用于治疗幼年特发性关节炎的抗IL-6受体抗体的制作方法
用于治疗幼年特发性关节炎的抗il-6受体抗体
1.相关申请的交叉引用
2.本技术要求2019年1月31日提交的美国临时申请号62/799,698;2019年5月22日提交的美国临时申请号62/851,474;2019年11月14日提交的美国临时申请号62/935,395;以及2019年12月3日提交的欧洲申请号19306553.9的权益;将其各自通过引用以其整体并入本文。
技术领域
3.本公开文本涉及幼年特发性关节炎的治疗领域,如全身型幼年特发性关节炎和多关节病程幼年特发性关节炎(其包括多关节和扩展性少关节幼年特发性关节炎)。本发明的某些方面涉及白细胞介素-6受体(il-6r)拮抗剂,如抗il-6r抗体,用于治疗全身型幼年特发性关节炎和多关节病程幼年特发性关节炎的用途。
背景技术:
4.幼年特发性关节炎(jia)是儿童最常见的风湿性疾病。国际风湿病学协会联盟(ilar)将jia定义为病因不明的关节炎,其发病于16岁前,并且在排除其他已知病症的情况下持续至少6周(petty,r.e.等人,2001.j rheumatol.2004;31(2):390-2;giannini,e.h.等人,1997arthritis rheum.40(7):1202-9;以及macaubas,c.等人,2009nat rev rheumatol.5(11):616-26)。所述病症包括如由ilar定义的七个亚型,包括多关节病程jia和全身型jia。参见petty,r.e.等人,2001.j rheumatol.2004;31(2):390-2,将其通过引用以其整体并入本文。
5.对jia没有已知的治愈方法。尽管常规治疗可以用于治疗受累及的个体,但仍需要更有效的治疗来治疗持续类型的jia。
技术实现要素:
6.本公开文本尤其提供了在有需要的受试者中治疗jia的方法,其包括给予有效量的特异性结合il-6r的抗体。
7.在一个实施方案中,jia是全身型jia(sjia)。
8.在一个实施方案中,特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列。
9.在各种实施方案中,所述抗体包含重链可变区(vh)和轻链可变区(vl),其中vh包含在seq id no:1序列中发现的三个互补决定区(cdr),并且其中vl包含在seq id no:2序列中发现的三个cdr。在各种实施方案中,抗il-6r抗体或其抗原结合片段包含三个重链互补决定区(hcdr;即hcdr1、hcdr2和hcdr3)和三个轻链互补决定区(lcdr,即lcdr1、lcdr2和lcdr3),其中hcdr1包含seq id no:3的氨基酸序列;hcdr2包含seq id no:4的氨基酸序列;hcdr3包含seq id no:5的氨基酸序列;lcdr1包含seq id no:6的氨基酸序列;lcdr2包含seq id no:7的氨基酸序列;并且lcdr3包含seq id no:8的氨基酸序列。
10.在一个实施方案中,所述抗体是萨瑞鲁单抗。
11.在各种实施方案中,没有其他改善疾病抗风湿药物(disease modifying antirheumatic drug,dmard)与所述抗体一起给予。在一些实施方案中,将至少一种其他dmard给予至受试者。在一个实施方案中,将至少一种其他dmard与抗体一起或并行给予至受试者。
12.在一些实施方案中,受试者患有sjia的至少一种症状,如至少1个关节的关节炎持续至少6周,伴有或之前有持续至少2周的发烧;主要发生在躯干和四肢上的短暂红斑疹;全身淋巴结病;肝肿大和/或脾肿大;多浆膜炎;体重减轻;疲劳;不适;发烧;外周血白细胞(wbc)计数升高(25 000至50 000/ml3);血小板计数增加(例如,》1x106),红细胞沉降率(esr)明显升高,为》100mm/h;贫血;和/或相对于健康受试者的高铁蛋白水平。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
13.如本文所用,与jia相关的“症状”包括与jia相关的任何临床或实验室(例如,诊断)表现,并且不限于受试者能够感觉或观察到的。
14.在各种实施方案中,受试者患有sjia的至少一种症状,如跛行;醒来时僵硬;受试者不愿使用手臂或腿;活动水平降低;每日高峰形热(例如,达到约40℃或至少40℃);关节肿胀;和/或精细运动活动困难。在所述方法的一个实施方案中,在给予所述抗体后,受试者中sjia的至少一种症状得到改善。
