基于深度学习的蛋白质结构预测方法及系统与流程
本发明涉及深度学习和生物信息领域,具体地,涉及一种基于深度学习的蛋白质结构预测方法及系统,尤其涉及一种基于人工智能的蛋白质结构预测、筛选与评估。
背景技术:
蛋白质是生命活动的主要承担者,生物体内许多重要生命过程都有蛋白质的参与。蛋白质由20种常见的氨基酸通过脱水缩合后形成的肽链连接而成。蛋白质的三维空间结构决定蛋白质的功能。从氨基酸序列预测蛋白质的三维结构是生物信息学中一个基础而尚未解决的问题。
迄今为止,对于确定蛋白质三维结构的研究方法主要分为两大类:一类是通过湿实验进行测定,一类是基于蛋白质序列进行预测。通过湿实验进行测定的方法包括x光衍射和核磁共振以及冷冻电镜技术等,这些方法存在一些显而易见的缺点,如耗时、昂贵等,在蛋白质序列快速累积的情况下,无法满足要求。因此如何提高从氨基酸序列直接预测蛋白质空间结构的准确率是蛋白质结构研究的关键问题。
对于从氨基酸序列直接预测蛋白质三维结构的研究,主要有两大类方法:一类是基于模板的的建模方法,一类是非模板的直接从头预测的方法。基于模板的建模方法受到蛋白质结构数据库中可用的结构模板数量和质量的限制,因此从头预测变得越来越重要。基于蛋白质序列片段的从头预测的工具如rosetta和quark等,虽然这些方法都在蛋白质结构预测方面取得了一定成果,但是他们也面临着结构预测中的一个重大挑战,即在无同源信息的情况下无法预测的问题。
针对上述问题,我们构建了基于人工智能的从头预测全自动流程,根据待预测蛋白质的序列信息进行特征计算,进而快速、准确的构建三维结构模型。
专利文献cn105184112a公开了一种基于改进小生境遗传算法的蛋白质结构预测方法。该方法将小生境遗传算法引入蛋白质结构预测中,并对遗传算法过程中的选择、变异进行了一定的改进。从实验得出的数据和与其他方法的比较结果来看,该方法可以更加全面的搜索出相应的蛋白质最小自由能量值,从而能得到更稳定的蛋白质结构;该方法的运行时间也大为缩短,说明了本方法具有良好的时间效率。该专利的流程和性能仍然有待完善的空间。
技术实现要素:
针对现有技术中的缺陷,本发明的目的是提供一种基于深度学习的蛋白质结构预测方法及系统。
根据本发明提供的一种基于深度学习的蛋白质结构预测方法,包括:
数据生成步骤:根据数据生成启动控制信息,获取原始多序列匹配数据,计算生成特征数据,用作以下网络输入;
网络结构搭建步骤:构建残基距离神经网络结构和角度神经网络结构,预测目标蛋白质残基间的距离及角度,用作以下结构生成算法的输入;
网络评价指标步骤:构建距离评价方法和角度评价方法,获取距离评价方法信息、角度评价方法信息;
结构生成和筛选步骤:构建蛋白质结构生成和筛选方案,获取蛋白质结构生成和筛选方案信息,用以输出目标蛋白质的三维结构预测结果;
结构评价指标步骤:构建结构评价指标,获取结构评价指标信息。在优化及测试中用以衡量蛋白质结构预测的准确度;
预测评价指标步骤:获取目标蛋白质预测结构的可信度评估,获取目标蛋白质预测结构的可信度评估信息。用以在应用实例中为用户提供指导;
根据距离评价方法信息、角度评价方法信息、蛋白质结构生成和筛选方案信息、结构评价指标信息、目标蛋白质预测结构的可信度评估信息,获取基于深度学习的蛋白质结构预测结果信息。
优选地,所述数据生成步骤包括:
数据生成第一子步骤:通过序列搜索算法在大规模序列数据库中搜索同源序列构建蛋白质多序列匹配数据集(multiplesequencealignment,msa);
在预测过程中将长度超过128的蛋白质序列依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集;
数据生成第二子步骤:生成特征数据集,所述特征数据集中的特征包括以下任意一种或者多种:
-序列热独码seq1hot;
-位置特异性打分矩阵pssm;
-位置特异性打分矩阵pssm;
蛋白质残基接触打分矩阵dca。
根据专业的计算方法生成特征数据集,特征包括:序列热独码seq1hot,位置特异性打分矩阵pssm(positionspecificscorematrix),蛋白质残基接触打分矩阵dca(directcouplinganalysis)。
优选地,所述网络结构搭建步骤包括:
网络结构搭建第一子步骤:搭建距离神经网络结构,定义残基距离神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构为残差网络,隐层神经单元为64,损失函数为交叉熵损失。
优选地,所述网络结构搭建步骤还包括:
