一种基于深度森林和PU学习的药物-靶标关系预测方法
一种基于深度森林和pu学习的药物
‑
靶标关系预测方法
技术领域
1.本发明涉及系统生物学领域,更具体地,涉及一种基于深度森林和pu学习 的药物
‑
靶标关系预测方法。
背景技术:
2.药物发现是一个复杂、昂贵、成功率低的过程。在过去的几十年里,尽管制 药公司在药物研发上增加了巨大的财政投资,但fda批准的新药仍然停滞不前。 药物重定位旨在发现现有药物的原始医学用途以外的新治疗线索的过程,能够加 快药物研发进程,从而降低药物研发的成本。药物重定位的关键步骤之一是寻找 可能的药物靶点相互作用。药物
‑
靶标通常与特定疾病有关,并可以通过调节靶 标的生理活性以有效地改善疾病症状,确定特定疾病有关的靶标分子是现代药物 发现的基础。尽管实验技术有了很大的进步,人们在分子、细胞等生物系统有了 很多的理解,但是生物湿实验方法用来确定这种关系仍然是昂贵和费时的。因此, 随着计算硬件和技术的发展,计算方法已经大量的被用来预测潜在的药物
‑
靶标 关联关系。
3.目前,用于预测药物
‑
靶标关联关系的计算方法主要分为3类:
4.(1)传统的药物设计方法,基于结构的药物设计是利用药物
‑
靶标的结构信 息开发其抑制剂的方法,因此受体结构是该方法的先决条件,需要使用实验技术 确定。分子对接能够预测小分子配体与适当的靶标结合位点的结合构象,以及产 生的信号的强度和类型,在设计药物和阐明基本的生物系统过程中起着重要作用。 但是当靶标蛋白的3d结构信息未知时,分子对接受到限制。基于配体的药物设 计是在缺少受体3d结构信息的情况下使用的一种方法,它依赖于与目标生物学 靶标结合的分子的知识。当识别具有不同于参考化合物时的新化学支架时,基于 配体的方法将不起作用。
5.(2)基于网络的预测方法,基于网络的方法是基于相似的药物共享相似的 靶标,反之亦然的假设。它能够集成药物和靶标的相似性网络信息、药物
‑
靶标 的网络拓扑结构信息、药物和副作用关联网络信息、药物和药物关联网络信息以 及靶标的功能相似性网络信息等各种相似性网络信息。由于网络可以使用矩阵表 示,可以通过简单的矩阵运算来描述,因此基于网络的方法可以在计算机上快速 的运行,可以成为预测药物
‑
靶标的关联关系的强大的工具。比如nrwrh方法 假设相似药物经常与相似的靶蛋白相互作用,并且将药物
‑
药物相似性网络,靶 标
‑
靶标相似性网络和已知的药物
‑
靶标相互作用网络整合为异质网络。开发了一 种有效的基于网络的随机游走模型,通过在异构网络上实现随机游走来预测潜在 的药物
‑
靶标相互作用。nrwrh充分利用了网络工具进行数据集成和药物
‑
靶标 相互作用的预测,随机游走是在异构网络上实现的,该网络由三个不同的网络组 成,即药物
‑
靶标相互作用网络,药物化学结构相似性网络和靶蛋白序列相似性 网络。即使所研究的药物没有已知的靶标,仍可以基于与该给定药物相似的已知 药物
‑
靶标预测该给定药物的潜在靶标。
6.还有daspfind方法使用异构网络上特定长度的简单路径进行药物
‑
靶标相 互作
用预测;sdtnbi方法使用化学子结构来弥合已知药物
‑
靶标相互作用网络 和新化学实体之间的鸿沟。为已知药物
‑
靶标相互作用网络和新化学实体中的药 物生成化学子结构后,可以通过将药物和新化学实体的子结构连接起来,将已知 药物
‑
靶标相互作用网络和新化学实体集成到结构
‑
药物
‑
目标网络中。bleakleyand yamanishi等人将已知的dti描述为二分图,并使用二分局部模型(blm) 预测新的dti。以上两种方法有效地预测了新的dtis,但是,它们无法发现新 药或靶标的潜在关联信息。虽然现有的基于网络的方法实现了很好的性能,但是 需要把更多的药物和靶标的其他信息集成到基于网络的方法中提升方法的性能。
