喹啉甲酰胺衍生物和其制造中间物的制造方法与流程
1.本发明涉及一种喹啉甲酰胺衍生物和其制造中间物的制造方法。
背景技术:
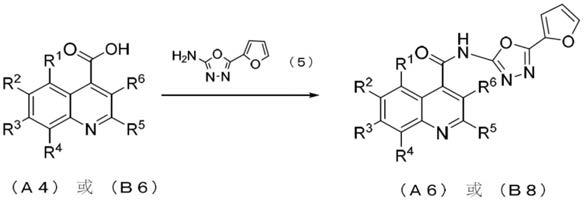
2.作为转录调节因子的stat(signal transducers and activators of transcription)是dna结合性蛋白质,作为将信号从细胞表面传递至核的路径中必需的介导因子密切参与细胞增殖或分化等。stat已知有7个不同的家族,其中,stat3在许多癌细胞中确认到其稳定的活化和过度表达,参与癌细胞的增殖或浸润。因此,可期待stat3的抑制剂作为抗癌剂,本技术人发现了包含下述式(a6)或式(b8)表示的化合物(式中的r1、r2、r3、r4、r5和r6如下所述)的特定的喹啉甲酰胺衍生物(专利文献1)。
3.式(a6)或(b8)表示的喹啉甲酰胺衍生物如下所述。
[0004][0005]
通过使5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺与式(a4)或(b6)表示的喹啉羧酸衍生物进行反应而制造,但以往报告了如下方法:式(a4)或(b6)表示这样的喹啉羧酸衍生物如下所示,在酸的存在下或不存在下使苯胺类、醛类和α
‑
酮酸类进行反应而制造(非专利文献a1、a2)。
[0006][0007]
[式中,r表示氢原子、甲基、硝基等]
[0008]
另外,已知式(a4)或(b6)表示这样的喹啉羧酸衍生物可由对应的靛红衍生物,使用普菲青格(pfitzinger)喹啉合成法而制造(非专利文献b1)。这里用作原料的靛红衍生物一般可通过如下使通过与苯胺类、水合氯醛和羟胺的反应所得到的产物在硫酸中进行环化而合成(非专利文献b2)。
[0009][0010]
然而,该方法存在使用具有爆炸性的羟胺的问题,另外,存在如下问题:在制造与式(a4)或(b6)表示的喹啉羧酸衍生物对应的靛红衍生物的情况下,如果以高浓度使用作为底物的苯胺类,则在合成中间物时生成副产物。
[0011]
另一方面,报告了如下方法:式(a6)或(b8)表示的喹啉甲酰胺衍生物的制造中使用的5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺以往如下所示通过使氨基脲与糠醛进行反应而得到缩氨基脲,将其分离后,使用碘作为氧化剂进行环化而制造(非专利文献c1)。
[0012][0013]
但是,该方法存在如下问题:由于分离缩氨基脲,因此费工夫,另外,使用毒性、腐蚀性、升华性高的碘,进而环化反应的收率低。
[0014]
现有技术文献
[0015]
专利文献
[0016]
专利文献1:日本专利第5650529号公报
[0017]
非专利文献
[0018]
非专利文献a1:synthetic comm.,2011,41,1435
‑
1443
[0019]
非专利文献a2:mod.chem.appl.,2016,4,195/1
‑
195/6
[0020]
非专利文献b1:heterocycles,2014,89,693
‑
707
[0021]
非专利文献b2:bioorg.med.chem.,2010,18,1482
‑
1496
[0022]
非专利文献c1:j.org.chem.,2015,80,1018
‑
1024
技术实现要素:
[0023]
第一,本发明涉及提供一种工业上有利地合成作为上述式(a6)表示的喹啉甲酰胺衍生物或其盐的制造中间物的式(a4)表示的喹啉羧酸衍生物或其盐的方法。
[0024]
另外,第二,本发明涉及提供一种工业上有利地合成可用作上述式(b8)表示的喹啉甲酰胺衍生物或其盐的制造原料的靛红衍生物(下述式(b4))或其盐的方法。
[0025]
另外,第三,本发明涉及提供一种工业上有利地合成可用于制造包含上述式(a6)或(b8)表示的喹啉甲酰胺衍生物的stat3抑制剂的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物的方法。
[0026]
本发明人等系鉴于这种实际情况进行了深入研究,结果发现在使用苯胺类、醛类和α
‑
酮酸类制造喹啉羧酸衍生物的情况下,如下所示通过使用三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物,能够以高收率制造式(a4)表示的喹啉羧酸衍生物,进而能够高效地制造式(a6)表示的喹啉甲酰胺衍生物或其盐。
[0027][0028]
[式中,r1、r2、r3和r4可以相同或不同,表示氢原子、卤素原子、低级烷基、硝基、羟基、氰基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、低级烷氧基、卤代低级烷氧基、低级烷氧基低级烷氧基、低级烷基羰基、低级烷氧基羰基、苄氧基、三苯甲氧基、叔丁基二甲基甲硅烷氧基、二低级烷基氨基、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基或低级烷硫基,r5表示氢原子、低级烷基、环低级烷基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、萘基、1,3
‑
苯并二茂基、苯乙烯基或
‑
cr
5a
or
5b
ch2or
5c
(这里,r
5a
表示甲基或叔丁基,r
5b
和r
5c
可以相同或不同,表示氢原子、低级烷基、低级烷酰基、叔丁基二甲基甲硅烷基、2
‑
(三甲基甲硅烷基)乙氧基甲基、苄氧基甲基、环上可以具有取代基的苯基或苄基、低级烯基或低级烷氧基甲基),r6表示氢原子、低级烷基、羟基、芳基或卤素原子]
[0029]
即,第1发明涉及以下的1)~3)。
[0030]
1)一种式(a4)表示的喹啉羧酸衍生物或其盐的制造方法,其特征在于,在三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物的存在下,使式(a2)表示的醛类与式(a1)表示的苯胺类进行反应,接下来使式(a3)表示的α
‑
酮酸类进行反应。
[0031]
2)根据1)的方法,其中,式(a1)表示的苯胺类为4
‑
(三氟甲氧基)苯胺,式(a2)表示的醛类为苯甲醛,式(a3)表示的α
‑
酮酸类为丙酮酸。
[0032]
3)一种式(a6)表示的喹啉甲酰胺衍生物或其盐的制造方法,其特征在于,使式(a5)表示的5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺与通过上述1)或2)的方法而制造的式(a4)表示的喹啉羧酸衍生物或其盐进行反应。
[0033]
另外,本发明人等发现在由苯胺类制造靛红衍生物的情况下,如下所示通过使用2
‑
(烷氧基亚氨基)乙酸类,能够以高收率制造式(b4)表示的靛红衍生物,进而,能够高效地制造式(b8)表示的喹啉甲酰胺衍生物或其盐。
[0034][0035]
[式中,r1、r2、r3和r4可以相同或不同,表示氢原子、卤素原子、低级烷基、硝基、羟基、氰基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、低级烷氧基、卤代低级烷氧基、低级烷氧基低级烷氧基、低级烷基羰基、低级烷氧基羰基、苄氧基、三苯甲氧基、叔丁基二甲基甲硅烷氧基、二低级烷基氨基、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基或低级烷硫基,r5表示氢原子、低级烷基、环低级烷基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、萘基、1,3
‑
苯并二茂基、苯乙烯基或
‑
cr
5a
or
5b
ch2or
5c
(这里,r
5a
表示甲基或叔丁基,r
5b
和r
5c
可以相同或不同,表示氢原子、低级烷基、低级烷酰基、叔丁基二甲基甲硅烷基、2
‑
(三甲基甲硅烷基)乙氧基甲基、苄氧基甲基、环上可以具有取代基的苯基或苄基、低级烯基或低级烷氧基甲基),r6表示氢原子、低级烷基、羟基、芳基或卤素原子,x表示低级烷基]
[0036]
即,第2发明涉及以下的1)~6)。
[0037]
1)一种式(b4)表示的靛红衍生物或其盐的制造方法,其特征在于,使式(b2)表示的2
‑
(烷氧基亚氨基)乙酸类与式(b1)表示的苯胺类进行反应,得到式(b3)表示的2
‑
(烷氧基亚氨基)
‑
乙酰胺类,将其在浓硫酸中进行加热。
[0038]
2)根据1)的方法,其中,式(b1)表示的苯胺类为4
‑
(三氟甲氧基)苯胺。
[0039]
3)一种式(b6)表示的喹啉羧酸衍生物或其盐的制造方法,其特征在于,在碱的存在下,使式(b5)表示的酮类与通过1)或2)的方法而制造的式(b4)表示的靛红衍生物或其盐进行反应。
[0040]
4)根据3)的方法,其中,式(b5)表示的酮类为苯乙酮。
[0041]
5)根据3)或4)的方法,其中,碱为氢氧化钠。
[0042]
6)一种式(b8)表示的喹啉甲酰胺衍生物或其盐的制造方法,其特征在于,使式(b7)表示的5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺与通过3)至5)中任一项的方法而制造的式(b6)表示的喹啉羧酸衍生物或其盐进行反应。
[0043]
另外,本发明人等发现通过使式(c2)表示的氨基脲或氨基硫脲或者其盐与式(c1)表示的醛类进行反应,不分离所生成的式(c3)表示的缩氨基脲类或缩氨基硫脲类,使用氯胺t进行环化,由此能够高效地制造式(c4)表示的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物。
[0044][0045]
[式中,r表示氢原子、低级烷基、低级烯基、环低级烷基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、苄基、萘基或苯乙烯基,x表示氧原子或硫原子]
[0046]
即,第3发明涉及以下的1)~2)。
[0047]
1)一种式(c4)表示的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物或其盐的制造方法,其特征在于,使式(c2)表示的氨基脲或氨基硫脲或者其盐与式(c1)表示的醛类进行反应,不分离所生成的式(c3)表示的缩氨基脲类或缩氨基硫脲类,使用氯胺t进行环化。
[0048]
2)根据1)的方法,其中,式(c1)表示的醛类为糠醛,式(c2)表示的氨基脲或氨基硫脲或者其盐为氨基脲或其盐。
[0049]
根据第1发明,能够以高收率制造式(a4)表示的喹啉羧酸衍生物或其盐,进而,能够工业上有利地制造作为stat3的抑制剂的式(a6)表示的喹啉甲酰胺衍生物或其盐。
[0050]
根据第2发明,能够以高收率制造式(b4)表示的靛红衍生物或其盐,进而,能够工业上有利地制造作为stat3的抑制剂的式(b8)表示的喹啉甲酰胺衍生物或其盐。
[0051]
根据第3发明,能够工业上有利地制造可用于制造包含式(a6)或(b8)表示的喹啉甲酰胺衍生物的stat3抑制剂的1,3,4
‑
(噻)二唑
‑2‑
胺或其盐。
具体实施方式
[0052]
本说明书中,”低级”的用语只要没有特别说明,则是指带有该用语的基团的烃部分的碳原子数在烃部分为链状的情况下为1~9个,在为环状的情况下为3~7个,是指链状烃部分可以为直链或支链的任一种。应予说明,本说明书中,烃部分的碳原子数(x~y个)简称为”c
x
‑
y”。
[0053]
另外,”可以具有取代基”是指作为对象的基团的氢原子可以被其它基取代,该取代基的数量可以为1或1个以上,在具有2个以上的取代基的情况下,该取代基可以相同或不同。
[0054]
本发明中的式(a1)~(a6)、(b1)~(b8)中,作为r1、r2、r3、r4和r6表示的卤素原子,可举出氟、氯、溴或碘,优选为氟、氯或溴。
[0055]
作为r1、r2、r3、r4、r5和r6表示的低级烷基,优选为甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基、庚基、辛基、壬基等c1‑9烷基,更优选为甲基、乙基、丙基、叔丁基、壬基。
[0056]
作为r1、r2、r3和r4表示的低级烷氧基,优选为甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等c1‑4烷氧基,更优选为甲氧基。
[0057]
作为r1、r2、r3和r4表示的卤代低级烷氧基,优选为卤代c1‑4烷氧基,例如可举出二氟甲氧基、三氟甲氧基、2,2,2
‑
三氟乙氧基等,优选为三氟甲氧基。
[0058]
作为r1、r2、r3和r4表示的低级烷氧基低级烷氧基,优选为c1‑4烷氧基c1‑4烷氧基,例如可举出甲氧基甲氧基、乙氧基甲氧基、甲氧基乙氧基、乙氧基乙氧基等,优选为甲氧基甲氧基、甲氧基乙氧基。
[0059]
作为r1、r2、r3和r4表示的低级烷基羰基,优选为c1‑4烷基羰基,例如可举出甲基羰基、乙基羰基、正丙基羰基、叔丁基羰基等,优选为甲基羰基。
[0060]
作为r1、r2、r3和r4表示的低级烷氧基羰基,优选为c1‑4烷氧基羰基,例如可举出甲氧基羰基、乙氧基羰基、正丙氧基羰基、叔丁氧基羰基等,优选为甲氧基羰基。
[0061]
作为r1、r2、r3和r4表示的二低级烷基氨基,优选为二c1‑4烷基氨基,例如可举出二甲基氨基、二乙基氨基、二正丙基氨基、二异丙基氨基等,优选为二甲基氨基。
[0062]
作为r1、r2、r3和r4表示的低级烷硫基,优选为c1‑4烷硫基,例如可举出甲硫基、乙硫基、正丙硫基、异丙硫基等,优选为甲硫基。
[0063]
在r1、r2、r3、r4和r5表示的可以具有取代基的苯基中,作为可取代于苯基的基团,例如可举出卤素原子(例如氯原子、溴原子等)、c1‑4烷基(例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等)、c2‑7烯基(例如乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等)、c1‑4烷氧基(例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等)、卤代c1‑4烷氧基(例如三氟甲氧基等)、羟基、硝基、氰基、c1‑4烷基羰基(例如甲基羰基等)、c1‑4烷氧基羰基(例如甲氧基羰基等)、二c1‑4烷基氨基(例如二甲基氨基等)、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基等。
[0064]
在r1、r2、r3、r4和r5表示的可以具有取代基的5~6元杂环式基团中,作为5~6元杂环式基团,例如可举出吡咯基、吡唑基、呋喃基、噻吩基、吡啶基、咪唑基、三唑基、四唑基、三嗪基、哒嗪基、嘧啶基、吡嗪基、异唑基、噻唑基、异噻唑基、噻二唑基、唑基、二唑基等,优选为吡啶基、呋喃基、噻吩基、吡咯基。
[0065]