网络结构搭建第二子步骤:搭建角度神经网络结构,定义角度神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构包块3部分,瓶颈网络,特征提取和输出网络,特征提取网络采取经典的resnet152架构,使用一维卷积替换2维卷积。输出网络为两个分类网络和回归网络,分类网络采取交叉熵(cross-entropy)损失函数,回归网络采取均方误差(mse,meansquareerror)损失函数。
以所述特征数据集中的特征作为距离神经网络输入,分别预测全链蛋白质序列及各子序列的残基间距离,并(均值)拼接为原全长蛋白质的距离分布;
以所述特征数据集中的特征作为上述角度神经网络输入,预测全长蛋白质的角度分布。
优选地,所述结构生成和筛选步骤包括:
结构生成和筛选子步骤:将预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型;并根据势能进行模型排序即筛选。
该流程优化中,预测模型相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)被用以衡量流程的准确度。
在具体实施方式中预测对象的真实结构不可知,因此proq3被用以评估预测结构的可信度。
根据本发明提供的一种基于深度学习的蛋白质结构预测系统,包括:
数据生成模块:根据数据生成启动控制信息,获取原始多序列匹配数据,计算生成特征数据,用作以下网络输入;
网络结构搭建模块:构建残基距离神经网络结构和角度神经网络结构,预测目标蛋白质残基间的距离及角度,用作以下结构生成算法的输入;
网络评价指标模块:构建距离评价方法和角度评价方法,获取距离评价方法信息、角度评价方法信息;
结构生成和筛选模块:构建蛋白质结构生成和筛选方案,获取蛋白质结构生成和筛选方案信息,用以输出目标蛋白质的三维结构预测结果;
结构评价指标模块:构建结构评价指标,获取结构评价指标信息。在优化及测试中用以衡量蛋白质结构预测的准确度;
预测评价指标模块:获取目标蛋白质预测结构的可信度评估,获取目标蛋白质预测结构的可信度评估信息。用以在应用实例中为用户提供指导;
优选地,所述数据生成模块包括:
数据生成第一子模块:通过序列搜索算法在大规模序列数据库中搜索同源序列构建蛋白质多序列匹配数据集(multiplesequencealignment,msa);
在预测过程中将长度超过128的蛋白质序列依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集;
数据生成第二子模块:生成特征数据集,所述特征数据集中的特征包括以下任意一种或者多种:
-序列热独码seq1hot;
-位置特异性打分矩阵pssm;
-位置特异性打分矩阵pssm;
蛋白质残基接触打分矩阵dca。
根据专业的计算系统生成特征数据集,特征包括:序列热独码seq1hot,位置特异性打分矩阵pssm(positionspecificscorematrix),蛋白质残基接触打分矩阵dca(directcouplinganalysis)。
优选地,所述网络结构搭建模块包括:
网络结构搭建第一子模块:搭建距离神经网络结构,定义残基距离神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构为残差网络,隐层神经单元为64,损失函数为交叉熵损失。
优选地,所述网络结构搭建模块还包括:
网络结构搭建第二子模块:搭建角度神经网络结构,定义角度神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构包块3部分,瓶颈网络,特征提取和输出网络,特征提取网络采取经典的resnet152架构,使用一维卷积替换2维卷积。输出网络为两个分类网络和回归网络,分类网络采取交叉熵(cross-entropy)损失函数,回归网络采取均方误差(mse,meansquareerror)损失函数。
以所述特征数据集中的特征作为距离神经网络输入,分别预测全链蛋白质序列及各子序列的残基间距离,并(均值)拼接为原全长蛋白质的距离分布;
以所述特征数据集中的特征作为上述角度神经网络输入,预测全长蛋白质的角度分布。
优选地,所述结构生成和筛选模块包括:
结构生成和筛选子模块:将预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型;并根据势能进行模型排序即筛选。
该流程优化中,预测模型相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)被用以衡量流程的准确度。