7.(3)基于机器学习的预测方法,基于机器学习的方法将各种生物数据和机 器学习模型有效地集成到一个统一的框架中,并获得了更好的预测性能。但是, 由于没有经过实验验证的负样本,因此大多数人随机选择一部分未知关系的药物
ꢀ‑
靶标对作为负样本,但是这些选定的负样本可能包含正样本,从而严重影响预 测准确性。更重要的是,这是一个关键问题,如何为基于机器学习的方法从各种 生物学数据中提取有用的特征。因此,3dgnn
‑
dti使用低级表示作为输入,并 探索了基于端到端神经网络的模型;lasso
‑
dnn提出了一种基于图的神经网络 算法来提取具有3d结构信息的图特征;zhao等人使用了图神经网络和深度神经 网络技术来预测dti。
8.以上方法从各个特征角度、集成各种网络信息来预测潜在的药物
‑
靶标关联 关系。虽然以上方法实现了很好的性能,但是有一些缺陷。首先,计算药物和靶 标的相似性上,有的方法忽略药物和靶标的生物属性,而采用药物
‑
靶标关联网 络的拓扑特征来计算药物和靶标的相似性。其次,有的采用算术均值方法融合不 同药物和靶标的数据,因此,可能会引入噪声。有的方法无法对一个新的药物和 一个新的靶标之间的关系进行预测。
技术实现要素:
9.本发明针对上述背景技术中现有技术的不足。提供一种基于深度森林和pu 学习的药物
‑
靶标关系预测方法,该方法预测准确率高,能够减少生物实验所需 要的时间和资源。
10.为实现上述目的,本发明采用以下技术方案:
11.一种基于深度森林和pu学习的药物
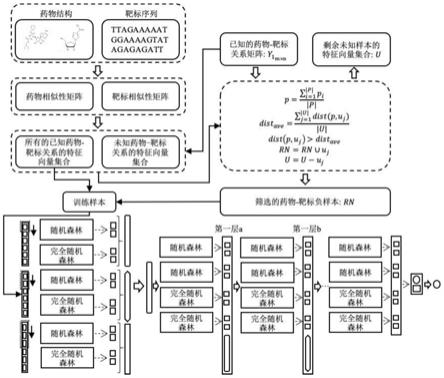
‑
靶标关系预测方法,包括以下步骤:
12.s1、获取药物的结构信息、靶标的序列信息和已知的药物
‑
靶标关系;
13.s2、采用pu学习来筛选潜在的药物
‑
靶标负相关关系;
14.根据s1中所述构建药物
‑
靶标正相关关系矩阵y1,计算药物相似性矩阵sim
d
, 计算靶标相似性矩阵sim
t
;在药物相似性矩阵sim
d
和靶标相似性矩阵sim
t
构成 的特征向量上进行计算,得到药物
‑
靶标得分矩阵,对分数进行排名;
15.s3、采用深度森林方法来预测潜在的药物
‑
靶标关系;
16.1)对于药物和靶标都是已知的情况,基于药物相似性矩阵sim
d
和靶标相似 性矩阵sim
t
,采用深度森林方法,得到药物
‑
靶标得分矩阵;
17.2)对于新的药物的情况,根据新的药物的结构信息,计算新的药物与已知 药物的结构相似性,基于靶标相似性矩阵sim
t
,采用深度森林方法,得到新的药 物
‑
靶标之间存在关系的概率值。
18.3)对于新的靶标的情况,根据新的靶标的氨基酸序列信息,计算新的靶标 与已知靶标的序列相似性,采用深度森林方法,得到新的靶标
‑
药物之间存在关 系的概率值。
19.进一步的,在步骤s1中构建药物
‑
靶标正相关关系矩阵y1,药物
‑
靶标矩阵 每一行对应一个药物,每一列对应一个靶标;若已知药物d
i
和靶标t
j
存在关系, 则y1(i,j)等于1;否则,y1(i,j)等于0;其中i=1,2,k,m;j=1,2,k,n;m和n分 别为已知的药物和靶标的个数。
20.进一步的,在步骤s1中的构建药物相似性矩阵的方法为:
21.1)基于药物的结构信息,使用一种基于图的方法simcomp,其中药物的 结构信息被视为由原子作为顶点以及共价键作为边的2d结构图;
22.2)根据药物的结构信息,计算药物drug
i
和药物drug
j
的结构相似性sim
d
, 计算公式如下:
[0023][0024]
其中,d
i
和d
j
分别表示药物drug
i
和药物drug
j
的结构信息,
[0025]
最后,为第i行第j列的元素值;由所有的构成的药物相似性矩阵 sim
d
。
[0026]
进一步的,在步骤s1中构建靶标相似性矩阵的方法为:
[0027]
首先,基于靶标的氨基酸序列信息,使用一种基于局部序列比对的算法史密 斯
‑
沃特曼算法;根据靶标的氨基酸序列信息,计算靶标protein
i
与靶标protein
j