作为可取代于该杂环式基团的基团,例如可举出卤素原子(例如氯原子、溴原子等)、c1‑4烷基(例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等)、c2‑7烯基(例如乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等)、c1‑4烷氧基(例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等)、卤代c1‑4烷氧基(例如三氟甲氧基等)、羟基、硝基、氰基、c1‑4烷基羰基(例如甲基羰基等)、c1‑4烷氧基羰基(例如甲氧基羰基等)、二c1‑4烷基氨基(例如二甲基氨基等)、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基等。
[0066]
作为r5表示的环低级烷基,优选为环c3‑7烷基,例如可举出环丙基、环丁基、环戊基、环己基等,优选为环己基。
[0067]
在r5表示的
‑
cr
5a
or
5b
ch2or
5c
中,作为r
5b
和r
5c
表示的低级烷基,可举出c1‑4烷基(例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等),作为低级烷酰基,可举出c1‑7烷酰基(例如乙酰基、丙酰基、丁酰基、戊酰基等),另外,作为环上可以具有取代基的苯基或苄基中的取代基,可举出c1‑4烷氧基(例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等)、卤素原子(例如氯原子、溴原子等)等。另外,作为低级烯基,可举出c2‑7烯基(例如乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等),作为低级烷氧基甲基,可举出c1‑4烷氧基甲基(例如甲氧基甲基、乙氧基甲基、丙氧基甲基、异丙氧基甲基、丁氧基甲基等)。
[0068]
作为r6表示的芳基,例如可举出苯基、萘基、茚基等c6‑
14
芳基等,优选可举出苯基。
[0069]
r1~r4优选r1为氢原子,r2为氢原子、卤素原子、低级烷基、硝基、羟基、氰基、可以具有取代基的苯基、可以具有取代基的5~6元杂环式基团、低级烷氧基、卤代低级烷氧基、低级烷氧基低级烷氧基、低级烷基羰基、低级烷氧基羰基、苄氧基、三苯甲氧基、叔丁基二甲基甲硅烷氧基、二低级烷基氨基、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基或低级烷硫基,r3和r4可以相同或不同,为氢原子、卤素原子、低级烷基或硝基。
[0070]
r1~r6特别优选r1、r3和r4为氢原子,r2为卤代c1‑4烷氧基(优选为三氟甲氧基),r5为可以具有取代基的苯基(优选为苯基),以及r6为氢原子。
[0071]
作为式(a4)表示的喹啉羧酸衍生物(以下,称为喹啉羧酸衍生物(a4))、式(a6)表示的喹啉甲酰胺衍生物(以下,称为喹啉甲酰胺衍生物(a6))的盐或式(b4)表示的靛红衍生物(以下,称为靛红衍生物(b4))、式(b6)表示的喹啉羧酸衍生物(以下,称为喹啉羧酸衍生物(b6))、式(b8)表示的喹啉甲酰胺衍生物(以下,称为喹啉甲酰胺衍生物(b8))的盐,例如可举出药理学上可允许的酸加成盐、金属盐、铵盐、有机胺加成盐、氨基酸加成盐等。作为药理学上可允许的酸加成盐,可举出盐酸、氢溴酸、硫酸、硝酸、磷酸、硼酸等各无机酸盐,以及作为有机酸的甲酸、乙酸、丙酸、富马酸、丙二酸、丁二酸、马来酸、酒石酸、柠檬酸、苯甲酸等羧酸类的盐,甲磺酸、对甲苯磺酸等磺酸类的盐,麸胺酸、天冬氨酸等氨基酸类的盐。作为药理学上可允许的金属盐,可举出锂、钠、钾等各碱金属盐,镁、钙等各碱土金属盐,铝、锌等各金属盐,作为药理学上可允许的铵盐,可举出铵、四甲基铵等各盐,作为药理学上可允许的有机胺盐,可举出三乙基胺、哌啶、吗啉、甲苯胺等各盐,作为药理学上可允许的氨基酸加成盐,可举出赖氨酸、甘氨酸、苯丙氨酸等加成盐。
[0072]
<第1发明>
[0073]
以下说明本发明的喹啉羧酸衍生物(a4)或其盐以及喹啉甲酰胺衍生物(a6)或其盐的制造方法的详细内容。应予说明,所有起始物质和制造化合物可以为盐,各反应中制造的化合物可通过常规方法转换为盐。
[0074]
a1.喹啉羧酸衍生物(a4)或其盐的制造
[0075]
本反应通过在三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物的存在下,使式(a2)表示的醛类(以下,称为醛类(a2))与式(a1)表示的苯胺类(以下,称为苯胺类(a1))进行反应,接下来使式(a3)表示的α
‑
酮酸类(以下,称为α
‑
酮酸类(a3))进行反应而进行。
[0076]
苯胺类(a1)与醛类(a2)的反应在三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物的存在下,在适当的溶剂中进行。
[0077]
该苯胺类(a1)相对于α
‑
酮酸类(a3),通常可以使用1.0~2.2倍摩尔,优选可以使用1.7~1.9倍摩尔。
[0078]
该醛类(a2)相对于α
‑
酮酸类(a3),通常可以使用1.0~2.4倍摩尔、优选可以使用1.9~2.1倍摩尔。
[0079]
本反应中使用的三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物相对于α
‑
酮酸类(a3),使用0.1~1.0倍摩尔,优选为0.4~1.0倍摩尔。
[0080]
上述反应在适当的溶剂中进行。作为所使用的溶剂,只要为不参与反应的溶剂就没有特别限制,例如可举出乙腈、异丁腈、丙腈、甲氧基乙腈等腈系溶剂,二乙醚、四氢呋喃(thf)、2
‑
甲基四氢呋喃、1,3
‑
或1,4
‑
二烷、叔丁基甲基醚(mtbe)、环戊基甲基醚(cpme)、1,2
‑
二甲氧基乙烷(dme)、二乙二醇二甲醚等醚系溶剂,n,n
‑
二甲基甲酰胺(dmf)、二甲基亚砜(dmso)等非质子性溶剂,二氯甲烷、氯仿、1,2
‑
二氯乙烷等卤代烃,甲苯、邻二甲苯、间二甲苯、对二甲苯等芳香族烃等。其中,优选腈系溶剂、醚系溶剂、非质子性溶剂、卤代烃,更优选乙腈。另外,这些溶剂也可以单独使用或组合而使用,溶剂的使用量没有特别限制。
[0081]
只要在反应温度为通常
‑
78℃~所使用的溶剂的沸点的范围内进行即可,优选为60~70℃。
[0082]
反应时间通常优选为0~24小时,更优选为10分钟~1小时。
[0083]
接下来,使α
‑
酮酸类(a3)与通过上述反应而得到的反应产物进行反应,得到喹啉羧酸衍生物(a4)或其盐。
[0084]
反应优选例如将溶解或悬浮于上述溶剂中的α
‑
酮酸类(a3)通常以0~24小时添加至上述反应的反应液中,更优选0~12小时。
[0085]
优选在反应温度为通常
‑
78℃~所使用的溶剂的沸点的范围、优选60~70℃,在搅拌下进行通常5分钟~45小时、优选为12~24小时。
[0086]
应予说明,苯胺类(a1)、醛类(a2)和α
‑
酮酸类(a3)可以以市售品的形式获得,或者可通过文献等中记载的方法或依据它们的方法而得到。
[0087]
反应结束后,在反应液中加入甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)等有机溶剂,适当地利用水进行清洗,利用碳酸氢钠、碳酸钠、氢氧化钠等碱性水溶液对有机层进行萃取操作,在所得到的水层中加入盐酸等酸性水溶液进行结晶化,滤取所产生的结晶,或者在反应液中加入水或盐水,利用甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)等有机溶剂进行萃取操作,依照常规方法对所得到的有机层进行干燥,或者滤取析出至反应液中的固体,由此能够分离喹啉羧酸衍生物(a4)或其盐。另外,也可以对喹啉羧酸衍生物(a4)或其盐通过硅胶柱色谱进行纯化操作,或者将丙酮、甲基乙基酮(mek)、甲基异丁基酮(mibk)、乙酸乙酯、乙酸丁酯、甲醇、乙醇、1
‑
或2
‑
丙醇、二氯甲烷、氯仿、乙腈、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷、庚烷、己烷等有机溶剂单独使用或组合使用,通过悬浮清洗或再结晶而进行纯化操作。
[0088]
以往,报告了通过在氨基磺酸等酸的存在下或不存在下使苯胺类、醛类和α
‑
酮酸类进行反应来制造喹啉羧酸衍生物(上述非专利文献a1、a2)。但是,该反应存在如下问题:根据苯胺类的取代基的种类,特别是在r2具有吸电子性取代基的情况下,收率降低。相对于此,根据本发明的使用三氟化硼
‑
四氢呋喃络合物或三氟化硼
‑
二乙醚络合物的方法,能够以高收率得到喹啉羧酸衍生物(a4)或其盐(参照下述比较例)。
[0089]
a2.喹啉甲酰胺衍生物(a6)或其盐的制造
[0090]
喹啉甲酰胺衍生物(a6)或其盐可依据上述专利文献1中记载的方法制造。
[0091]
即,可通过如下操作而得到:在碱和缩合剂的存在下,在适当的非活性溶剂、例如氯仿、二氯甲烷等卤代烃、苯、甲苯等芳香族烃、二乙醚、四氢呋喃(thf)、1,4
‑
二烷等醚系溶剂、n,n
‑
二甲基甲酰胺(dmf)、n
‑
甲基吡咯啶酮(nmp)、二甲基亚砜(dmso)等非质子性极性溶剂、吡啶、喹啉等碱性溶剂或它们的混合溶剂中,在
‑
78℃~所使用的溶剂的沸点之间的温度下,使上述得到的喹啉羧酸衍生物(a4)或其盐与5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺(a5)反应5分钟~48小时。
[0092]
作为碱,例如可举出三乙胺、n,n
‑
二异丙基乙基胺(dipea)、n
‑
甲基吗啉(nmm)、吡啶等有机碱;碳酸钾、碳酸氢钾、磷酸三钾、氢氧化钠、氢化钠等无机碱;甲醇钠、叔丁醇钾等金属醇盐等。
[0093]
作为缩合剂,例如可使用dcc、wsci等碳二酰亚胺系缩合剂,bop等系缩合剂,hatu、hbtu等铵/脲系缩合剂,dmt
‑
mm等三嗪系缩合剂,cdi等咪唑系缩合剂,dpp
‑
cl等次膦酰氯系缩合剂,t3p等膦酸酐系缩合剂等。
[0094]
反应结束后,利用盐水或水等对反应液进行清洗后,依照常规方法进行干燥,或者在反应液中添加盐水或水,滤取所产生的固体,由此能够得到喹啉甲酰胺衍生物(a6)或其盐。另外,也可以对喹啉甲酰胺衍生物(a6)或其盐通过硅胶柱色谱进行纯化操作,或者将丙酮、甲基乙基酮(mek)、甲基异丁基酮(mibk)、乙酸乙酯、乙酸丁酯、甲醇、乙醇、1
‑
或2
‑
丙醇、二氯甲烷、氯仿、乙腈、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷、庚烷、己烷等有机溶剂单独使用或组合使用,通过悬浮清洗或再结晶而进行纯化操作。
[0095]
<第2发明>
[0096]
以下说明本发明的靛红衍生物(b4)或其盐、喹啉羧酸衍生物(b6)或其盐以及喹啉甲酰胺衍生物(b8)或其盐的制造方法的详细内容。应予说明,所有起始物质和制造化合物可以为盐,各反应中所制造的化合物可通过常规方法转换为盐。
[0097]
b1.靛红衍生物(b4)或其盐的制造
[0098]
本反应系通过使式(b2)表示的2
‑
(烷氧基亚氨基)乙酸类(以下,称为2
‑
(烷氧基亚氨基)乙酸类(b2))与式(b1)表示的苯胺类(以下,称为苯胺类(b1))进行反应,得到式(b3)表示的2
‑
(烷氧基亚氨基)
‑
乙酰胺类(以下,称为2
‑
(烷氧基亚氨基)
‑
乙酰胺类(b3)),将其在浓硫酸中进行加热而进行。
[0099]
苯胺类(b1)与2
‑
(烷氧基亚氨基)乙酸类(b2)的反应使用缩合剂,在适当的溶剂中进行。
[0100]
作为缩合剂,只要为用于酰胺键形成的缩合剂即可,例如可举出dcc、wsci等碳二酰亚胺系缩合剂,bop等系缩合剂,hatu、hbtu等铵/脲系缩合剂,dmt
‑
mm等三嗪系缩合剂,cdi等咪唑系缩合剂,dpp
‑
cl等次膦酰氯系缩合剂,t3p等膦酸酐系缩合剂等。
[0101]
该缩合剂相对于苯胺类(b1),使用1.05~1.30倍摩尔,优选为1.15倍摩尔。
[0102]
上述反应在适当的溶剂中进行。作为所使用的溶剂,只要为不参与反应的溶剂就没有特别限制,例如可举出乙腈、异丁腈、丙腈、甲氧基乙腈等腈系溶剂,二乙醚、四氢呋喃(thf)、2
‑
甲基四氢呋喃、1,3
‑
或1,4
‑
二烷、叔丁基甲基醚(mtbe)、环戊基甲基醚(cpme)、1,2
‑
二甲氧基乙烷(dme)、二乙二醇二甲醚等醚系溶剂,乙酸乙酯、乙酸丁酯等酯系溶剂,n,
n
‑
二甲基甲酰胺(dmf)、二甲基亚砜(dmso)等非质子性溶剂,二氯甲烷、氯仿、1,2
‑
二氯乙烷等卤代烃,甲苯、邻二甲苯、间二甲苯、对二甲苯等芳香族烃等。其中,优选腈系溶剂、醚系溶剂、酯系溶剂、卤代烃、非质子性溶剂,更优选乙腈。另外,这些溶剂也可以单独使用或组合使用,溶剂的使用量没有特别限制。
[0103]
反应只要在非活性气体下,在通常
‑
78℃~所使用的溶剂的沸点的范围内进行即可,优选为0℃~40℃。反应时间通常优选为5分钟~24小时,更优选为10分钟~6小时。
[0104]
接下来,使2
‑
(烷氧基亚氨基)乙酸类(b2)与通过上述反应而得到的反应产物进行反应,得到2
‑
(烷氧基亚氨基)
‑
乙酰胺类(b3)。
[0105]
该2
‑
(烷氧基亚氨基)乙酸类(b2)相对于苯胺类(b1),通常可以使用1.10~1.35倍摩尔,优选可以使用1.25倍摩尔。
[0106]
反应只要在非活性气体下,在通常
‑
78℃~所使用的溶剂的沸点的范围内进行即可,优选为15℃~所使用的溶剂的沸点。反应时间通常优选为5分钟~24小时,更优选为30分钟~8小时。
[0107]
应予说明,作为本反应中所使用的2
‑
(烷氧基亚氨基)乙酸类(b2),可优选举出2
‑
(甲氧基亚氨基)乙酸。2
‑
(甲氧基亚氨基)乙酸可如下述的参考例所示,由乙醛酸和作为肟化试剂所通用的甲氧基胺盐酸盐,通过常规方法而合成。
[0108]
将通过上述反应而得到的2
‑
(烷氧基亚氨基)
‑
乙酰胺类(b3)在浓硫酸中进行加热,由此得到靛红衍生物(b4)或其盐。
[0109]
本反应优选在通常15~290℃的范围、优选60~80℃下,在搅拌下进行通常5分钟~42小时、优选3~24小时。
[0110]
应予说明,苯胺类(b1)和2
‑
(烷氧基亚氨基)乙酸类(b2)可以以市售品的形式获得,或者可通过文献等中记载的方法或依据它们的方法而得到。
[0111]
反应结束后,可通过滤取等公知的分离方法分离反应产物,适当地利用水进行清洗,得到靛红衍生物(b4)或其盐。
[0112]
如此,根据上述方法,能够在不使用具有爆炸性的羟胺的情况下,并且即便以高浓度使用苯胺类(b1)也不会产生来自该化合物的副产物,以高收率得到靛红衍生物(b4)或其盐。
[0113]
b2.喹啉羧酸衍生物(b6)或其盐的制造
[0114]
喹啉羧酸衍生物(b6)或其盐可由对应的靛红衍生物,使用普菲青格(pfitzinger)喹啉合成法而制造。
[0115]
即,可通过在碱性条件下使上述得到的靛红衍生物式(b4)或其盐与式(b5)表示的酮类(称为酮类(b5))进行反应而制造。
