一种基于二代测序的转录组数据自动化分析方法与流程
本发明涉及一种基于二代测序的转录组数据自动化分析方法。
背景技术:
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有rna的总和,主要包括mrna和非编码rna。转录组研究是基因功能及结构研究的基础和出发点,通过新一代高通量测序,能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息,已广泛应用于基础研究、临床诊断和药物研发等领域。
随着新一代测序平台的市场化,rna测序(rnasequenclng,rna-seq)技术已成为了转录组学研究的重要手段之一。该技术利用新一代高通量测序平台对基因组cdna测序,通过统计相关reads(用于测序的cdna小片段)数计算出不同mrna的表达量,分析转录本的结构和表达水平,同时发现未知转录本和稀有转录本,精确地识别可变剪切位点以及编码序列单核苷酸多态性,提供最全面的转录组信息。
目前转录组测序数据日益增多,涉及到的分析软件也是琳琅满目,但是大多分析都是只能分析其中的一个小部分内容,需要手动一步一步的进行串联,步骤非常繁琐且输入输出文件格式转换非常麻烦,因此很容易给分析过程及结果带来不必要的错误。同时,转录组测序数据量较大,而且一般会进行大批量样本比较分析,传统算法流程计算十分耗时耗资源,需要在高端服务器上分析较长时间才能得到相关分析结果及报告。
为了满足人们日益增长的分析需求,公开号为cn109637588a的中国专利公开了一种基于全转录组高通量测序构建基因调控网络的方法,该方法主要针对得到的差异表达基因结果进行后续分析,如mrna、lncrna和mirna的共表达关系,构建相关竞争性内源rna调控关系,以及关键基因的通路富集分析等,但未在对原始数据的高效处理上进行重点关注。
目前国内尚缺少一个使用场景全面、合理高效且自动化程度高的转录组测序数据分析方法,因此,设计了一种基于二代测序的转录组数据自动化分析方法,适用于多样本的转录组高通量测序数据的分析,提升了数据分析处理的自动化程度,功能多样、适用性强,具有现实意义和良好的应用前景。
技术实现要素:
针对上述背景技术中的不足,本发明的目的在于提供一种可以避免上述技术缺陷的一种基于二代测序的转录组数据自动化分析方法,适用于多样本的转录组高通量测序数据的分析、自动化程度高、功能多样且适用性强。
为实现上述目的,本发明提供如下技术方案:
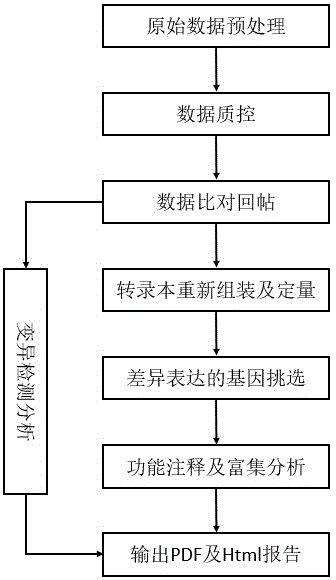
一种基于二代测序的转录组数据自动化分析方法,该自动化分析方法包括以下步骤:
1)对原始测序数据进行数据预处理;
2)对预处理后测序数据进行质控分析;
3)对通过质控的测序数据进行map比对分析;
4)基于比对结果进行变异检测分析;
5)基于比对后的结果进行重新组装及定量分析;
6)基于重新组装及定量分析结果进行差异基因的挑选;
7)基于挑选得到的差异基因进行功能注释及富集分析;
8)根据以上步骤得到的结果进行整理统计及画图等操作,从而输出pdf报告及html报告供客户查看。
进一步地,所述步骤1)具体为:对得到的测序原始数据进行相应的过滤预处理,包括过滤低质量和接头相关序列,同时在此基础上过滤含有rrna的序列,从而得到cleandata。
进一步地,所述步骤2)具体为:对通过过滤预处理后的cleandata进行相应的质量控制,包括质量值的质控,不同碱基含量的质控,重复序列的质控等。
进一步地,所述步骤3)具体为:对以上通过质控的测序数据进行map比对处理,根据是否有参考基因组(genome)序列分别进行基因组(genome)序列以及基因(gene)序列的map比对。
进一步地,所述步骤4)具体为:对以上步骤3)中基于基因组(genome)序列的map比对结果进行变异检测分析。
进一步地,所述步骤5)具体为:对以上步骤3)中基于基因组(genome)或者基因(gene)序列的map比对结果进行转录本的重新组装,并根据重新组装结果进行定量分析,得到每个转录本的定量结果,并根据后续需要对转录本进行合并,从而进一步得到相关基因的定量结果。
进一步地,所述步骤6)具体为:在得到重新组装及定量结果后,使用相关算法对每个基因或者转录本进行差异表达分析,并根据筛选条件得到用于后续分析的差异基因列表。