的 序列相似性sim
t
,计算公式如下所示:
[0028][0029]
其中,p
i
和p
j
分别表示靶标protein
i
与靶标protein
j
的序列信息;
[0030]
最后,为第i行第j列的元素值;由所有的构成的靶标相似性矩阵 sim
t
。
[0031]
进一步的,在步骤s2中采用pu学习来筛选潜在的药物
‑
靶标负相关关系的 方法具体步骤为为:
[0032]
a)构建药物
‑
靶标正相关关系矩阵y1,药物
‑
靶标矩阵每一行对应一个药物, 每一列对应一个靶标;若已知药物d
i
和靶标t
j
存在关系,则y1(i,j)等于1;否则,y1(i,j)等于0;其中i=1,2,k,m;j=1,2,k,n;m和n分别为已知的药物和靶标的 个数;
[0033]
b)定义{p
k
,k=1,2,k,|p|}为所有的已知药物
‑
靶标正相关关系的特征向量集 合,{u
z
,z=1,2,k,|u|}为所有的未知药物
‑
靶标关系的特征向量集合,|p|和|u|分 别为已知和未知药物
‑
靶标关系的数量;其中若已知药物d
i
和靶标t
j
存在关系, 则药物相似性矩阵与靶标相似性矩阵连结之后的特征向量为特征向量 p
k
,维度为w,即n+m;若已知药物d
i
和靶标t
j
未知关系,则药物相似性矩阵 与靶标相似性矩阵连结之后的特征向量为特征向量u
z
,维度为w,即 n+m;其中药物相似性矩阵表示药物结构相似性矩阵sim
d
的第i行,靶标 相似性矩阵表示靶标相似性矩阵sim
t
的第j列;
[0034]
c)根据所有的已知药物
‑
靶标关系的特征向量集合{p
k
,k=1,2,k,|p|},计算 正
样本标准化向量p,计算公式如下:
[0035]
p=p+p
k
(k=1,2,...,|p|)
[0036]
对正样本向量p进行标准化,公式为:
[0037]
p=p/|p|
[0038]
其中|p|为已知的药物
‑
靶标正相关关系的数量;
[0039]
d)根据正样本标准化向量p,计算所有的未知药物
‑
靶标关系的特征向量与 正样本标准化向量p的平均距离并标准化,计算公式如下:
[0040][0041]
对平均距离进行标准化,公式为:
[0042]
dist
ave
=dist/|u|
[0043]
其中|u|为未知的药物
‑
靶标关系的数量;w为药物相似性矩阵与靶标 相似性矩阵连结之后的特征向量的维度;
[0044]
e)根据标准化平均距离dist
ave
,计算所有的未知药物
‑
靶标关系的特征向量 {u
z
,z=1,2,k,|u|}与正样本标准化向量p的欧式距离,计算公式如下:
[0045][0046]
其中|u|为未知的药物
‑
靶标关系的数量;w为药物相似性矩阵与靶标 相似性矩阵连结之后的特征向量的维度;
[0047]
若某个未知药物
‑
靶标关系的特征向量u
z
与标准化平均距离p的欧式距离 dist
z
大于标准化平均距离dist
ave
,则将这个未知药物
‑
靶标关系的特征向量u
z
合并 到可靠的负样本集rn中;对可靠的负样本集rn从大到小进行排序,从可靠的 负样本集rn中选择负样本与正样本的比例为n:1的数量作为潜在的药物
‑
靶标 负相关关系,构成筛选的潜在药物
‑
靶标负相关关系的特征向量集合 {rn
k
,k=1,2,k,n*|p|};
[0048]
f)基于构建的药物
‑
靶标正相关关系矩阵y1,根据筛选的潜在的药物
‑
靶标负 相关关系,构建药物
‑
靶标关系矩阵y,每一行对应一个药物,每一列对应一个 靶标;若药物d
i
和靶标t
j
为筛选的潜在的负相关关系,则y1(i,j)等于
‑
1;否则, y1(i,j)等于0;其中i=1,2,k,m;j=1,2,k,n;m和n分别为已知的药物和靶标的 个数。
[0049]
进一步的,在步骤s3中采用深度森林方法来预测潜在的药物
‑
靶标关系,具 体步骤为:
[0050]
a)结合所有的已知药物
‑