在具体实施方式中预测对象的真实结构不可知,因此proq3被用以评估预测结构的可信度。
与现有技术相比,本发明具有如下的有益效果:
1、本发明构建了基于人工智能的从头预测全自动流程,能够根据待预测蛋白质的序列信息进行特征计算,进而快速、准确的构建三维结构模型;
2、本发明流程设计合理,使用方便,能够克服现有技术的缺陷;
3、本发明提供了一种基于深度学习和生物信息理论的蛋白质三维结构模拟方法,该方法包括:蛋白质同源矩阵的搜索步骤、相关特征数据的计算步骤、蛋白质残基间距离与角度预测的网络构建步骤、距离与角度的预测准确度评估步骤、基于距离及角度的三维模型快速生成及优化步骤、三维结构模型的筛选步骤、及预测结果的评估步骤。该流程与传统方法相比,具有预测准确、快速的优势,可进行高通量的宏蛋白质组模拟。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为根据本发明实施例的基于人工智能的蛋白质结构预测的全流程示意性框图;
图2为根据本发明实施例的基于深度学习的残基间预测模型的示意性结构框图;
图3为根据本发明实施例的基于深度学习的角度预测模型的示意性结构框图。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变化和改进。这些都属于本发明的保护范围。
根据本发明提供的一种基于深度学习的蛋白质结构预测方法,包括:
数据生成步骤:根据数据生成启动控制信息,获取原始多序列匹配数据,计算生成特征数据,用作以下网络输入;
网络结构搭建步骤:构建残基距离神经网络结构和角度神经网络结构,预测目标蛋白质残基间的距离及角度,用作以下结构生成算法的输入;
网络评价指标步骤:构建距离评价方法和角度评价方法,获取距离评价方法信息、角度评价方法信息;
结构生成和筛选步骤:构建蛋白质结构生成和筛选方案,获取蛋白质结构生成和筛选方案信息,用以输出目标蛋白质的三维结构预测结果;
结构评价指标步骤:构建结构评价指标,获取结构评价指标信息。在优化及测试中用以衡量蛋白质结构预测的准确度;
预测评价指标步骤:获取目标蛋白质预测结构的可信度评估,获取目标蛋白质预测结构的可信度评估信息。用以在应用实例中为用户提供指导;
优选地,所述数据生成步骤包括:
数据生成第一子步骤:通过序列搜索算法在大规模序列数据库中搜索同源序列构建蛋白质多序列匹配数据集(multiplesequencealignment,msa);
在预测过程中将长度超过128的蛋白质序列依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集;
数据生成第二子步骤:生成特征数据集,所述特征数据集中的特征包括以下任意一种或者多种:
-序列热独码seq1hot;
-位置特异性打分矩阵pssm;
-位置特异性打分矩阵pssm;
蛋白质残基接触打分矩阵dca。
根据专业的计算方法生成特征数据集,特征包括:序列热独码seq1hot,位置特异性打分矩阵pssm(positionspecificscorematrix),蛋白质残基接触打分矩阵dca(directcouplinganalysis)。
优选地,所述网络结构搭建步骤包括:
网络结构搭建第一子步骤:搭建距离神经网络结构,定义残基距离神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构为残差网络,隐层神经单元为64,损失函数为交叉熵损失。
优选地,所述网络结构搭建步骤还包括:
网络结构搭建第二子步骤:搭建角度神经网络结构,定义角度神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构包块3部分,瓶颈网络,特征提取和输出网络,特征提取网络采取经典的resnet152架构,使用一维卷积替换2维卷积。输出网络为两个分类网络和回归网络,分类网络采取交叉熵(cross-entropy)损失函数,回归网络采取均方误差(mse,meansquareerror)损失函数。
以所述特征数据集中的特征作为距离神经网络输入,分别预测全链蛋白质序列及各子序列的残基间距离,并(均值)拼接为原全长蛋白质的距离分布;
以所述特征数据集中的特征作为上述角度神经网络输入,预测全长蛋白质的角度分布。
优选地,所述结构生成和筛选步骤包括:
结构生成和筛选子步骤:将预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型;并根据势能进行模型排序即筛选。
该流程优化中,预测模型相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)被用以衡量流程的准确度。