进一步地,所述步骤7)具体为:在得到相关差异基因列表后,使用相关算法及数据库对这些差异基因进行相应的功能注释,并对这些基因进行富集分析(go/kegg等),从而得到不同样本组织间差异基因的对应功能、通路等。
进一步地,所述步骤8)具体为:在得到以上所有相关结果的同时,会自动进行相关结果的整理和统计,并根据统计结果进行相关图表的绘制,并进一步整理成最终的pdf以及html报告供客户查看。
本发明提供的一种基于二代测序的转录组数据自动化分析方法与现有技术相比,具备以下有益效果:
1.使用更为高效的算法软件进行分析,不仅分析速度快,而且结果准确率也更高;
2.本发明提供的方法由计算机程序自动控制,简化了数据处理分析质控及报告生成环节,并降低了人工成本,提升了数据分析处理效率。
附图说明
图1为一种基于二代测序的转录组数据自动化分析方法的流程图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
如图1所示,一种基于二代测序的转录组数据自动化分析方法,该自动化分析方法,包括以下步骤:
1)对原始测序数据进行数据预处理;
2)对预处理后测序数据进行质控处理;
3)对通过质控的测序数据进行map比对分析;
4)基于比对结果进行变异检测分析;
5)基于比对后的结果进行重新组装及定量分析;
6)基于重新组装及定量分析结果进行差异基因的挑选;
7)基于挑选得到的差异基因进行功能注释及富集分析;
8)根据以上步骤得到的结果进行整理统计及画图等操作,从而输出pdf报告及html报告供客户查看。
所述步骤1)具体为:对得到的测序原始数据进行相应的过滤预处理,包括使用trim_galore软件过滤低质量和接头相关序列,同时在此基础上使用sortmerna过滤含有rrna的序列,从而得到cleandata。
所述步骤2)具体为:对通过过滤预处理后的cleandata进行相应的质量控制,包括使用fastqc以及multiqc对cleandata进行质量值的质控,不同碱基含量的质控,重复序列的质控等。
所述步骤3)具体为:对以上通过质控的测序数据进行map比对处理,根据是否有参考基因组(genome)序列分别进行基因组(genome)序列以及基因(gene)序列的map比对,使用更为高效的比对软件hisat2,比传统比对软件速度快10-100倍,同时得到的比对结果更为准确。
所述步骤4)具体为:对以上步骤3)中基于基因组(genome)序列的map比对结果使用最新版的gatk4进行变异检测分析。
所述步骤5)具体为:对以上步骤3)中基于基因组(genome)或者基因(gene)序列的map比对结果使用stringtie软件进行转录本的重新组装,并根据重新组装结果进行定量分析,得到每个转录本的定量结果,并根据后续需要对转录本进行合并,从而进一步得到相关基因的定量结果。
所述步骤6)具体为:在得到重新组装及定量结果后,使用相关算法(如deseq2、edger等)对每个基因或者转录本进行差异表达分析,并根据筛选条件(如logfoldchange>=1.5&&p-value<0.05等)得到用于后续分析的差异基因列表。
所述步骤7)具体为:在得到相关差异基因列表后,使用相关算法及数据库对这些差异基因进行相应的功能注释,并对这些基因进行富集分析(go/kegg等),从而得到不同样本组织间差异基因的对应功能、通路等。
所述步骤8)具体为:在得到以上所有相关结果的同时,会自动进行相关结果的整理和统计,并根据统计结果进行相关图表的绘制,并进一步整理成最终的pdf以及html报告供客户查看。
本发明提供一种基于二代测序的转录组数据自动化分析方法,使用更为高效准确的软件对转录组测序数据进行分析,从原始数据的预处理及质控,数据的map比对,差异基因的获取,以及后续差异基因相关分析,分析算法及参数可根据实际应用需求进行选择和调整,能更好地满足实际应用场景;同时本发明提供的方法由程序自动控制,简化了数据分析处理环节并降低了人工成本,提升了数据分析处理的效率。
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
以上所述仅为本发明的优选实例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!