靶标正相关关系的特征向量集合{p
k
,k=1,2,k,|p|} 和筛选的潜在的药物
‑
靶标负相关关系的特征向量集合{rn
k
,k=1,2,k,n*|p|},构 建药物
‑
靶标关系的正负相关关系的特征向量集合{i
k
,k=1,2,...,(n+1)*|p|};
[0051]
b)使用三个不同大小的滑动窗口机制对药物
‑
靶标关系的正负相关关系的 特征向量集合{i
k
,k=1,2,...,(n+1)*|p|}进行处理,得到三个不同大小类型的子特 征向量,并将三个不同大小类型的子特征向量分别用于训练随机森林和完全随机 森林,得到三个不同大小类型的类概率向量;其中,滑动窗口的三个不同大小分 别是
步长为l;
[0052]
c)基于步骤b)产生的三个不同大小类型的类概率向量,使用级联森林结构 对类概率向量进行分类;其中,每一层级联层中都包括三层结构,且每个结构都 是由n3个随机森林和n4个完全随机森林构成;在级联森林结构中,为了降低过 拟合的风险,每个森林生成的类向量是通过k折交叉验证产生的;即每个样本都 会被当作训练数据训练k
‑
1次,生成k
‑
1个类c维向量,然后对其取平均值即为 这个森林最终特征向量,将这n3+n4个森林的c维特征向量连在一起,作为下一 层的增强特征向量;在扩展一个新的层后,整个级联森林的性能将在验证集上进 行评估,如果没有显着的性能提升,训练过程将终止;因此,级联森林中层的数 量是自动确定的;最后,产生n3+n4个c维的类向量d
i
(i=1,2,...,(n3+n4));
[0053]
d)基于步骤c)产生的n3+n4个c维的类向量d
i
(i=1,2,...,(n3+n4)),计算最终 的分类类别,计算公式如下:
[0054][0055]
进一步的,在步骤c)中,具体过程为:
[0056]
c1)在级联森林结构中,由前一层的输入数据和输出结果进行连结操作并作 为下一层的输入;首先,将类概率向量d1用于训练两个随机森林和两个完全随机 森林,产生c*(n3+n4)维的类向量;
[0057]
c2)在第一层a中,基于步骤c1)产生的c*(n3+n4)维的类向量,与类概率向 量d1连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0058]
c3)在第一层b中,基于步骤c2)产生的c*(n3+n4)维的类向量,与类概率向 量d2连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0059]
c4)在第一层c中,基于步骤c3)产生的c*(n3+n4)维的类向量,与类概率向 量d3连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0060]
c5)在以后的级联层结构中,以此类推。
[0061]
进一步的,步骤a)、b)、c)中,具体为:
[0062]
a1)基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和 筛选的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测未知的药物
‑
靶标关 系的概率值,产生药物
‑
靶标关系概率矩阵y;
[0063]
a2)对于任意两个已知的药物和靶标,根据y的值判断药物与靶标存在关联 关系的可能性。
[0064]
b1)计算新的药物与已知的药物的结构相似性,并分别与靶标相似性矩阵 连结,定义为新的药物
‑
已知的靶标特征向量集;
[0065]
b2)基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和 筛选的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测新的药物
‑
已知的靶 标关系的概率值,产生新的药物
‑
已知的靶标关系概率值向量;