[0116]
反应优选在氢氧化钠或氢氧化钾等碱的存在下,在醇(例如甲醇、乙醇、1
‑
或2
‑
丙醇等)/水溶剂中进行。
[0117]
该酮类(b5)相对于靛红衍生物(b4)或其盐,通常可以使用1.00~5.00倍摩尔,优选可以使用1.05~3.15倍摩尔。
[0118]
反应优选在通常
‑
78℃~所使用的溶剂的沸点的范围、优选60℃~所使用的溶剂的沸点下,在搅拌下进行通常5分钟~58小时、优选6~30小时。
[0119]
反应结束后,将反应液适当地在减压下蒸馏去除溶剂,在残渣中加入甲苯、二乙
醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)等有机溶剂,利用水或碳酸氢钠、碳酸钠、氢氧化钠等碱性水溶液进行萃取操作,在所得到的水层中加入盐酸等酸性水溶液进行结晶化,滤取所产生的结晶,或者在残渣中加入盐酸等酸性水溶液,利用甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)、乙酸乙酯、二氯甲烷、氯仿等有机溶剂进行萃取操作,依照常规方法对所得到的有机层进行干燥,或者滤取析出至反应液中的固体,由此能够分离喹啉羧酸衍生物(b6)或其盐。另外,也可以对喹啉羧酸衍生物(b6)或其盐通过硅胶柱色谱进行纯化操作,或者将丙酮、甲基乙基酮(mek)、甲基异丁基酮(mibk)、乙酸乙酯、乙酸丁酯、甲醇、乙醇、1
‑
或2
‑
丙醇、二氯甲烷、氯仿、乙腈、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷、庚烷、己烷等有机溶剂单独使用或组合使用,通过悬浮清洗或再结晶而进行纯化操作。
[0120]
b3.喹啉甲酰胺衍生物(b8)或其盐的制造
[0121]
喹啉甲酰胺衍生物(b8)或其盐可依据上述专利文献1中记载的方法制造。
[0122]
即,可通过如下操作而得到:在碱和缩合剂的存在下,在适当的惰性溶剂、例如氯仿、二氯甲烷等卤代烃、苯、甲苯等芳香族烃、二乙醚、四氢呋喃(thf)、1,4
‑
二烷等醚系溶剂、n,n
‑
二甲基甲酰胺(dmf)、n
‑
甲基吡咯啶酮(nmp)、二甲基亚砜(dmso)等非质子性极性溶剂、吡啶、喹啉等碱性溶剂或这些的混合溶剂中,在
‑
78℃~所使用的溶剂的沸点之间的温度下,使上述得到的喹啉羧酸衍生物(b6)或其盐与5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺(b7)反应5分钟~48小时。
[0123]
作为碱,例如可举出三乙基胺、n,n
‑
二异丙基乙基胺(dipea)、n
‑
甲基吗啉(nmm)、吡啶等有机碱,碳酸钾、碳酸氢钾、磷酸三钾、氢氧化钠、氢化钠等无机碱,甲醇钠、叔丁醇钾等金属醇盐等。
[0124]
作为缩合剂,例如可使用dcc、wsci等碳二酰亚胺系缩合剂,bop等系缩合剂,hatu、hbtu等铵/脲系缩合剂,dmt
‑
mm等三嗪系缩合剂,cdi等咪唑系缩合剂,dpp
‑
cl等次膦酰氯系缩合剂,t3p等膦酸酐系缩合剂等。
[0125]
反应结束后,利用盐水或水等对反应液进行清洗后,依照常规方法进行干燥,或者在反应液中添加盐水或水,滤取所产生的固体,由此能够得到喹啉甲酰胺衍生物(b8)或其盐。另外,也可以对喹啉甲酰胺衍生物(b8)或其盐通过硅胶柱色谱进行纯化操作,或者将丙酮、甲基乙基酮(mek)、甲基异丁基酮(mibk)、乙酸乙酯、乙酸丁酯、甲醇、乙醇、1
‑
或2
‑
丙醇、二氯甲烷、氯仿、乙腈、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)、四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷、庚烷、己烷等有机溶剂单独使用或组合使用,通过悬浮清洗或再结晶而进行纯化操作。
[0126]
<第3发明>
[0127]
本发明中的式(c1)~(c4)中,作为r表示的低级烷基,优选为甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基、庚基、辛基、壬基等c1‑9烷基,更优选为甲基、乙基、丙基、叔丁基、壬基。
[0128]
作为r表示的低级烯基,例如优选为乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等,更优选为乙烯基、2
‑
甲基
‑1‑
丙烯基。
[0129]
作为r表示的环低级烷基,优选为环c3‑7烷基,例如可举出环丙基、环丁基、环戊基、环己基等,优选为环己基。
[0130]
在r表示的可以具有取代基的苯基中,作为可取代于苯基的基团,例如除卤素原子(例如氯原子、溴原子等)、c1‑4烷基(例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等)、c2‑7烯基(例如乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等)、c1‑4烷氧基(例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等)、羟基、硝基、氰基、c1‑4烷基羰基(例如甲基羰基等)、c1‑4烷氧基羰基(例如甲氧基羰基等)、氨基、二c1‑4烷基氨基(例如二甲基氨基等)、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基以外,还可举出作为使环上的2个碳原子结合的二价基团的氧亚乙基、氧亚乙基氧基等。
[0131]
在r表示的可以具有取代基的5~6元杂环式基团中,作为5~6元杂环式基团,例如可举出吡咯基、吡唑基、呋喃基、噻吩基、吡啶基、咪唑基、三唑基、四唑基、三嗪基、哒嗪基、嘧啶基、吡嗪基、异唑基、噻唑基、异噻唑基、噻二唑基、唑基、二唑基等,优选为吡啶基、呋喃基、噻吩基,更优选为2
‑
呋喃基、3
‑
呋喃基。
[0132]
作为可取代于该杂环式基团的基团,例如可举出卤素原子(例如氯原子、溴原子等)、c1‑4烷基(例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等)、c2‑7烯基(例如乙烯基、丙烯基、2
‑
甲基
‑1‑
丙烯基、1
‑
甲基
‑1‑
丙烯基等)、c1‑4烷氧基(例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等)、羟基、硝基、氰基、c1‑4烷基羰基(例如甲基羰基等)、c1‑4烷氧基羰基(例如甲氧基羰基等)、氨基、二c1‑4烷基氨基(例如二甲基氨基等)、叔丁氧基羰基氨基、苄氧基羰基氨基、2
‑
硝基苯磺酰氨基等。
[0133]
作为r表示的萘基,具体而言,可举出1
‑
萘基或2
‑
萘基。
[0134]
本发明中的化学式中,x可举出氧原子或硫原子。
[0135]
本发明中,r更优选为呋喃基(优选为2
‑
呋喃基),x更优选为氧原子。
[0136]
作为式(c2)表示的氨基脲或氨基硫脲(以下,称为氨基脲或氨基硫脲(c2))、式(c4)表示的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(以下,称为1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4))的盐,例如可举出药理学上可允许的酸加成盐,例如可举出盐酸、氢溴酸、硫酸、硝酸、磷酸、硼酸等各无机酸盐,以及作为有机酸的甲酸、乙酸、丙酸、富马酸、丙二酸、丁二酸、马来酸、酒石酸、柠檬酸、苯甲酸等羧酸类的盐,甲磺酸、对甲苯磺酸等磺酸类的盐,谷氨酸、天冬氨酸等氨基酸类的盐。
[0137]
以下说明本发明的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)的制造方法的详细内容。应予说明,原料物质和制造化合物可以为盐,各反应中所制造的化合物可通过常规方法转换为盐。
[0138]
本发明的1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)或其盐的制造通过使式(c1)表示的醛类(以下,称为醛类(c1))与氨基脲或氨基硫脲(c2)或其盐进行脱水缩合,不分离生成的式(c3)表示的缩氨基脲类或缩氨基硫脲类(以下,称为缩氨基脲类或缩氨基硫脲类(c3)),使用氯胺t进行环化而进行。
[0139]
醛类(c1)与氨基脲或氨基硫脲(c2)或者其盐的缩合反应通常可使用酸作为缩合促进剂,在水或极性溶剂中进行。
[0140]
醛类(c1)与氨基脲或氨基硫脲(c2)或者其盐的使用比率以摩尔比计,优选为1:1~1:1.20,更优选为1:1.03~1:1.10。
[0141]
只要在反应温度为通常
‑
78℃~所使用的溶剂的沸点的范围内进行即可,优选为0~50℃。
[0142]
作为进行反应的氛围,可以为空气氛围,也可以在氮气、氩气等非活性气体氛围下进行。
[0143]
关于反应压力,从经济性的方面出发,优选在大气压下进行,也可以在加压或减压条件下进行。
[0144]
作为上述极性溶剂,可举出甲醇、乙醇、丙醇、异丙醇、丁醇等醇系溶剂,乙腈、异丁腈、丙腈、甲氧基乙腈等腈系溶剂,四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷等醚系溶剂等,更优选为使用水或水与四氢呋喃(thf)的混合液,使用量没有特别限制。
[0145]
作为上述缩合促进剂中使用的酸,可举出盐酸、硫酸、乙酸、碳酸、对甲苯磺酸、硝酸、草酸、磷酸、氢溴酸、氢碘酸、氨基磺酸、高氯酸等,优选为盐酸、乙酸、碳酸。
[0146]
作为优选的样态,例如可举出在溶解有乙酸钠的水溶液中进行反应。
[0147]
应予说明,醛类(c1)可以以市售品的形式获得,或者可通过文献等中记载的方法或依据它们的方法而得到。另外,对于氨基脲或氨基硫脲(c2)或其盐,氨基脲盐酸盐、氨基硫脲可分别以市售品的形式获得。
[0148]
接下来,不分离通过上述反应而生成的缩氨基脲类或缩氨基硫脲类(c3),在含水溶剂中,在碱性条件下,使用氯胺t进行氧化环化。即,上述缩合反应结束后,在反应液中添加溶剂、碱、氯胺t,通过一锅(one
‑
pot)法进行环化反应。
[0149]
作为所使用的溶剂,只要是不参与反应的溶剂就没有特别限制,例如可举出甲醇、乙醇、丙醇、异丙醇、丁醇等醇系溶剂,乙腈、异丁腈、丙腈、甲氧基乙腈等腈系溶剂,四氢呋喃(thf)、1,3
‑
或1,4
‑
二烷等醚系溶剂等。其中,更优选为四氢呋喃(thf)。另外,这些溶剂也可以单独使用或组合使用,溶剂的使用量没有特别限制。
[0150]
作为碱,可举出氢氧化钾、碳酸钾、碳酸氢钾、磷酸三钾、氢氧化钠、碳酸钠、碳酸氢钠等,更优选为碳酸钾。
[0151]
该碱相对于醛类使用2.00~4.00倍摩尔,可以为2.50~3.00倍摩尔。
[0152]
氯胺t是指对甲苯磺酰氯酰胺钠,可使用市售的氯胺t三水合物等。
[0153]
氯胺t相对于醛类使用1.10~2.00倍摩尔,可以为1.20~1.70倍摩尔。
[0154]
只要在反应温度为通常
‑
78℃~所使用的溶剂的沸点的范围内进行即可,优选为0℃~所使用的含水溶剂的沸点。
[0155]
反应时间通常优选为5分钟~48小时,更优选为1~30小时。
[0156]
反应结束后,利用硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液对反应液进行清洗后,加入甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机溶剂,利用盐酸水与盐水的混合液进行萃取操作,在所得到的水层中加入氢氧化钠等碱性水溶液进行结晶化,进行滤取;或者利用硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液对反应液进行清洗后,加入溶解于二乙醚、乙酸乙酯、1,4
‑
二烷等有机溶剂中的盐酸,滤取所产生的固体,使所得到的固体悬浮于水,加入氢氧化钠等碱性水溶液后,进行滤取;或者在上述滤液中添加甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机溶剂,利用盐酸水与盐水的混合液进行萃取操作,在所得到的水层中加入氢氧化钠等碱性水溶液进行结晶化,进行滤取;或者在反应液中加入硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液,滤取析出的固体;或者分离上述滤液的有机层,在所得到的有机层中加入甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机
溶剂,利用盐酸水与盐水的混合液进行萃取操作,在所得到的水层中加入氢氧化钠等碱性水溶液进行结晶化,进行滤取;或者反应结束后,利用硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液对反应液进行清洗后,加入甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机溶剂,利用盐酸水与盐水的混合液进行萃取操作,在所得到的水层中加入氢氧化钠等碱性水溶液而使其为碱性,进行减压浓缩;或者反应结束后,利用硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液对反应液进行清洗后,加入盐酸水与盐水的混合液,利用甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机溶剂进行萃取操作,利用碳酸氢钠、氢氧化钠等碱性水溶液对所得到的有机层进行清洗,进行减压浓缩;或者利用硫代硫酸钠、亚硫酸钠、亚硫酸氢钠等还原剂的水溶液与盐水的混合液对反应液进行清洗后,加入甲苯、二乙醚、二异丙醚(ipe)、叔丁基甲基醚(mtbe)等有机溶剂以及溶解于二乙醚、乙酸乙酯、1,4
‑
二烷等有机溶剂中的盐酸,利用盐酸水与盐水的混合液进行萃取操作,在所得到的水层中加入氢氧化钠等碱性水溶液进行结晶化,进行滤取;由此能够分离1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)或其盐。另外,也可以通过硅胶柱色谱对1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)或其盐进行纯化操作。
[0157]
以往报告了如下方法:1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)、例如5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺通过使氨基脲(c2)与糠醛进行反应而得到缩氨基脲(c3),将其分离后,使用碘作为氧化剂进行环化而制造(非专利文献c1)。但是,该方法存在如下问题:需要分离缩氨基脲,因此耗费一定的劳力,另外,使用毒性、腐蚀性、升华性高的碘,进而环化反应的收率低。
[0158]