15.在所述方法的各种实施方案中,所述抗体改善了至少一个得分或度量,如jia acr(例如,jia acr30、jia acr50、jia acr70、jia acr90和jia acr100)、jia acr组分(例如,患有活动性关节炎的关节的计数、具有有限运动范围的关节的计数、疾病活动性的医师全面评估、急性期反应物如红细胞沉降率或c反应蛋白(crp)、儿童健康评估问卷、患者(或父母)总体健康状况的全面评估)和/或退烧(例如,在首次给予所述抗体时发烧的受试者中)、皮质类固醇(例如,糖皮质激素)使用量自基线的减少和/或幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27)。在所述方法的各种实施方案中,所述改善表征为至少一个得分或度量,如jia acr30、jia acr50、jia acr70、jia acr90和/或jia acr100。在所述方法的各种实施方案中,所述改善表征为至少一个得分或度量,如疾病活动性得分的医师全面评估、总体健康状况的患者或父母评估、儿童健康评估问卷、患有活动性关节炎的关节的数量、活动受限的关节的数量、高敏c反应蛋白和/或退烧(例如,在首次给予所述抗体时发烧的受试者中)。在各种实施方案中,所述改善表征为至少一种生物标记物。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如华莱士标准所定义。华莱士标准被定义为医师全面vas《1/10,无活动性关节炎,无活动性葡萄膜炎,以及crp《10mg/l。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如由幼年关节炎疾病活动性得分-27-crp≤1所定义。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病或低疾病活性,如由幼年关节炎疾病活动性得分-27-crp≤3.8所定义。
16.在所述方法的一个实施方案中,受试者对当前的治疗没有足够的响应,并且被认为是生物类改善疾病抗风湿药物的候选者。
17.在所述方法的一个实施方案中,受试者年龄在约1岁与约17岁之间。在其他实施方案中,受试者的年龄在约4岁与约6岁之间,或者在约12岁与约18岁之间。
18.在所述方法的一个实施方案中,受试者具有5个或更多个活动关节,或者2个活动关节和全身症状(例如发烧)。在所述方法的一个实施方案中,尽管糖皮质激素的剂量稳定了至少3天,受试者在任何连续7天中的至少3天中具有5个或更多个活动关节或者2个或更多个活动关节,以及sjia发烧高于约37.5℃。
[0019]“活动关节”是(i)不是由于畸形导致的关节内肿胀,和/或(ii)伴随疼痛或压痛的活动受限的关节。
[0020]
在所述方法的一个实施方案中,jia是多关节病程jia(pcjia)。在某些实施方案中,pcjia是扩展性少关节jia。在各种实施方案中,pcjia是类风湿因子(rf)阳性多关节jia。在一些实施方案中,pcjia是rf阴性多关节jia。在所述方法的一个实施方案中,受试者的年龄在约12岁与约14岁之间,或者在约7岁与约9岁之间。
[0021]
在一个实施方案中,特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列。
[0022]
在各种实施方案中,所述抗体包含vh和vl,其中vh包含在seq id no:1序列中发现的三个cdr,并且其中vl包含在seq id no:2序列中发现的三个cdr。在各种实施方案中,抗il-6r抗体或其抗原结合片段包含三个hcdr(即hcdr1、hcdr2和hcdr3)和三个lcdr(即lcdr1、lcdr2和lcdr3),其中hcdr1包含seq id no:3的氨基酸序列;hcdr2包含seq id no:4的氨基酸序列;hcdr3包含seq id no:5的氨基酸序列;lcdr1包含seq id no:6的氨基酸序列;lcdr2包含seq id no:7的氨基酸序列;并且lcdr3包含seq id no:8的氨基酸序列。
[0023]
在一个实施方案中,所述抗体是萨瑞鲁单抗。
[0024]
在各种实施方案中,没有其他dmard与所述抗体一起给予。在一些实施方案中,将至少一种其他dmard给予至受试者。在一个实施方案中,将至少一种其他dmard与抗体一起或并行给予至受试者。
[0025]
在一些实施方案中,受试者患有rf阳性多关节jia的至少一种症状。在一些实施方案中,受试者患有rf阳性多关节jia的至少一种症状,如可发展为关节半脱位(例如,在手腕和/或拇指中)的变形性对称性多关节炎;关节挛缩(例如,近端和远端指间、近端指间的骨过度生长和手指畸形,如鹅颈或钮孔状畸形);慢性滑膜炎症;关节软骨丧失和关节旁骨侵蚀;正球性慢性贫血;esr和c反应蛋白升高;白细胞计数;无症状的颈椎关节炎;和/或小颌畸形。在各种实施方案中,受试者具有关节半脱位(例如,在手腕和/或拇指中)和/或关节挛缩(例如,近端和远端指间、近端指间的骨过度生长和手指畸形,如鹅颈或钮孔状畸形)。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。在所述方法的一个实施方案中,受试者年龄在约12岁与约14岁之间。