[0066]
b3)对于新的药物和已知的靶标,根据新的药物
‑
已知的靶标关系概率值向量的 值判断新的药物与已知的靶标存在关联关系的可能性。
[0067]
c1)计算新的靶标与已知的靶标的序列相似性,并分别与药物相似性矩阵 连
结,定义为新的靶标
‑
已知的药物特征向量集;
[0068]
c2)基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和 筛选的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测新的靶标
‑
已知的药 物关系的概率值,产生新的靶标
‑
已知的药物关系概率值向量;
[0069]
c3)对于新的靶标和已知的药物,根据新的靶标
‑
已知的药物关系概率值向 量的值判断新的靶标与已知的药物存在关联关系的可能性。
[0070]
本发明的有益效果为:通过结合深度森林和pu学习算法,来预测潜在的药 物
‑
靶标关联关系,能够更准确的预测药物
‑
靶标关系,能够减少生物实验所需的 时间和资源,为药物发现和药物重定位提供了研究基础。
附图说明
[0071]
图1为本发明dfpu
‑
dtis流程图;
[0072]
图2为滑动窗口机制流程图。
具体实施方式
[0073]
下面结合具体实施方式对本发明作进一步的说明。
[0074]
一种基于深度森林和pu学习的药物
‑
靶标关系预测方法:具体流程如图一所 示。
[0075]
一.药物相似性和靶标相似性计算
[0076]
1)药物
‑
靶标矩阵的构建;基于已知的药物
‑
靶标正相关关系,本发明首先 构建了药物
‑
靶标矩阵每一行对应一个药物,每一列对应一个靶标;若已知药物d
i
和靶标t
j
存在关系,则y1(i,j)等于1;否则,y1(i,j)等于0;其中i=1,2,k,m; j=1,2,k,n;m和n分别为已知的药物和靶标的个数。
[0077]
2)药物相似性计算
[0078]
基于药物的结构信息,使用一种基于图的方法simcomp,其中药物的结构 信息被视为由原子作为顶点以及共价键作为边的2d结构图;该方法通过比较两 个药物的化学结构的共同子结构大小来比较两个药物的结构相似性,即两个药物 的化学结构的共同子结构越小,两个药物的相似性越小;
[0079]
然后,根据根据药物的结构信息,计算药物drug
i
和药物drug
j
的结构相似性 sim
d
,计算公式如下:
[0080][0081]
其中,d
i
和d
j
分别表示药物drug
i
和药物drug
j
的结构信息,
[0082]
最后,为第i行第j列的元素值;由所有的构成的药物相似性矩阵 sim
d
。
[0083]
3)靶标相似性计算
[0084]
基于靶标的氨基酸序列信息,使用一种基于局部序列比对的算法史密斯
‑
沃 特曼算法;根据靶标的氨基酸序列信息,计算靶标protein
i
与靶标protein
j
的序 列相似性sim
t
,计算公式如下所示:
[0085][0086]
其中,p
i
和p
j
分别表示靶标protein
i
与靶标protein
j
的序列信息;
[0087]
最后,为第i行第j列的元素值;由所有的构成的靶标相似性矩阵sim
t
。
[0088]
二pu学习(positive
‑
unlabeled learning,pu)筛选潜在的药物
‑
靶标负相关关 系
[0089]
本发明采用pu学习来筛选潜在的药物
‑
靶标负相关关系。本发明使用pu学 习的理由如下:由于没有经过实验验证的负样本,因此大多数人随机选择一部分 未知关系的药物
‑
靶标对作为负样本,但是这些选定的负样本可能包含正样本, 从而严重影响预测准确性。因此,本发明采用pu学习筛选潜在的药物
‑
靶标负 相关关系以提高预测的准确度。
[0090]
a)构建药物
‑
靶标正相关关系矩阵y1,药物
‑
靶标矩阵每一行对应一个药物, 每一列对应一个靶标;若已知药物d