相对于此,根据本发明的方法,不耗费分离缩氨基脲的工夫,另外,不使用毒性、腐蚀性、挥发性高的碘,能够有效地制造5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
(噻)二唑
‑2‑
胺衍生物(c4)或其盐。
[0159]
以下,通过实施例和比较例详细地说明本发明,但本发明并不受这些任何限定。
[0160]
实施例
[0161]
实施例a1 2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0162]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,将丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液用3小时滴加至反应液中。21小时后,将反应液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以微黄白色固体的形式得到标题化合物(1.55g、收率82%)。
[0163]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.74(1h,d,j=1.5hz),8.33
‑
8.30(3h,m),7.87(1h,dd,j=9.2,2.7hz),7.63
‑
7.54(3h,m)
[0164]
esi
‑
ms(m/z):334(m+h)
+
[0165]
实施例a2 2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0166]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
二乙醚络合物(357μl、2.84mmol),加热至65℃。1小时后,将丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液用3小时滴加至反应液中。21小时后,将反应
液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以微黄白色固体的形式得到标题化合物(1.62g、收率86%)。
[0167]
实施例a3 2
‑
苯基喹啉
‑4‑
羧酸的制造
[0168]
在苯胺(0.952g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。通过硅胶柱色谱(己烷/乙酸乙酯)对所得到的残渣进行纯化,以淡黄色固体的形式得到标题化合物(0.614g、收率43%)。
[0169]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.0(1h,s),8.67(1h,d,j=8.5hz),8.47(1h,s),8.32
‑
8.30(2h,m),8.18(1h,d,j=7.9hz),7.89
‑
7.85(1h,m),7.74
‑
7.70(1h,m),7.61
‑
7.53(3h,m)
[0170]
esi
‑
ms(m/z):250(m+h)
+
[0171]
实施例a4 6
‑
氟
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0172]
在4
‑
氟苯胺(1.14g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以淡黄色固体的形式得到标题化合物(1.10g、收率72%)。
[0173]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.1(1h,s),8.56(1h,s),8.48(1h,dd,j=11.0,2.7hz),8.30
‑
8.27(2h,m),8.26(1h,dd,j=9.3,6.0hz),7.83
‑
7.78(1h,m),7.61
‑
7.53(3h,m)
[0174]
esi
‑
ms(m/z):268(m+h)
+
[0175]
实施例a5 6
‑
氯
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0176]
在4
‑
氯苯胺(1.30g、10.2mmol)的乙腈(9.5ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(5.7ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以黄褐色固体的形式得到标题化合物(1.27g、收率79%)。
[0177]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.1(1h,s),8.79(1h,d,j=2.4hz),8.56(1h,s),8.32
‑
8.28(2h,m),8.19(1h,d,j=9.2hz),7.89(1h,dd,j=9.2,2.4hz),7.60
‑
7.55(3h,m)
[0178]
esi
‑
ms(m/z):284,286(m+h)
+
[0179]
实施例a6 6
‑
溴
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0180]
在4
‑
溴苯胺(1.76g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、
11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以白色固体的形式得到标题化合物(1.32g、收率71%)。
[0181]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.95(1h,d,j=2.1hz),8.55(1h,s),8.31
‑
8.29(2h,m),8.12(1h,d,j=8.9hz),7.99(1h,dq,j=8.9,1.1hz),7.62
‑
7.54(3h,m)
[0182]
esi
‑
ms(m/z):328,330(m)
+
[0183]
实施例a7 6
‑
甲基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0184]
在对甲苯胺(1.10g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以淡黄色固体的形式得到标题化合物(1.17g、收率78%)。
[0185]1h
‑
nmr(400mhz,dmso
‑
d6)δ:8.44(2h,s),8.28(2h,t,j=4.0hz),8.09(1h,d,j=8.5hz),7.72(1h,dd,j=8.5,1.8hz),7.59
‑
7.54(3h,m),2.56(3h,s)
[0186]
esi
‑
ms(m/z):264(m+h)
+
[0187]
实施例a8 6
‑
硝基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0188]
在4
‑
硝基苯胺(1.41g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以棕色固体的形式得到标题化合物(1.48g、收率89%)。
[0189]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.4(1h,s),9.70(1h,d,j=2.4hz),8.68(1h,s),8.54(1h,dd,j=9.2,2.4hz),8.37
‑
8.36(2h,m),8.34
‑
8.32(1h,m),7.64
‑
7.61(3h,m)
[0190]
esi
‑
ms(m/z):295(m+h)
+
[0191]
实施例a9 6
‑
羟基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0192]
在4
‑
氨基苯酚(1.12g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以淡黄色固体的形式得到标题化合物(0.907g、收率60%)。
[0193]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.8(1h,s),10.3(1h,s),8.39(1h,s),8.24
‑
8.21(2h,m),8.04(1h,d,j=2.7hz),8.02(1h,d,j=9.2hz),7.57
‑
7.47(3h,m),7.40(1h,dd,j=9.2,2.7hz)
[0194]
esi
‑
ms(m/z):266(m+h)
+
[0195]
实施例a10 6
‑
氰基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0196]
在4
‑
氨基苯甲腈(0.483g、4.09mmol)的乙腈(2.3ml)溶液中加入苯甲醛(0.482g、4.54mmol)、三氟化硼
‑
四氢呋喃络合物(125μl、1.14mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.200g、2.27mmol)的乙腈(3.8ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以淡黄色固体的形式得到标题化合物(0.455g、收率73%)。
[0197]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.4(1h,s),9.20(1h,d,j=1.8hz),8.62(1h,s),8.35
‑
8.33(2h,m),8.29(1h,d,j=8.5hz),8.15(1h,dd,j=8.5,1.8hz),7.62
‑
7.59(3h,m)
[0198]
esi
‑
ms(m/z):275(m+h)
+
[0199]
实施例a11 6
‑
甲氧基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0200]
在4
‑
甲氧基苯胺(1.26g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以黄色固体的形式得到标题化合物(0.824g、收率52%)。
[0201]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.9(1h,s),8.47(1h,s),8.26
‑
8.25(2h,m),8.15(1h,d,j=2.4hz),8.09(1h,d,j=9.2hz),7.59
‑
7.49(4h,m),3.93(3h,s)
[0202]
esi
‑
ms(m/z):280(m+h)
+
[0203]
实施例a12 6
‑
甲氧基甲氧基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0204]
(实施例a12
‑
1)
[0205]
在4
‑
甲氧基甲氧基苯胺(1.57g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以褐色固体的形式得到标题化合物(0.499g、收率28%)。
[0206]
(实施例a12
‑
2)
[0207]
在4
‑
甲氧基甲氧基苯胺(1.57g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入甲苯,利用1mol/l氢氧化钠水溶液进行萃取。将所得到的水层进行冰冷,加入浓盐酸而将ph调整为4左右后,搅拌0.5小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,由此以褐色固体的形式得到标题化合物(1.14g、收率65%)。
[0208]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.9(1h,s),8.46(1h,s),8.28
‑
8.21(3h,m),8.12(1h,d,j=9.2hz),7.62
‑
7.49(4h,m),5.37(2h,s),3.45(3h,s)
[0209]
esi
‑
ms(m/z):310(m+h)
+
[0210]
实施例a13 6
‑
乙酰基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0211]
在4
’‑
氨基苯乙酮(1.38g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时
滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥。利用氯仿对所得到的固体进行悬浮清洗,在减压下进行干燥后,以褐色固体的形式得到标题化合物(0.602g、收率36%)。
[0212]1h
‑
nmr(400mhz,dmso
‑
d6)δ:9.34(1h,d,j=1.8hz),8.57(1h,s),8.36
‑
8.30(3h,m),8.24(1h,d,j=8.7hz),7.64
‑
7.58(3h,m),2.73(3h,s)
[0213]
esi
‑
ms(m/z):292(m+h)
+
[0214]
实施例a14 6
‑
甲氧基羰基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0215]
在4
‑
氨基苯甲酸甲酯(1.55g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以淡黄色固体的形式得到标题化合物(1.38g、收率79%)。
[0216]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),9.39(1h,t,j=1.1hz),8.58(1h,s),8.36
‑
8.24(4h,m),7.64
‑
7.56(3h,m),3.96(3h,s)
[0217]
esi
‑
ms(m/z):308(m+h)
+
[0218]
实施例a15 6
‑
苄氧基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0219]
在4
‑
苄氧基苯胺(2.04g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以淡黄色固体的形式得到标题化合物(1.32g、收率66%)。
[0220]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.9(1h,s),8.46(1h,s),8.26
‑
8.24(3h,m),8.10(1h,d,j=9.5hz),7.61
‑
7.49(6h,m),7.45
‑
7.34(3h,m),5.28(2h,s)
[0221]
esi
‑
ms(m/z):356(m+h)
+
[0222]
实施例a16 6