在具体实施方式中预测对象的真实结构不可知,因此proq3被用以评估预测结构的可信度。
根据本发明提供的一种基于深度学习的蛋白质结构预测系统,包括:
数据生成模块:根据数据生成启动控制信息,获取原始多序列匹配数据,计算生成特征数据,用作以下网络输入;
网络结构搭建模块:构建残基距离神经网络结构和角度神经网络结构,预测目标蛋白质残基间的距离及角度,用作以下结构生成算法的输入;
网络评价指标模块:构建距离评价方法和角度评价方法,获取距离评价方法信息、角度评价方法信息;
结构生成和筛选模块:构建蛋白质结构生成和筛选方案,获取蛋白质结构生成和筛选方案信息,用以输出目标蛋白质的三维结构预测结果;
结构评价指标模块:构建结构评价指标,获取结构评价指标信息。在优化及测试中用以衡量蛋白质结构预测的准确度;
预测评价指标模块:获取目标蛋白质预测结构的可信度评估,获取目标蛋白质预测结构的可信度评估信息。用以在应用实例中为用户提供指导;
优选地,所述数据生成模块包括:
数据生成第一子模块:通过序列搜索算法在大规模序列数据库中搜索同源序列构建蛋白质多序列匹配数据集(multiplesequencealignment,msa);
在预测过程中将长度超过128的蛋白质序列依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集;
数据生成第二子模块:生成特征数据集,所述特征数据集中的特征包括以下任意一种或者多种:
-序列热独码seq1hot;
-位置特异性打分矩阵pssm;
-位置特异性打分矩阵pssm;
蛋白质残基接触打分矩阵dca。
根据专业的计算系统生成特征数据集,特征包括:序列热独码seq1hot,位置特异性打分矩阵pssm(positionspecificscorematrix),蛋白质残基接触打分矩阵dca(directcouplinganalysis)。
优选地,所述网络结构搭建模块包括:
网络结构搭建第一子模块:搭建距离神经网络结构,定义残基距离神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构为残差网络,隐层神经单元为64,损失函数为交叉熵损失。
优选地,所述网络结构搭建模块还包括:
网络结构搭建第二子模块:搭建角度神经网络结构,定义角度神经网络结构中网络层类别和层的参数,以及网络的损失函数;
神经网络结构包块3部分,瓶颈网络,特征提取和输出网络,特征提取网络采取经典的resnet152架构,使用一维卷积替换2维卷积。输出网络为两个分类网络和回归网络,分类网络采取交叉熵(cross-entropy)损失函数,回归网络采取均方误差(mse,meansquareerror)损失函数。
以所述特征数据集中的特征作为距离神经网络输入,分别预测全链蛋白质序列及各子序列的残基间距离,并(均值)拼接为原全长蛋白质的距离分布;
以所述特征数据集中的特征作为上述角度神经网络输入,预测全长蛋白质的角度分布。
优选地,所述结构生成和筛选模块包括:
结构生成和筛选子模块:将预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型;并根据势能进行模型排序即筛选。
该流程优化中,预测模型相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)被用以衡量流程的准确度。
在具体实施方式中预测对象的真实结构不可知,因此proq3被用以评估预测结构的可信度。
在本发明一个实施例中,所述数据生成步骤包括:
通过序列搜索算法在大规模序列数据库中搜索同源序列构建蛋白质多序列匹配数据集(multiplesequencealignment,msa)。根据专业计算方法生成特征数据集,特征包括:序列热独码seq1hot,位置特异性打分矩阵pssm(positionspecificscorematrix),蛋白质残基接触打分矩阵dca(directcouplinganalysis)。在预测过程中将长度超过128的蛋白质序列依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集。
在本发明一个实施例中,所述网络结构搭建步骤包括:
搭建距离神经网络结构,定义残基距离神经网络结构中网络层类别和层的参数,以及网络的损失函数。神经网络结构为残差网络,隐层神经单元为64,损失函数为交叉熵损失。