i
和靶标t
j
存在关系,则y1(i,j)等于1;否则, y1(i,j)等于0;其中i=1,2,k,m;j=1,2,k,n;m和n分别为已知的药物和靶标的 个数;
[0091]
b)定义{p
k
,k=1,2,k,|p|}为所有的已知药物
‑
靶标正相关关系的特征向量集 合,{u
z
,z=1,2,k,|u|}为所有的未知药物
‑
靶标关系的特征向量集合,|p|和|u|分 别为已知和未知药物
‑
靶标关系的数量;其中若已知药物d
i
和靶标t
j
存在关系, 则药物相似性矩阵与靶标相似性矩阵连结之后的特征向量为特征向量 p
k
,维度为w,即n+m;若已知药物d
i
和靶标t
j
未知关系,则药物相似性矩阵 与靶标相似性矩阵连结之后的特征向量为特征向量u
z
,维度为w,即 n+m;其中药物相似性矩阵表示药物结构相似性矩阵sim
d
的第i行,靶标 相似性矩阵表示靶标相似性矩阵sim
t
的第j列;
[0092]
c)根据所有的已知药物
‑
靶标关系的特征向量集合{p
k
,k=1,2,k,|p|},计算 正样本标准化向量p,计算公式如下:
[0093]
p=p+p
k
(k=1,2,...,|p|)
[0094]
对正样本向量p进行标准化,公式为:
[0095]
p=p/|p|
[0096]
其中|p|为已知的药物
‑
靶标正相关关系的数量;
[0097]
d)根据正样本标准化向量p,计算所有的未知药物
‑
靶标关系的特征向量与 正样本标准化向量p的平均距离并标准化,计算公式如下:
[0098][0099]
对平均距离进行标准化,公式为:
[0100]
dist
ave
=dist/|u|
[0101]
其中|u|为未知的药物
‑
靶标关系的数量;w为药物相似性矩阵与靶标 相似性矩阵连结之后的特征向量的维度;
[0102]
e)根据标准化平均距离dist
ave
,计算所有的未知药物
‑
靶标关系的特征向量 {u
z
,z=1,2,k,|u|}与正样本标准化向量p的欧式距离,计算公式如下:
[0103][0104]
其中|u|为未知的药物
‑
靶标关系的数量;w为药物相似性矩阵与靶标 相似性矩阵连结之后的特征向量的维度;
[0105]
若某个未知药物
‑
靶标关系的特征向量u
z
与标准化平均距离p的欧式距离 dist
z
大于标准化平均距离dist
ave
,则将这个未知药物
‑
靶标关系的特征向量u
z
合并 到可靠的负样本集rn中;对可靠的负样本集rn从大到小进行排序,从可靠的 负样本集rn中选择负样本与正样本的比例为n:1的数量作为潜在的药物
‑
靶标 负相关关系,构成筛选的潜在药物
‑
靶标负相关关系的特征向量集合{rn
k
,k=1,2,k,n*|p|};
[0106]
f)基于构建的药物
‑
靶标正相关关系矩阵y1,根据筛选的潜在的药物
‑
靶标负 相关关系,构建药物
‑
靶标关系矩阵y,每一行对应一个药物,每一列对应一个 靶标;若药物d
i
和靶标t
j
为筛选的潜在的负相关关系,则y1(i,j)等于
‑
1;否则, y1(i,j)等于0;其中i=1,2,k,m;j=1,2,k,n;m和n分别为已知的药物和靶标的 个数。
[0107]
至此,本发明得到了所有的已知药物
‑
靶标正相关关系的特征向量集合 {p
k
,k=1,2,k,|p|},筛选的潜在药物
‑
靶标负相关关系的特征向量集合 {rn
k
,k=1,2,k,n*|p|};为了避免正负样本数量的差异带来额外的噪声,本发明 实施中,n的取值被确定为1。即筛选的负样本数量和正样本的数量是相等的。
[0108]
三.采用深度森林方法来预测潜在的药物
‑
靶标关系
[0109]
a)结合所有的已知药物
‑
靶标正相关关系的特征向量集合{p
k
,k=1,2,k,|p|} 和筛选的潜在的药物
‑
靶标负相关关系的特征向量集合{rn
k
,k=1,2,k,n*|p|};构 建药物
‑
靶标关系的正负相关关系的特征向量集合{i