‑
苄氧基羰基氨基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0223]
在(4
‑
氨基苯基)氨基甲酸苄酯(2.48g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以浅黄色固体的形式得到标题化合物(1.46g、收率64%)。
[0224]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.9(1h,s),10.3(1h,s),8.92(1h,d,j=2.4hz),8.41(1h,s),8.27
‑
8.24(2h,m),8.09(1h,d,j=9.2hz),7.90(1h,dd,j=9.3,2.3hz),7.59
‑
7.34(8h,m),5.23(2h,s)
[0225]
esi
‑
ms(m/z):399(m+h)
+
[0226]
实施例a17 6
‑
甲基硫
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0227]
在4
‑
甲硫基苯胺(1.42g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、
11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以淡黄色固体的形式得到标题化合物(1.31g、收率78%)。
[0228]1h
‑
nmr(400mhz,dmso
‑
d6)δ:8.50(1h,j=1.8hz),8.48(1h,s),8.28
‑
8.27(2h,m),8.08(1h,d,j=8.5hz),7.77
‑
7.75(1h,m),7.58
‑
7.53(3h,m),2.62(3h,s)
[0229]
esi
‑
ms(m/z):296(m+h)
+
[0230]
实施例a18 7
‑
氟
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0231]
在3
‑
氟苯胺(1.14g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以浅粉色固体的形式得到标题化合物(0.582g、收率38%)。
[0232]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.1(1h,s),8.77(1h,dd,j=9.5,6.4hz),8.47(1h,s),8.32
‑
8.29(2h,m),7.92(1h,dd,j=10.4,2.7hz),7.69
‑
7.54(4h,m)
[0233]
esi
‑
ms(m/z):268(m+h)
+
[0234]
实施例a19 8
‑
氟
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0235]
在2
‑
氟苯胺(1.14g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以淡黄色固体的形式得到标题化合物(0.604g、收率40%)。
[0236]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.56(1h,s),8.49
‑
8.44(1h,m),8.34
‑
8.31(2h,m),7.74
‑
7.68(2h,m),7.63
‑
7.54(3h,m)
[0237]
esi
‑
ms(m/z):268(m+h)
+
[0238]
实施例a20 7
‑
氯
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0239]
在3
‑
氯苯胺(1.30g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以浅粉色固体的形式得到标题化合物(1.02g、收率63%)。
[0240]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.72(1h,d,j=9.2hz),8.51(1h,s),8.32
‑
8.29(2h,m),8.23(1h,d,j=2.1hz),7.76(1h,dd,j=9.2,2.1hz),7.62
‑
7.54(3h,m)
[0241]
esi
‑
ms(m/z):284,286(m+h)
+
[0242]
实施例a21 8
‑
氯
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0243]
在2
‑
氯苯胺(0.522g、4.09mmol)的乙腈(2.3ml)溶液中加入苯甲醛(0.482g、4.54mmol)、三氟化硼
‑
四氢呋喃络合物(125μl、1.14mmol),加热至65℃。10分钟后,用3小时
滴加丙酮酸(0.200g、2.27mmol)的乙腈(3.8ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。通过硅胶柱色谱(氯仿/甲醇)对残渣进行纯化。利用氯仿对所得到的粗产物进行悬浮清洗,在减压下进行干燥后,以浅黄色固体的形式得到标题化合物(0.232g、收率36%)。
[0244]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.61(1h,dd,j=8.5,1.2hz),8.58(1h,s),8.40
‑
8.38(2h,m),8.07(1h,dd,j=7.3,1.2hz),7.68(1h,dd,j=7.3,1.2hz),7.64
‑
7.55(3h,m)
[0245]
esi
‑
ms(m/z):284,286(m+h)
+
[0246]
实施例a22 5,7
‑
二氯
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0247]
在3,5
‑
二氯苯胺(1.66g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以白色固体的形式得到标题化合物(1.40g、收率78%)。
[0248]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.1(1h,s),8.37
‑
8.35(2h,m),8.33(1h,s),8.24(1h,d,j=1.8hz),7.97(1h,d,j=1.8hz),7.62
‑
7.55(3h,m)
[0249]
esi
‑
ms(m/z):318,320(m+h)
+
[0250]
实施例a23 7
‑
甲基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0251]
在间甲苯胺(1.10g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以白色固体的形式得到标题化合物(0.554g、收率37%)。
[0252]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.0(1h,s),8.56(1h,d,j=8.5hz),8.39(1h,s),8.30
‑
8.26(2h,m),7.97(1h,m),7.60
‑
7.51(4h,m),2.56(3h,s)
[0253]
esi
‑
ms(m/z):264(m+h)
+
[0254]
实施例a24 8
‑
甲基
‑2‑
苯基喹啉
‑4‑
羧酸的制造
[0255]
在邻甲苯胺(1.10g、10.2mmol)的乙腈(5.7ml)溶液中加入苯甲醛(1.21g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,利用氯仿对粗产物进行悬浮清洗,在减压下进行干燥后,以浅黄色固体的形式得到标题化合物(0.379g、收率25%)。
[0256]1h
‑
nmr(400mhz,dmso
‑
d6)δ:13.9(1h,s),8.47
‑
8.44(1h,m),8.46(1h,s),8.36
‑
8.32(2h,m),7.72(1h,d,j=6.7hz),7.61
‑
7.51(4h,m),2.85(3h,s)
[0257]
esi
‑
ms(m/z):264(m+h)
+
[0258]
实施例a25 2
‑
(4
‑
溴苯基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0259]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入4
‑
溴苯甲醛(2.10g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥。在滤液中加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入之前得到的固体,利用浓盐酸将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以白色固体的形式得到标题化合物(1.66g、收率71%)。
[0260]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.3(1h,s),8.73(1h,dd,j=2.7,1.2hz),8.62(1h,s),8.31(1h,d,j=9.2hz),8.28(2h,d,j=8.5hz),7.88(1h,dd,j=8.9,2.4hz),7.79(2h,d,j=8.9hz)
[0261]
esi
‑
ms(m/z):412,414(m+h)
+
[0262]
实施例a26 2
‑
(4
‑
硝基苯基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0263]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入4
‑
硝基苯甲醛(1.81g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以淡黄色固体的形式得到标题化合物(1.52g、收率71%)。
[0264]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.3(1h,s),8.76(1h,d,j=1.5hz),8.71(1h,s),8.59(2h,d,j=8.2hz),8.41(2h,d,j=8.2hz),8.36(1h,d,j=9.2hz),7.91(1h,dd,j=9.5,2.7hz)
[0265]
esi
‑
ms(m/z):379(m+h)
+
[0266]
实施例a27 2
‑
(4
‑
甲氧基苯基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0267]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入4
‑
甲氧基苯甲醛(1.55g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以微黄白色固体的形式得到标题化合物(1.57g、收率76%)。
[0268]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.70(1h,dd,j=2.6,1.1hz)8.56(1h,s),8.29(2h,d,j=8.5hz),8.26(1h,d,j=9.2hz),7.83(1h,dd,j=9.2,2.7hz),7.14(2h,d,j=8.9hz),3.87(3h,s)
[0269]
esi
‑
ms(m/z):364(m+h)
+
[0270]
实施例a28 2
‑
(呋喃
‑2‑
基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0271]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入糠醛(1.09g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室
温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用4%碳酸氢钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥。通过硅胶柱色谱(己烷/乙酸乙酯)对所得到的粗产物进行纯化,以微黄白色固体的形式得到标题化合物(0.403g、收率22%)。
[0272]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.73(1h,d,j=1.2hz),8.44(1h,s),8.22(1h,d,j=9.2hz),8.01(1h,d,j=1.2hz),7.84(1h,dd,j=9.2,3.1hz),7.52(1h,d,j=3.1hz),6.78(1h,dd,j=3.4,1.2hz)
[0273]
esi
‑
ms(m/z):324(m+h)
+
[0274]
实施例a29 2
‑
(噻吩
‑2‑
基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0275]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入2
‑
噻吩醛(1.27g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入水,利用甲苯进行萃取。利用水对所得到的有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以黄褐色固体的形式得到标题化合物(1.51g、收率78%)。
[0276]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.3(1h,s),8.66(1h,d,j=1.5hz),8.57(1h,s),8.18(1h,d,j=9.2hz),8.15(1h,dd,j=3.8,1.1hz),7.83(1h,dd,j=5.0,1.1hz),7.83
‑
7.80(1h,m),7.26(1h,dd,j=4.9,3.7hz)
[0277]
esi
‑
ms(m/z):340(m+h)
+
[0278]
实施例a30 2
‑
(萘
‑2‑
基)
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0279]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入2
‑
萘甲醛(1.77g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,滤取析出的固体。利用乙腈进行清洗后,在减压下进行干燥,由此以黄色固体的形式得到标题化合物(1.70g、收率78%)。