搭建角度神经网络结构,定义角度神经网络结构中网络层类别和层的参数,以及网络的损失函数。神经网络结构包块3部分,瓶颈网络,特征提取和输出网络,特征提取网络采取经典的resnet152架构,使用一维卷积替换2维卷积。输出网络为两个分类网络和回归网络,分类网络采取交叉熵(cross-entropy)损失函数,回归网络采取均方误差(mse,meansquareerror)损失函数。
以上述特征为输入,根据距离神经网络分别预测全链蛋白质序列及各子序列的残基间距离,并(均值)拼接为原全长蛋白质的距离分布;根据角度神经网络预测全长蛋白质的角度分布。
在本发明一个实施例中,所述网络评价指标包括:
构建距离网络评价指标,定义距离预测准确度的评估方法。
在本发明一个实施例中,所述结构生成和筛选包括:
将预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型。并根据势能进行模型排序及筛选。
在本发明一个实施例中,所述结构评价指标包括:
预测模型相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)被用以在测试中衡量流程的准确度。
在本发明一个实施例中,所述预测结构评价指标包括:
在具体实施方式中预测对象的真实结构不可知,因此proq3被用以评估预测结构的可信度。
本发明的具体技术细节、训练及实施/测试方法将在具体实施方式部分具体阐述。
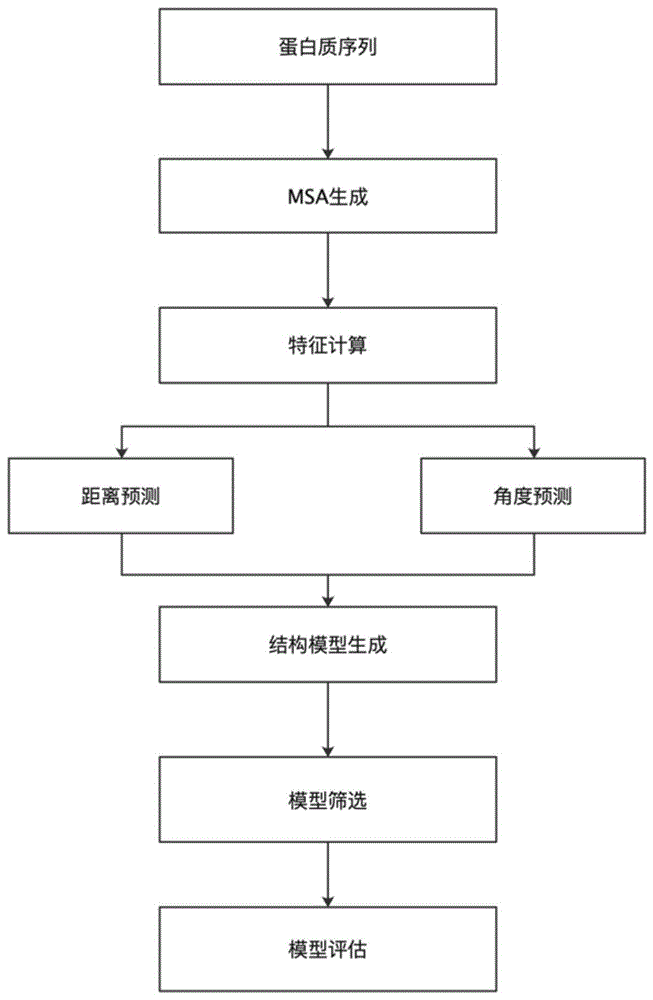
首先,参照图1来用于描述在蛋白质预测中本发明的主要流程步骤,其中包括:根据蛋白质序列进行msa的生成及特征计算、蛋白质残基间距离与角度的预测、基于距离及角度的结构模型生成、模型的优化筛选、及最终模型的评估。
1.步骤101,蛋白质多序列匹配数据集(msa)的搜索是指在考虑到突变、插入、删除、重组等进化事件的前提下,把多个氨基酸序列对齐并逐列进行比较,从而构建数据库中与被预测蛋白质序列相似的数据集。如果把多序列比对当作一张二维表,其中每一行表示一个氨基酸序列,每一列表示一个残基的位置,然后将进行比对的序列依照下列规则填入表中:(a)一个序列中所有残基的相对位置保持不变;(b)将不同序列中相同或相似的残基放入同一列,使得序列间相同或相似残基尽可能上下对齐。在一个实例中,将采取hhblits/jackhmmer/hmmsearch的方法从uniprot30/uniprot100/metagenomenr序列数据库中进行逐步搜索,直至得到信息充足的msa或搜索结束。
在一个实例中,长度超过128的蛋白质序列将被依次切割成64,128及256的子序列,并分别单独构建上述多序列匹配数据集。
2.步骤102,特征中被预测蛋白质的序列独热编码seq1hot,是使用n位状态寄存器对n个状态进行编码,每个状态都由他独立寄存器位,并且在任意时候,只有一位有效。这里的n是指氨基酸的种类(n=21)。
特征中被预测蛋白质的位置特异性打分矩阵pssm,通过以下步骤计算得到:
第一步,构建位置频度矩阵pfm(positionfrequencymatrix):
pfm矩阵大小应该为n*l,其中n是氨基酸的种类(n=21),l是序列的长度。通过msa多序列匹配矩阵(m*l),计算每一列的各个氨基酸的数量,然后存入pfm矩阵的相应位置,即得到pfm矩阵x。
第二步,构建位置概率矩阵ppm(positionprobabilitymatrix):
通过下面公式,可以由pfm计算位置矩阵ppm,
其中,k为不同氨基酸种类,i为行号,j为列号,即:
i∈(1,2,....,n),j∈(1,2,…,l)
且i是指示函数,即:
第三步,构建位置特异性打分矩阵pssm(positionspecificscorematrix)
这里引入参数b,b=1/k,对于蛋白质序列k=20,那么对于相同位置上的ppm和pssm的矩阵元素关系为:
特征中被预测蛋白质的残基接触打分矩阵,是采用直接耦合分析矩阵dca来分析msa中的协同进化模式。
直接耦合分析矩阵dca的目的是通过协同进化的耦合来解释残基之间的相关性。使用的模型为广义波茨模型(pottsmodel):
其中,h(a1,a2,...al)为哈密顿量,表述了蛋白质残基间交互系统的总能量。hi(ai)和jij(ai,aj)分别为位置i对应的ai残基类型的局部场参数以及位置i和位置j分别对应ai残基和aj残基的耦合参数。该模型不仅包含了局部场hi,还考虑了残基间的耦合参数jij。据此,可以在整个msa上构建一个全局统计模型p(a1,a2,...al),在全局模型中,msa的一个序列的概率可以定义为:
其中,z为归一化常数,保证