k
,k=1,2,...,(n+1)*|p|}
[0110]
b)如图2所示,使用三个不同大小的滑动窗口机制对药物
‑
靶标关系的正负 相关关系的特征向量集合{i
k
,k=1,2,...,(n+1)*|p|};进行处理,得到三个不同大小 类型的子特征向量,并将三个不同大小类型的子特征向量分别用于训练随机森林 和完全随机森林,得到三个不同大小类型的类概率向量;其中,滑动窗口的三个 不同大小分别是步长为l;
[0111]
c)基于步骤b)产生的三个不同大小类型的类概率向量,使用级联森林结构 对类概率向量进行分类;其中,每一层级联层中都包括三层结构,且每个结构都 是由n3个随机森林和n4个完全随机森林构成;在级联森林结构中,为了降低过 拟合的风险,每个森林生成的类向量是通过k折交叉验证产生的;即每个样本都 会被当作训练数据训练k
‑
1次,生成k
‑
1个c维的类向量,然后对其取平均值即 为这个森林最终特征向量,将这n3+n4个森林的c维特征向量连在一起,作为下 一层的增强特征向量;在扩展一个新的层后,整个级联森林的性能将在验证集上 进行评估,如果没有显着的性能提升,训练过程将终止;因此,级联森林中层的 数量是自动确定的;最后,产生n3+n4c维的类向量d
i
(i=1,2,...,(n3+n4));
[0112]
d)基于步骤c)产生的n3+n4个c维的类向量d
i
(i=1,2,...,(n3+n4)),计算最终 的分类类别,计算公式如下:
[0113][0114]
四.滑动窗口机制结构具体为:
[0115]
b1)使用步长为l、大小为大小为s
i
(i=1,2,3)的滑动窗口去扫描药物
‑
靶标关 系的正负相关关系的特征向量集合{i
k
,k=1,2,...,(n+1)*|p|},特征向量的维度为 w,每个特征向量将会得到个子特征向量,每个子特征向量 的维度是s
i
(i=1,2,3);
[0116]
其中,窗口大小s1、s2、s3分别为
[0117]
b2)基于步骤b1)产生的个子特征向量,它们被用来训练 随机森林和完全随机森林,得到类概率向量,计算公式如下:
[0118][0119]
其中,c是类别标签的数量;n1和n2分别是随机森林和完全随机森林的数量。在 本发明中,滑动窗口的步长被设置为1,随机森林n1的值被设置为1,完全随机 森林n2的值被设置为1;
[0120]
以一个300维的特征向量被一个滑动窗口大小为150和步长为1扫描为例进 行详细说明,在扫描之后,将会得到151个150维的子特征向量;将151个150 维的子特征向量用于训练随机森林和完全随机森林,在本发明中是二分类问题, 因此每个森林会得到151个2维的向量,对其进行连结操作得到604维的类概率 向量;
[0121]
五.级联森林结构具体为:
[0122]
c1)在级联森林结构中,由前一层的输入数据和输出结果进行连结操作并作 为下一层的输入;首先,将类概率向量d1用于训练两个随机森林和两个完全随机 森林,产生c*(n3+n4)维的类向量;
[0123]
c2)在第一层a中,基于步骤c1)产生的c*(n3+n4)维的类向量,与类概率向 量d1连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0124]
c3)在第一层b中,基于步骤c2)产生的c*(n3+n4)维的类向量,与类概率向 量d2连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0125]
c4)在第一层c中,基于步骤c3)产生的c*(n3+n4)维的类向量,与类概率向 量d3连结,用于训练两个随机森林和两个完全随机森林,产生c*(n3+n4)维的类 向量;
[0126]
c5)在以后的级联层结构中,以此类推。
[0127]
六.基于深度森林和pu学习预测药物
‑
靶标关系,具体为:
[0128]
1)预测已知的药物与已知的靶标的关联关系