[0280]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.92(1h,d,j=1.8hz),8.81(1h,s),8.76(1h,d,j=1.8hz),8.51(1h,dd,j=8.9,1.5hz),8.36(1h,d,j=9.2hz),8.18
‑
8.17(1h,m),8.12(1h,d,j=9.2hz),8.02
‑
8.01(1h,m),7.89(1h,dd,j=9.2,1.8hz),7.63
‑
7.61(2h,m)
[0281]
esi
‑
ms(m/z):384(m+h)
+
[0282]
实施例a31 3
‑
甲基
‑2‑
苯基
‑6‑
三氟甲氧基喹啉
‑4‑
羧酸的制造
[0283]
在4
‑
(三氟甲氧基)苯胺(1.56g、8.82mmol)的乙腈(4.9ml)溶液中加入苯甲醛(1.04g,9.80mmol)、三氟化硼
‑
四氢呋喃络合物(270μl、2.45mmol),加热至65℃。10分钟后,用3小时滴加2
‑
氧代丁酸(0.500g、4.90mmol)的乙腈(8.2ml)溶液。21小时后,将反应液冷却至室温,加入甲苯,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下后,搅拌1小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥。利用氯仿对所得到的粗体进行悬浮清洗,在减压下进行干燥后,以白色固体的形式得到标题化合物(0.640g、收率38%)。
[0284]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.5(1h,s),8.21(1h,d,j=9.2hz),7.80(1h,dd,j=
9.2,2.4hz),7.70
‑
7.68(1h,m),7.64
‑
7.61(2h,m),7.55
‑
7.51(3h,m),2.42(3h,s)
[0285]
esi
‑
ms(m/z):348(m+h)
+
[0286]
实施例a32 2
‑
(叔丁基)
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0287]
在4
‑
(三氟甲氧基)苯胺(1.81g、10.2mmol)的乙腈(5.7ml)溶液中加入新戊醛(0.978g、11.4mmol)、三氟化硼
‑
四氢呋喃络合物(313μl、2.84mmol),加热至65℃。10分钟后,用3小时滴加丙酮酸(0.500g、5.68mmol)的乙腈(9.5ml)溶液。21小时后,将反应液冷却至室温,加入饱和盐水,利用四氢呋喃进行萃取。利用饱和盐水对有机层进行清洗,利用无水硫酸镁进行干燥后,进行过滤,在减压下蒸馏去除溶剂。利用氯仿对所得到的残渣进行悬浮清洗,在减压下进行干燥后,以浅黄色固体的形式得到标题化合物(1.32g、收率74%)。
[0288]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.1(1h,s),8.69(1h,d,j=2.4hz),8.19(1h,d,j=9.2hz),8.18(1h,s),7.80(1h,dd,j=9.2,2.4hz),1.44(9h,s)
[0289]
esi
‑
ms(m/z):314(m+h)
+
[0290]
比较例a1 2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0291]
将苯甲醛(300mg、2.83mmol)、4
‑
(三氟甲氧基)苯胺(551mg、3.11mmol)、丙酮酸(299mg、3.39mmol)加入至水(14ml)中进行搅拌。向其中加入氨基磺酸(8.2mg、0.085mmol)进行加热回流。18小时后,将反应液冷却至室温,利用甲苯进行萃取。利用水对有机层进行清洗后,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下并搅拌1小时。滤取所产生的固体,在减压下进行干燥,以黄色固体的形式得到标题化合物(44mg、收率5%)。
[0292]
比较例a2 2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0293]
在苯甲醛(600mg、5.65mmol)与丙酮酸(299mg、3.39mmol)的乙醇(5ml)溶液中加入4
‑
(三氟甲氧基)苯胺(551mg、3.11mmol)的乙醇(5ml)溶液,进行加热回流。21小时后,将反应液冷却至室温,加入甲苯,利用1mol/l氢氧化钠水溶液进行萃取。在所得到的水层中加入浓盐酸而将ph调整为1以下并搅拌1小时。滤取所产生的粘性固体,在减压下进行干燥,以橙色的非晶固体的形式得到标题化合物(0.285g、收率15%)。
[0294]
实施例a33 n
‑
(5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
基)
‑2‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
甲酰胺的制造
[0295]
在氩气氛围下,将实施例1中得到的2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸(1.00g、3.00mmol)和5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺(0.567g、3.75mmol)加入至四氢呋喃(12ml)的悬浮液中,并加入o
‑
(1h
‑
苯并三唑
‑1‑
基)
‑
n,n,n’,n
’‑
四甲基脲六氟磷酸盐(hbtu)(1.54g、4.05mmol)、n
‑
甲基吗啉(nmm)(0.455g、4.50mmol),在45℃进行搅拌。24小时后,将反应液冷却至室温,加入水进行搅拌。1小时后,滤取所产生的固体,利用水进行清洗后,在减压下进行干燥。在90℃使所得到的粗体(1.63g)溶解于1
‑
丙醇(54ml)中后,进行冰冷,搅拌2小时。滤取所产生的固体,利用经冰冷的1
‑
丙醇进行清洗,将所得到的固体在减压下进行干燥,以白色固体的形式得到标题化合物(1.19g、收率85%)。
[0296]1h
‑
nmr(400mhz,dmso
‑
d6)δ:12.9(1h,s),8.65(1h,s),8.39
‑
8.37(2h,m),8.34(1h,d,j=9.2hz),8.31
‑
8.29(1h,m),8.09(1h,d,j=1.8hz),7.90(1h,dd,j=9.2,1.8hz),7.63
‑
7.58(3h.m),7.33(1h,d,j=3.7hz),6.83(1h,dd,j=3.7,1.8hz)
[0297]
esi
‑
ms(m/z):467(m+h)
+
[0298]
实施例b1 5
‑
(三氟甲氧基)靛红的制造
[0299]
(1)在氩气氛围下,将羰基二咪唑(cdi)(8.42g、51.9mmol)分成4份并用1小时添加至2
‑
(甲氧基亚氨基)乙酸(5.82g、56.5mmol)的乙腈(44ml)溶液中,在室温下进行搅拌。30分钟后,加入4
‑
(三氟甲氧基)苯胺(8.00g、45.2mmol),在室温下进行搅拌。1.5小时后,在反应液中加入水并搅拌30分钟。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以白色固体的形式得到2
‑
(甲氧基亚氨基)
‑
n
‑
(4
‑
(三氟甲氧基)苯基)乙酰胺(11.5g、收率97%、纯度99.5%(hplc))。
[0300]1h
‑
nmr(400mhz,dmso
‑
d6)δ:10.5(1h,s),7.81
‑
7.77(2h,m),7.74(1h,s),7.36(2h,dd,j=9.0,0.8hz),3.99(3h,s)
[0301]
esi
‑
ms(m/z):263(m+h)
+
[0302]
(2)将(1)中得到的2
‑
(甲氧基亚氨基)
‑
n
‑
(4
‑
(三氟甲氧基)苯基)乙酰胺(10.0g、38.1mmol)溶解于浓硫酸(48ml)中,加热至70℃。6.5小时后,将反应液进行冰冷,加入经冰冷的水,搅拌30分钟。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以黄褐色固体的形式得到标题化合物(7.36g、收率83%、纯度98.0%(hplc))。
[0303]1h
‑
nmr(400mhz,dmso
‑
d6)δ:11.2(1h,s),7.61
‑
7.58(1h,m),7.54(1h,d,j=2.1hz),7.00(1h,d,j=8.5hz)
[0304]
esi
‑
ms(m/z):232(m+h)
+
[0305]
比较例b1 5
‑
(三氟甲氧基)靛红的制造
[0306]
(1)在4
‑
(三氟甲氧基)苯胺(1.00g、5.65mmol)的乙腈(51ml)溶液中加入水(5.7ml)、硫酸钠(0.401g、2.82mmol)、水合氯醛(1.49g、9.03mmol)、羟胺盐酸盐(0.628g、9.03mmol),进行加热回流。20小时后,将反应液冷却至室温,在减压下蒸馏去除溶剂。在所得到的残渣中加入水并搅拌2小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以淡黄色固体的形式得到2
‑
(羟基亚氨基)
‑
n
‑
(4
‑
(三氟甲氧基)苯基)乙酰胺(1.09g、收率78%、纯度93.0%(hplc))。
[0307]1h
‑
nmr(400mhz,dmso
‑
d6)δ:12.2(1h,s),10.4(1h,s),7.81
‑
7.79(2h,m),7.65(1h,s),7.34(2h,d,j=7.9hz)
[0308]
esi
‑
ms(m/z):249(m+h)
+
[0309]
(2)将(1)中得到的2
‑
(羟基亚氨基)
‑
n
‑
(4
‑
(三氟甲氧基)苯基)乙酰胺(1.09g、5.12mmol)溶解于浓硫酸(11ml)中,加热至50℃。23小时后,将反应液进行冰冷,加入经冰冷的水,搅拌3小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以黄褐色固体的形式得到标题化合物(0.760g、收率75%、纯度92.2%(hplc))。
[0310]
实施例b2 2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸的制造
[0311]
在5
‑
(三氟甲氧基)靛红(3.00g、13.0mmol)的甲醇(52ml)溶液中加入5mol/l氢氧化钠水溶液(6.5ml、32.4mmol)、苯乙酮(2.34g、19.5mmol),进行加热回流。24小时后,将反应液冷却至室温,在减压下蒸馏去除溶剂。在所得到的残渣中加入甲苯,利用水进行萃取。利用1mol/l氢氧化钠水溶液对有机层进行再萃取,合并水层,利用浓盐酸将ph调整为1以下后,在室温下搅拌2小时。滤取所产生的固体,利用水进行清洗后,在减压下进行干燥,以褐色固体的形式得到标题化合物(3.84g、收率89%)。
[0312]1h
‑
nmr(400mhz,dmso
‑
d6)δ:14.2(1h,s),8.74(1h,d,j=1.5hz),8.33
‑
8.30(3h,
m),7.87(1h,dd,j=9.2,2.7hz),7.63
‑
7.54(3h m)
[0313]
esi
‑
ms(m/z):334(m+h)
+
[0314]
实施例b3 n
‑
(5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
基)
‑2‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
甲酰胺的制造
[0315]
在氩气氛围下,将实施例2中得到的2
‑
苯基
‑6‑
(三氟甲氧基)喹啉
‑4‑
羧酸(3.50g、10.5mmol)和5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺(1.98g、13.1mmol)加入至四氢呋喃(42ml)的悬浮液中,并加入o
‑
(1h
‑
苯并三唑
‑1‑
基)
‑
n,n,n’,n
’‑
四甲基脲六氟磷酸盐(hbtu)(5.38g、14.2mmol)、n
‑
甲基吗啉(nmm)(1.59g、15.8mmol),在45℃进行搅拌。24小时后,将反应液冷却至室温,加入水进行搅拌。1小时后,滤取所产生的固体,利用水进行清洗后,在减压下进行干燥。在90℃使所得到的粗体(5.71g)溶解于1
‑
丙醇(190ml)中后,进行冰冷,搅拌2小时。滤取所产生的固体,利用经冰冷的1
‑
丙醇进行清洗后,在减压下进行干燥,以白色固体的形式得到标题化合物(3.69g、收率75%)。
[0316]1h
‑
nmr(400mhz,dmso
‑
d6)δ:12.9(1h,s),8.65(1h,s),8.39
‑
8.37(2h,m),8.34(1h,d,j=9.2hz),8.31
‑
8.29(1h,m),8.09(1h,d,j=1.8hz),7.90(1h,dd,j=9.2,1.8hz),7.63
‑
7.58(3h,m),7.33(1h,d,j=3.7hz),6.83(1h,dd,j=3.7,1.8hz)
[0317]
esi
‑
ms(m/z):467(m+h)
+
[0318]
实施例c1 5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0319]
将氨基脲盐酸盐(12.2g、109mmol)与乙酸钠(8.97g、109mmol)的水(104ml)溶液进行冰冷,加入糠醛(10.0g、104mmol)、四氢呋喃(thf)(30ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(267ml)、碳酸钾(36.0g、260mmol)、氯胺t三水合物(36.6g、130mmol),升温至室温并进行搅拌。21小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(11.2g、收率71%)的白色固体。
[0320]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.92
‑
7.91(1h,m),7.31(2h,brs),6.99(1h,d,j=3.1hz),6.71(1h,dd,j=3.1,1.8hz)
[0321]
esi
‑
ms(m/z):152(m+h)
+
[0322]
实施例c2 5
‑
苯基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0323]
(实施例c2
‑
1)