一旦从msa通过直接耦合分析算法获得参数j,就可以通过进一步处理获得残疾对的打分,参数j的大小为21l*21l的矩阵,需要通过下式得到l*l的蛋白质残基接触打分矩阵:
该打分矩阵可能带来遗传进化过程中的背景噪声,我们通过平均乘积矫正(averageproductcorrection,apc)法去除背景噪声。apc的矫正公式为:
其中,si:为打分矩阵在第i行的平均值,s:j为打分矩阵在第j行的平均值,s::为打分矩阵的总体平均值。平均乘积矫正可以保证矫正后的打分矩阵的行列式均值为0,其依据为假设为,由于每个残基只跟小部分其他残基产生接触,原始打分矩阵每一行或者每一列的均值就会由系统偏差造成而不是该残基跟其他小部分残基形成的交互打分。apc还可以理解为对于原始打分矩阵最大主成分的一种近似,因此消除了原始打分矩阵来自背景偏差的最高可变性。
3.步骤103,被预测蛋白质残基间的距离预测由距预测网络完成。在蛋白质三维结构文件中(proteindatabank,pdb),每个氨基酸原子都有着相应的三维坐标,本发明的距离预测网络预测两两氨基酸的cb原子之间的距离分布:按照
本领域的科技人员知道cropping是非常有效的数据增强方法,本发明采取64x64的方式对蛋白质的特征进行cropping处理。考虑到训练有效性,要求在cropping后的每个tile间有效数据至少超过四分之一,根据这个要求约束cropping时偏移的值,对行偏移和列偏移分开取随机值并保留。在网络训练中,groundtruth的行偏移和列偏移将与特征对齐。
残基间的距离预测网络由160个图2所示的block及最后的projectdown和softmax层构成。网络采用focalloss,考虑到训练时会采用梯度累积,无法对累积的每个batch进行数据统计,采用较为naive的方式进行weightbalance,设置最后一个bin(即超过
本领域的技术人员了解极深网络的训练十分困难,因为我们将具体描述在训练过程中采取的策略:
学习率衰减:每5个epoch后学习率衰减为原学习率的0.33倍;
梯度下降:每64个step的梯度累积后求平均值,使用该均值进行梯度更新。该方式称为梯度累积,相当于消耗64倍的时间进行batchsize64的训练。
网络深度递增:在训练过程中逐步增加网络深度,具体过程如下:
a.训练主体具有60个blocks的模型,开始训练直至loss稳定;
b.与原始模型中主体的blocks后添加20个block,冻结原始模型中主体blocks的参数,继续训练直到loss稳定;
c.取消原始模型中所有的参数冻结,继续训练直到loss稳定;
d.若此时训练的模型准确率低于原模型,则删除该模型,再次进行第b、c步;
e.重复b、c、d步,直到主体blocks数量到达160个blocks。
上述残基间距离神经网络根据4所述的评估标准进行训练优化。在本发明的一个实施例中所预测蛋白质的特征:序列独热编码,位置特异性打分矩阵及接触打分矩阵为输入,根据上述距离神经网络进行距离分布的预测。本领域的技术人员应该注意:
由于网络固定输入为64x64大小,因此在预测时同样需要对数据进行cropping;
按照训练数据预处理方式,对预测数据进行多次cropping,不打乱顺序,生成多组特征切片(featuretiles);
对每个切片进行预测,生成多组距离预测切片(distancepredictiontiles);
按照原顺序,对各组距离预测切片进行组合,生成多个距离预测;
平均各个距离预测,得到最终的距离预测输出。
在本发明的实施例应用中,全链蛋白质序列及各子序列的残基间距离被分别单独预测并再次平均,拼接为原全长蛋白质的距离分布。
被预测蛋白质残基间的角度预测由角度预测网络完成,角度决定了其折叠后在3维空间的结构。氨基酸的角度指的两个值phi和psi角,即φ,ψ角,其变化范围为[-π,π],分成36个bin,每个bin指的是15度的区间,参照图3所示,将角度预测分为分类任务和回归两个任务,分类任务输出概率分布,回归任务输出phi和psi的离散值。角度神经网络为多头网络,一共分为3个部分,第一部分为普通的3x3的瓶颈卷积层,该层的作用将特征(feature)的通道数(channel)数降低到64维,其作用和传统的残差网络瓶颈层保持一致。第二部分为特征提取层,为了保证网络不出现梯度消失和梯度爆炸,这里采取残差网络提取特征,该层一共分为4个stage,每一个stage包括多个block,每一个block为标准的残差结构,每一个卷积层后接一层bn层和relu激活层。特征提取网络一共包块4个stage,分别包块3,8,36,3个残差块,每一个stage都保存固定的channel数,分别为64,128,256,512。特征提取层的网络输出为bx32x512,其中b指的是batch_size的大小。第三部分为3个3x3的卷积层,phi负责预测角度中的φ角,psi预测角度中的ψ角,phi和psi为分类网络,经过softmax后输出为3个bin的概率分布即bx32x36,第三个为回归网络,输出为单独的离散值,每个残基对应φ、ψ两个角度,共输出两个值即bx32x2。氨基酸序列为一维线性序列且长度不固定,传统的2维卷积不适应于角度预测场景,这里采取一维卷积。