[0129]
基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和筛选 的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测未知的药物
‑
靶标关系的 概率值,产生药物
‑
靶标关系概率矩阵y;
[0130]
对于任意两个已知的药物和靶标,根据y的值判断药物与靶标存在关联关系 的可
能性。
[0131]
2)预测新的药物与已知的靶标的关联关系
[0132]
计算新的药物与已知的药物的结构相似性,并分别与靶标相似性矩阵连结,定义为新的药物
‑
已知的靶标特征向量集;
[0133]
基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和筛选 的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测新的药物
‑
已知的靶标关 系的概率值,产生新的药物
‑
已知的靶标关系概率值向量;
[0134]
对于新的药物和已知的靶标,根据新的药物
‑
已知的靶标关系概率值向量的 值判断新的药物与已知的靶标存在关联关系的可能性。
[0135]
预测已知的药物与新的靶标的关联关系
[0136]
计算新的靶标与已知的靶标的序列相似性,并分别与药物相似性矩阵连结,定义为新的靶标
‑
已知的药物特征向量集;
[0137]
基于构建的药物
‑
靶标关系矩阵y,采用已知的药物
‑
靶标正相关关系和筛选 的潜在的药物
‑
靶标负相关关系训练深度森林模型,预测新的靶标
‑
已知的药物关 系的概率值,产生新的靶标
‑
已知的药物关系概率值向量;
[0138]
对于新的靶标和已知的药物,根据新的靶标
‑
已知的药物关系概率值向量的 值判断新的靶标与已知的药物存在关联关系的可能性。
[0139]
七.实验验证
[0140]
本发明使用了四个药物
‑
靶标数据集,分别是酶、离子通道、gpcrs和核受 体,并且在这四个数据集上采用十折交叉验证进行验证;详细情况如下表所示。
[0141]
表1药物
‑
靶标相互作用数据集
[0142][0143]
为了评估基于深度森林和pu学习模型预测的性能,使用两个评价指标来评 判模型方法的鲁棒性,分别是auc(the areas under roc curves)和aupr(the areasunder precision
‑
recall curves),并且使用三种不同类型的交叉验证验证模型,分别 为cv1、cv2和cv3。cv1是为了测试对于新的药物的预测能力,随机的将药 物
‑
靶标关系矩阵y的90%行作为训练集,其余作为测试集;cv2是为了测试对 于新的靶标的预测能力,随机的将药物
‑
靶标关系矩阵y的90%列作为训练集, 其余作为测试集;cv3是为了测试对于药物
‑
靶标关联对的预测能力,随机的将 药物
‑
靶标关系矩阵y的90%药物
‑
靶标关系作为训练集,其余作为测试集。
[0144]
本发明将dfpu
‑
dtis和其他药物
‑
靶标预测方法进行比较(blm
‑
nii,cmf, nrlmf,duallaprls),各项指标均高于现有的方法,各个方法的详细的性能指 标如下表所示。通过与其他方法在三种不同类型的交叉验证情况下的比较,表明 了本发明比现有的方法更准
确,性能更好,为药物发现和药物重定位提供了研究 基础。
[0145]
三种不同类型的五折交叉验证的算法性能指 标
[0146][0147][0148]
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保 护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换, 均在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1