄和磷酸,搅拌至溶液均匀澄清透明即得。
步骤2至步骤7同实施例1。
对比例1:二氧化硅用量高于5%。
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤3同实施例1。
结果:步骤3制得的软材过湿、过粘,无法制粒,不满足步骤4工序要求,表明过多的二氧化硅用量影响产品工艺的可实现性。
对比例2:二氧化硅用量低于1%。
处方:
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤7同实施例1。
对比例3:
处方:参考实施例2,用滑石粉代替二氧化硅,用量相同。
制备方法:
步骤1.外加辅料制备方法:搅拌下向处方量纯化水中加入处方量苹果酸,搅拌均匀即得。
步骤2至步骤7同实施例1。
试验例1.根据药典规定的检测方法对实施例1-7产品及对比例2-3产品进行检测,数据记录于表1。
表1实施例与对比例检验结果汇总
说明:表1溶出度为第30分钟,6片的分别溶出度。
表1数据说明:除对比例1工艺无法实现外,本专利所展示的所有实施例和对比例均可顺利得到符合内控要求的头孢克洛颗粒。
试验例2.头孢克洛颗粒稳定性样品检测
将实施例1-7产品、对比例2-3产品、生产日期相近的参比制剂希克劳®同步进行稳定性考察,考察结果对比见表2和表3。
考察条件:加速条件(0月、1月、2月、3月和6月),结果见表2;
中间条件(0月、3月、6月、9月和12月),结果见表3。
考察项目:含量、有关物质(最大单个杂质、总杂质)。
含量检验方法:参考usp42头孢克洛干混悬剂含量检测方法,色谱柱:岛津ods-3v4.6*250mm,5μm,流动相:以缓冲液(用780ml水与10ml三乙胺混合,加入1g戊烷磺酸钠溶解,用磷酸调节ph值至2.5±0.1)与甲醇220ml的混合溶液为流动相,检测波长:265nm。
含量要求:90.0~110.0%。
有关物质检测方法:参考美国usp42头孢克洛干混悬剂有关物质检测方法,色谱柱:岛津ods-3v4.6*250mm,5μm、流动相a:取二水合磷酸二氢钠7.8g,加水溶解并稀释至1000ml,用磷酸调节ph值至4.0;流动相b:流动相a-乙腈(55:45),检测波长:220nm。
有关物质要求:最大单杂≤2.0%;总杂≤3.0%。
表2头孢克洛颗粒的加速条件考察结果汇总
注:“/”表示未进行;
表2数据表明:
6个月加速考察期间,实施例1~7产品的含量降解无显著差异;有关物质最大单杂均未超过0.3%,总杂均未超过1.0%;而对比例2和3产品的最大单杂0.39%、0.49%,总杂2.7%、3.77%;实施例1~7产品与市售希刻劳®相比,含量降低更少、杂质增长更慢,有明显质量优势。
实施例1~7产品与对比例2产品相比可知,二氧化硅用量低于1%重量比时,加速条件下放置6个月含量明显相对0月时降低,总杂质显著增长,表明二氧化硅用量低于1%时不能起到提高头孢克洛稳定性的作用;与对比例3相比可知,不使用二氧化硅作为助流剂时,其它助流剂(如滑石粉)没有提高头孢克洛稳定性的作用;如前所述,对比例1表明,二氧化硅用量高于5%重量比时,软材过湿,有碍于工艺顺利进行。
上述数据表明,采用占药物组合物1%~5%的二氧化硅、1%-3%的崩解剂和粘合剂、65%~90%的填充剂、0.2%~2%的矫味剂以及如有必要可添加的适量ph调节剂和/或着色剂制备的样品稳定性好,产品质量明显提升。
表3头孢克洛颗粒的中间条件考察结果汇总
注:0月-12月变化率=(12月数值-0月数值)/0月数值x100%
表3数据表明:
12个月中间条件考察期间,二氧化硅用量占组合物总重2%~4%的实施例2,杂质增长变化率为﹢50%,而二氧化硅用量占组合物总重分别为1%和5%%的实施例1和例4总杂质增长变化率为﹢160%,故本发明采用的二氧化硅用量占组合物总重的1~5%,优选占单位包装内容物的2~4%的结论是成立的;实施例1、2、4最大单杂和总杂增长变化率均显著优于对比例3。
试验例3.高湿挑战试验
取实施例1、2、4、6、7和对比例3产品各约20g,均匀平铺于培养皿中,分别敞口裸露放置于92.5%rh饱和硝酸钾环境,于下述时间取样检测颗粒水分(快速水分测定仪80℃检测),并分别对放置第47小时的产品检测含量、有关物质,结果记录于表4。
表4高湿挑战试验结果汇总
表4结果显示:在92.5%rh湿度条件下,各样品47小时水分均达到4.0%左右,从0时→47时各样品吸水速率增量无显著差异,说明相对于对比例3,采用本发明技术所得产品的抗吸湿特性并未获得改善。
实施例产品放置47h后含量、有关物质几乎无变化,与实施例1~7稳定性0月检验结果(试验例2)相当;对比例3产品放置47h含量呈下降趋势,有关物质呈升高趋势。说明本发明产品明显改善了头孢克洛颗粒的降解趋势。
- 还没有人留言评论。精彩留言会获得点赞!