氨基酸长度可变带来的问题在于无法进行批量训练,批量训练对于提升网络的学习效果非常明显,为了让网络保持批量训练,采取crop的方式进行特征切割。假设长度为l的一条氨基酸链,一共包括l个氨基酸残基,每个氨基酸残基的feature维度为526维,整个链的feature为l×526维,这里固定切割长度为32,即将长度为l的链切割成若干32个氨基酸残基组成的小片段,每个小片段的feature维度为32x526。为了保证样本的丰富性,切割片段能覆盖整条氨基酸链且l不一定能完全整除32,这里对氨基酸链首部特征进行填充(padding),填充值为0,填充长度为32-lmod32。因此一条长度为l的氨基酸链经过切割后形成
上述角度神经网络根据5所述的评估标准进行训练优化。在本发明的一个实施例中,所预测蛋白质的特征:序列独热编码,位置特异性打分矩阵及接触打分矩阵为输入,根据上述角度神经网络进行角度分布的预测。
4.上述距离预测准确度评估方法:距离预测准确度的评估主要是评估残基接触图。根据两个残基在序列中的间距,残基间距接触可以分为短程,中程和远程接触,分别对应序列距离在6到11、12到23和24以上的残基间接触。由于中程和远程接触对蛋白质结构至关重要,在评价过程中只有这两部分的残基接触参与评价。评价指标为:
其中,tp(truepositive)和fp(falsepositive)分别表示正样本和负样本被预测为正样本的数目。
5.上述角度预测准确度评估方法:根据3中的角度神经网络结构设计,其输出值分为3部分,两个分类网络和一个回归网络,分别负责输出phi和psi角度的36个bin的概率分布及其两个离散值。氨基酸残基的角度值对于在折叠后的3维空间坐标起着至关重要的作用,氨基酸是线性序列,前面的序列角度错位直接影响后续序列在折叠后的3维坐标,这里不直接预测某个角度的值而是指定一个范围,可以减小折叠后残基在3维空间的坐标误差。单纯指定范围又可能使其偏离真实值太远,而且分36个bin是根据试验得来,这里加一个回归任务预测某个残疾角度的离散值,配合分类网络一起工作学习合适的网络参数。分类网络的评估指标采取传统的多分类交叉熵(cross-entropy)作为损失函数,分别为lossphi和losspsi,回归任务的损失函数lossreg采取传统的均方误差(mean-square-error,mse),交叉熵的作用在于监控网络输出和真实值在概率分布的误差,均方误差的作用在于使网络的输出不断逼近角度的真实值。最终的loss函数是3者之和即:
loss=lossphi+losspsi+lossreg
loss体现了网络输出和真实值的误差,经过神经网络的反向传播算法(bp)迫使网络将误差转化为梯度去更新网络权重从而达到学习的目的。这里回归loss的作用主要在于辅助phi和psi两个分类网络学习正确分类,让loss下降得更快更稳定。分类任务的loss对于回归网络也起到一定的制约作用使其不至于偏离真实值太远。这里使用loss来评价网络的性能,loss越低表明网络学习效果越好,loss低表明网络学到了合适的参数,网络作为黑盒,对于一个非凸优化问题来说起到很好的拟合效果。
网络的3个输出值中,取phi和psi分类网络的bin区间的中心值作为网络的最终输出,也是下游任务所需要的值,这里采平均取绝对值误差(mae,meanabsoluteerror)来评价网络输出和真实值之间的误差,mae的计算公式为:
其中i指的是第i个残基。
6.步骤104,在本发明的一个实施例中,将4中预测的残基间距离与角度分布转换为可约的平滑能量势能,通过梯度下降的方式快速获取在此约束下的势能最小化模型。并根据势能进行模型排序即筛选。
距离势能v(dij)的设计如下:
其中pk为残基i与j之间的距离预测dij在第个k个bin的概率,dk为第k个bin所指定距离区间的中心点,n为算入距离势能的bin的总数。根据蛋白质结构的特性及距离预测准确度随长度衰减的实验结果,我们在距离势能中只保留其中的
φ角度势能v(φi)的设计如下:
其中pk为残基i的phi角度预测φi在第k个bin的概率,n为角度预测中bin的总数。ψ角度势能v(ψi)设计同上。因此,全长为l的蛋白质q的势能为距离与角度的势能总和:
上面的势能为离散函数,为方便计算,这个势能被转换为相应的三次仿样函数。可微分化的操作使得这个势能的最小化可以通过梯度下降(gd)的方式进行计算。为了加快计算速度,我们采取先粗粒建模再侧链优化的分布式建模。
首先,蛋白质结构被简化为主链加侧链重心点的粗粒模式,我们根据预测φ、ψ角度生成粗粒的初始模型,rosetta内置的l-bfgs算法被用来最小化上面设计的势能及范德华力(vdw)、氢键相互作用(hb)、ramachandran效应(rama)等蛋白质势能。5,000个gd迭代后最小势能相对应的结构将进入下一轮的侧链优化。这一轮的优化结合了距离势能与rosetta最新的talaris势能,根据训练集上的优化其比例被设定为1:5。200个优化迭代后的最小势能模型被选取为此轮计算的结果。
由于gd的确定性限制无法跨过局部极小值,我们对每个蛋白质重复约2,000次gd及优化过程,并在其中加入如下的随机策略:
1.在粗粒模型初始化时对φ、ψ角度加入5°-10°的随机采样范围;
2.在计算势能时只采纳预测概率高于ε的bin,其中ε∈[0,0.5];
3.在添加势能限制时依此添加a)short-,medium-,long-限制;b)short-+medium-,long-限制;c)short-+medium-+long-限制。
步骤105,2,000个结构模型的排序为根据其总势能进行,最小势能的前5个模型被认定为被模拟蛋白质的最可靠预测。
上述模型生成步骤的优化根据7中的模型打分进行。
7.上述步骤104的优化根据生成模型的准确度进行,而预测模型的准确度通过其相对于真实实验结构的模型打分(tm-score)与均方根差(rmsd)进行评估。
rmsd直接测量了预测模型与实验结构排列中的平均分子间距离,对于预测结构a与实验结构b,其定义为:
其中ra为结构a的回转半径,
tm-score是常用的衡量蛋白质模型与其真实结构整体拓扑相似度的金标准,其定义如下:
其中l为蛋白质实验结构的长度,n为预测模型与实验结构排列中对应的残基数量,di为排列中第i个模型残基与实验结构残基间的距离,而max为最大值,即最优化排列下的分值。由此公式可知tm-score∈(0,1],且分值越高代表预测模型越准确。pdb的数据分析表明,tm-score大于0.5的两个结构可大致被视作同一scop/cathfold,即当tm-score大于0.5时,模型通常被视为成功的预测。
在本发明的训练中,上述rmsd和tm-score被用来衡量6中生成的模型结构与所模拟的蛋白质真实结构的相似度,并以此为标准优化6中的结构生成方式及相关参数。
8.步骤106,在本发明的一个实施例中,7中提到的真实实验结构不存在,因此为了给用户提供预测结构的全模型及残基的可信度,评估将由proq3完成:根据所预测蛋白质的序列独热编码及位置特异性打分矩阵,通过计算预测结构模型的rosetta势能、二级结构、相对可触表面积来给予模型的准确度打分。由于proq3为本领域技术人员所熟知的常用模型评估方法,其具体技术内容这里将不做赘述。预测结构的打分范围为[0,1],分值越高代表模型可信度越高。
本领域技术人员知道,除了以纯计算机可读程序代码方式实现本发明提供的系统及其各个装置、模块、单元以外,完全可以通过将方法步骤进行逻辑编程来使得本发明提供的系统及其各个装置、模块、单元以逻辑门、开关、专用集成电路、可编程逻辑控制器以及嵌入式微控制器等的形式来实现相同功能。所以,本发明提供的系统及其各项装置、模块、单元可以被认为是一种硬件部件,而对其内包括的用于实现各种功能的装置、模块、单元也可以视为硬件部件内的结构;也可以将用于实现各种功能的装置、模块、单元视为既可以是实现方法的软件模块又可以是硬件部件内的结构。
在本申请的描述中,需要理解的是,术语“上”、“下”、“前”、“后”、“左”、“右”、“竖直”、“水平”、“顶”、“底”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本申请和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本申请的限制。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变化或修改,这并不影响本发明的实质内容。在不冲突的情况下,本申请的实施例和实施例中的特征可以任意相互组合。
- 还没有人留言评论。精彩留言会获得点赞!