[0324]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入苯甲醛(1.11g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(3.67g、13.0mmol),进行加热回流。2.5小时后,追加氯胺t三水合物(1.17g、4.16mmol)。4小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。30分钟后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。使所得到的固体悬浮于水(30ml),加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.800g、收率48%)的白色固体。
[0325]
(实施例c2
‑
2)
[0326]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入苯甲醛(1.11g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。30分钟后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。在滤液中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.13g、收率68%)的白色固体。
[0327]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.81
‑
7.80(2h,m),7.56
‑
7.51(3h,m),7.27(2h,brs)
[0328]
esi
‑
ms(m/z):162(m+h)
+
[0329]
实施例c3 5
‑
(4
‑
甲氧基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0330]
(实施例c3
‑
1)
[0331]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入对茴香醛(1.42g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(3.67g、13.0mmol),在冰冷下搅拌1小时后,在室温下搅拌1小时后,进行加热回流。19小时后,将反应液冷却至室温,加入20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液,滤取析出物。分离滤液的有机层,在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.49g、收率75%)的白色固体。
[0332]
(实施例c3
‑
2)
[0333]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入对茴香醛(1.42g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)和甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.59g、收率80%)的白色固体。
[0334]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.73(2h,d,j=9.2hz),7.13(2h,brs),7.08(2h,d,j=8.5hz),3.82(3h,s)
[0335]
esi
‑
ms(m/z):192(m+h)
+
[0336]
实施例c4 5
‑
(呋喃
‑3‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0337]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入3
‑
糠醛(1.00g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,
在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(3.67g、13.0mmol),在室温下进行搅拌。2.5小时后,追加氯胺t三水合物(1.17g、4.16mmol)。4小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.970g、收率62%)的白色固体。
[0338]1h
‑
nmr(400mhz,dmso
‑
d6)δ:8.24
‑
8.23(1h,m),7.86(1h,t,j=1.8hz),7.15(2h,brs),6.83(1h,d,j=1.8hz)
[0339]
esi
‑
ms(m/z):152(m+h)
+
[0340]
实施例c5 5
‑
(噻吩
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0341]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入噻吩
‑2‑
甲醛(1.17g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(3.67g、13.0mmol),在室温下进行搅拌。2.5小时后,追加氯胺t三水合物(1.17g、4.16mmol)。4小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.895g、收率51%)的白色固体。
[0342]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.76(1h,dd,j=5.2,1.2hz),7.51(1h,dd,j=3.7,1.2hz),7.28(2h,brs),7.20(1h,dd,j=5.2,3.7hz)
[0343]
esi
‑
ms(m/z):168(m+h)
+
[0344]
实施例c6 5
‑
(萘
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0345]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2
‑
萘甲醛(1.63g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(3.67g、13.0mmol),进行加热回流。5小时后,追加氯胺t三水合物(1.17g、4.16mmol)。5.5小时后,将反应液冷却至室温,加入20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液,滤取析出物。分离滤液的有机层,在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.26g、收率57%)的白色固体。
[0346]1h
‑
nmr(400mhz,dmso
‑
d6)δ:8.31(1h,d,j=1.2hz),8.09
‑
8.06(2h,m),8.00
‑
7.99(1h,m),7.95(1h,dd,j=8.5,1.2hz),7.63
‑
7.59(2h,m),7.33(2h,brs)
[0347]
esi
‑
ms(m/z):212(m+h)
+
[0348]
实施例c7(e)
‑5‑
苯乙烯基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0349]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入反式肉桂醛(1.38g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物
(3.67g、13.0mmol),进行加热回流。2.5小时后,追加氯胺t三水合物(1.17g、4.16mmol)。4小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。1小时后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。使所得到的固体悬浮于水(30ml),加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.865g、收率44%)的淡黄色固体。
[0350]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.69
‑
7.68(2h,m),7.43
‑
7.33(3h,m),7.26(2h,brs),7.17(1h,d,j=16.5hz),7.09(1h,d,j=16.5hz)
[0351]
esi
‑
ms(m/z):188(m+h)
+
[0352]
实施例c8 5
‑
乙基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0353]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入丙醛(0.605g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下搅拌1小时后,进行加热回流。2小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上后,在减压下将该溶液进行浓缩。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以白色固体的形式得到标题化合物(0.485g、收率41%)。
[0354]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.81(2h,brs),2.63(2h,q,j=7.7hz),1.17(3h,t,j=7.7hz)
[0355]
esi
‑
ms(m/z):114(m+h)
+
[0356]
实施例c9 5
‑
(叔丁基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0357]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入新戊醛(0.897g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.826g、收率56%)的白色固体。
[0358]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.83(2h,brs),1.27(9h,s)
[0359]
esi
‑
ms(m/z):142(m+h)
+
[0360]
实施例c10 5
‑
环己基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0361]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入环己基甲醛(1.17g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、
31.2mol)和甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.04g、收率60%)的白色固体。
[0362]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.81(2h,brs),2.72
‑
2.69(1h,m),1.92
‑
1.88(2h,m),1.76
‑
1.69(2h,m),1.63
‑
1.61(1h,m),1.47
‑
1.17(5h,m)
[0363]
esi
‑
ms(m/z):168(m+h)
+
[0364]
实施例c11 5
‑
苄基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0365]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2
‑
苯基乙醛(1.25g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下搅拌30分钟后,进行加热回流。2小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.541g、收率30%)的白色固体。
[0366]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.35
‑
7.34(2h,m),7.28
‑
7.26(3h,m),6.89(2h,brs),4.03(2h,s)
[0367]
esi
‑
ms(m/z):176(m+h)
+
[0368]
实施例c12 5
‑
(4
‑
氯苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0369]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入4
‑
氯苯甲醛(1.46g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。30分钟后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。在滤液中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.25g、收率61%)的白色固体。
[0370]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.81
‑
7.78(2h,m),7.62
‑
7.59(2h,m),7.31(2h,brs)
[0371]
esi
‑
ms(m/z):196,198(m+h)
+
[0372]
实施例c13 5
‑
(3
‑
甲氧基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0373]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入3
‑
甲氧基苯甲醛(1.42g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)、甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固
体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.20g、收率60%)的白色固体。
[0374]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.45(1h,t,j=7.9hz),7.38
‑
7.36(1h,m),7.30
‑
7.28(1h,m),7.26(2h,brs),7.11
‑
7.06(1h,m)3.82(3h,s)
[0375]
esi
‑
ms(m/z):192(m+h)
+
[0376]
实施例c14 5
‑
(2
‑
甲氧基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0377]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2
‑
甲氧基苯甲醛(1.42g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)、甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.39g、收率70%)的白色固体。
[0378]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.64(1h,dd,j=7.3,1.8hz),7.53
‑
7.48(1h,m),7.20(1h,d,j=8.5hz),7.12(2h,brs),7.09
‑
7.04(1h,m),3.85(3h,s)
[0379]
esi
‑
ms(m/z):192(m+h)
+
[0380]
实施例c15 5
‑
(2,4
‑
二甲氧基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0381]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2,4
‑
二甲氧基苯甲醛(1.73g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下搅拌1小时后,进行加热回流。2小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。1小时后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。在滤液中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.63g、收率71%)的白色固体。
[0382]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.56(1h,d,j=8.5hz),7.02(2h,brs),6.70(1h,d,j=2.4hz),6.65(1h,dd,j=8.5,2.4hz),3.84(3h,s),3.83(3h,s)
[0383]
esi
‑
ms(m/z):222(m+h)
+
[0384]
实施例c16 5
‑
(对甲苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0385]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入4
‑
甲基苯甲醛(1.25g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。30分钟后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。在滤液中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅
拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.38g、收率76%)的白色固体。
[0386]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.68(2h,d,j=7.9hz),7.34(2h,d,j=7.9hz),7.20(2h,brs),2.36(3h,s)
[0387]
esi
‑
ms(m/z):176(m+h)
+
[0388]
实施例c17 5
‑
(2,3
‑
二氢苯并呋喃
‑5‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0389]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2,3
‑
二氢苯并呋喃
‑5‑
甲醛(1.54g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)、甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.72g、收率81%)的白色固体。
[0390]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.66(1h,d,j=1.8hz),7.53(1h,dd,j=8.5,1.8hz),7.09(2h,s),6.89(1h,d,j=8.5hz),4.60(2h,t,j=8.9hz),3.24(2h,t,j=8.9hz)
[0391]
esi
‑
ms(m/z):204(m+h)
+
[0392]
实施例c18 5
‑
壬基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0393]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入癸醛(1.63g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下搅拌30分钟后,进行加热回流。2小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入甲苯后,利用2mol/l盐酸与30%盐水的4:1的混合液进行清洗。进一步利用饱和碳酸氢钠水溶液对有机层进行清洗后,减压蒸馏去除溶剂。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以白色固体的形式得到标题化合物(0.874g、收率40%)。
[0394]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.81(2h,brs),2.60(2h,t,j=7.3hz),1.62
‑
1.55(2h,m),1.29
‑
1.26(12h,m),0.86(3h,t,j=7.3hz)
[0395]
esi
‑
ms(m/z):212(m+h)
+
[0396]
实施例c19 5
‑
(萘
‑1‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0397]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入1
‑
萘甲醛(1.63g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol),在室温下进行搅拌。30分钟后,滤取所产生的固体,利用四氢呋喃(thf)进行清洗。在滤液中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入之前滤取的固体,进一步加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.19g、收率
54%)的白色固体。
[0398]1h
‑
nmr(400mhz,dmso
‑
d6)δ:9.13(1h,dd,j=8.7,0.9hz),8.10(1h,d,j=8.2hz),8.05(1h,dd,j=7.6,1.4hz),7.98(1h,dd,j=7.3,1.4hz),7.72
‑
7.62(3h,m),7.37(2h,m)
[0399]
esi
‑
ms(m/z):212(m+h)
+
[0400]
实施例c20 5
‑
(2
‑
甲基丙
‑1‑
烯
‑1‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0401]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入3
‑
甲基丁
‑2‑
烯醛(0.876g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。4.5小时后,将反应液进行加热回流。18小时后,加入氯胺t三水合物(1.17g、4.2mmol)。1小时后,将反应液冷却至室温,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.837g、收率58%)的白色固体。
[0402]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.99(2h,brs),5.98
‑
5.97(1h,m),2.06(3h,s),1.91(3h,d,j=1.2hz)
[0403]
esi
‑
ms(m/z):140(m+h)
+
[0404]
实施例c21 5
‑
丙基
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0405]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入丁醛(0.751g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上后,在减压下将该溶液进行浓缩。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以白色固体的形式得到标题化合物(0.626g、收率47%)。
[0406]1h
‑
nmr(400mhz,dmso
‑
d6)δ:6.83(2h,brs),2.59(2h,t,j=7.3hz),1.64
‑
1.60(2h,m),0.92(3h,t,j=7.6hz)
[0407]
esi
‑
ms(m/z):128(m+h)
+
[0408]
实施例c22 5
‑
(4
‑
硝基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0409]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入4
‑
硝基苯甲醛(1.57g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.84g、17.2mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)、甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(0.398g、收率19%)的黄色固体。
[0410]1h
‑
nmr(400mhz,dmso
‑
d6)δ:8.37(2h,d,j=8.5hz),8.03(2h,d,j=8.5hz),7.55
(2h,brs)
[0411]
esi
‑
ms(m/z):207(m+h)
+
[0412]
实施例c23 4
‑
(5
‑
氨基
‑
1,3,4
‑
二唑
‑2‑
基)苯酚的制造
[0413]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入4
‑
羟基苯甲醛(1.27g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上后,在减压下进行浓缩。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以淡黄色固体的形式得到标题化合物(0.353g、收率19%)。
[0414]1h
‑
nmr(400mhz,dmso
‑
d6)δ:10.6(1h,brs),7.62
‑
7.61(2h,m),7.07(2h,brs),6.89
‑
6.88(2h,m)
[0415]
esi
‑
ms(m/z):178(m+h)
+
[0416]
实施例c24 5
‑
(2,6
‑
二甲氧基苯基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0417]
将氨基脲盐酸盐(1.22g、10.9mmol)与乙酸钠(0.897g、10.9mmol)的水(10ml)溶液进行冰冷,加入2,6
‑
二甲氧基苯甲醛(1.73g、10.4mmol)、四氢呋喃(thf)(3.0ml)并进行搅拌。1.5小时后,在反应液中加入四氢呋喃(thf)(27ml)、碳酸钾(3.60g、26.0mmol)、氯胺t三水合物(4.11g、14.6mmol),在室温下进行搅拌。3小时后,利用20%亚硫酸氢钠水溶液与30%盐水的2:1的混合液对反应液进行清洗。在所得到的有机层中加入4mol/l盐酸乙酸乙酯(7.8ml、31.2mol)、甲苯,利用2mol/l盐酸与30%盐水的4:1的混合液进行萃取。在所得到的水层中加入10mol/l氢氧化钠水溶液而将ph调整为12以上。搅拌整夜后,滤取所产生的固体,利用水进行清洗后,进行减压干燥,得到标题化合物(1.39g、收率70%)的淡黄色固体。
[0418]1h
‑
nmr(400mhz,dmso
‑
d6)δ:7.49(1h,dd,j=8.5,7.9hz),6.98(2h,brs),6.76(2h,d,j=8.5hz),3.74(6h,s)
[0419]
esi
‑
ms(m/z):222(m+h)
+
[0420]
比较例c1 5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺的制造
[0421]
通过以往的制造方法制造5
‑
(呋喃
‑2‑
基)
‑
1,3,4
‑
二唑
‑2‑
胺,与本发明的制造方法比较收率。具体而言,在氨基脲盐酸盐(55.8mg、0.500mmol)与乙酸钠(41.0mg、0.500mmol)的水(1.0ml)溶液中加入糠醛(48.0mg、0.500mmol)的甲醇(1.0ml)溶液,在室温下进行搅拌。30分钟后,将反应液进行减压浓缩。使所得到的残渣悬浮于1,4
‑
二烷(5.0ml),加入碳酸钾(207mg、1.50mmol)、碘(152mg、0.600mmol),密封反应容器,加热至80℃。4小时后,将反应液冷却至室温,加入5%硫代硫酸钠水溶液,利用二氯甲烷/甲醇=10/1的溶液进行萃取。利用无水硫酸钠将所得到的有机层进行干燥后,进行过滤,在减压下蒸馏去除溶剂。通过硅胶柱色谱(氯仿/甲醇)对所得到的残渣进行纯化,以白色固体的形式得到标题化合物(12.6mg、收率17%)。
[0422]
以往的制造方法与本发明的制造方法相比,收率低。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1