[0026]
在各种实施方案中,受试者患有rf阴性多关节jia的至少一种症状。在各种实施方案中,受试者患有rf阴性多关节jia的至少一种症状,如伴运动减少的对称性多关节炎、肌肉无力和/或身体机能降低。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。在所述方法的一个实施方案中,受试者年龄在约7岁与约9岁之间。
[0027]
在一个实施方案中,受试者患有扩展性少关节jia的至少一种症状。在各种实施方案中,受试者患有扩展性少关节jia的至少一种症状,如在疾病的最初6个月后累及超过4个
关节(例如,在较大的关节如膝盖、脚踝、手腕中)的无菌性炎性滑膜炎;跛行走动;慢性前葡萄膜炎;膝盖或脚踝慢性关节炎,导致该肢体过度生长,从而导致后续的腿部长度差异;肌肉萎缩(例如,当膝盖受到累及时,在伸肌如股外侧肌、股四头肌中);和/或膝盖或手腕的屈曲挛缩。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
[0028]
在各种实施方案中,受试者患有pcjia的至少一种症状,如跛行;醒来时僵硬;受试者不愿使用手臂或腿;活动水平降低;关节肿胀;和/或精细运动活动困难。
[0029]
在所述方法的一个实施方案中,在给予所述抗体后,受试者中pcjia的至少一种症状得到改善。在所述方法的各种实施方案中,所述改善表征为至少一个得分或度量,如定义为以下核心组变量中的≥3/6有改善且不超过1/6恶化的jia acr30、jia acr50、jia acr70、jia acr90和jia acr 100:(i)疾病活动性的医师全面评估(例如,通过视觉模拟量表(vas)),(ii)总体健康状况的患者或父母评估,(iii)儿童健康评估问卷(例如,通过儿童健康评估问卷残疾指数(chaq-di))、(iv)患有活动性关节炎的关节数量、(v)活动受限的关节数量,和(vi)炎症指数(例如,高敏c反应蛋白)。在一个实施方案中,jia acr30响应是当≥3/6核心组变量自基线改善≥30%且不超过1/6的恶化≥30%时。
[0030]
在所述方法的各种实施方案中,疾病活动性的改善表征为至少一个幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27,其包括4个测量:疾病活动性的医师全面评估、健康状况的父母/患者全面评估、患有活动性疾病的关节的计数和炎症指数(hs-crp或esr水平))。
[0031]
在所述方法的各种实施方案中,所述改善表征为至少一个得分或度量,如疾病活动性的医师全面评估、总体健康状况的患者或父母评估、儿童健康评估问卷、患有活动性关节炎的关节的数量、活动受限的关节的数量、高敏c反应蛋白和/或幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27)。
[0032]
在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如华莱士标准所定义。华莱士标准被定义为医师全面vas《1/10,无活动性关节炎,无活动性葡萄膜炎,以及crp《10mg/l。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如由幼年关节炎疾病活动性得分-27-crp≤1所定义。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病或低疾病活性,如由幼年关节炎疾病活动性得分-27-crp≤3.8所定义。
[0033]
在所述方法的一个实施方案中,受试者年龄在约2岁与约17岁之间。
[0034]
在各种实施方案中,受试者已患有jia至少6个月。在一个实施方案中,受试者已患有关节炎,其在疾病的至少前6个月期间累及了多达4个关节,并且然后在至少前6个月之后发展为累及了5个或更多个关节。在一些实施方案中,受试者已患有关节炎,其在疾病的前6个月期间累及了多达4个关节,并且然后在前6个月之后发展为累及了5个或更多个关节。
[0035]
在所述方法的一个实施方案中,受试者具有至少5个患有活动性关节炎的关节。在所述方法的一个实施方案中,受试者患有在疾病的前6个月期间累及5个或更多个关节的关节炎。在所述方法的一个实施方案中,关节炎是类风湿因子阴性的。可替代地,在一个实施方案中,关节炎是类风湿因子阳性的。
[0036]
在所述方法的一个实施方案中,将所述抗体皮下给予。在所述方法的各种实施方
案中,将所述抗体每周或每两周给予。
[0037]
在所述方法的一个实施方案中,将所述抗体以约2mg/kg至约4mg/kg的剂量给予。在一个实施方案中,将所述剂量每周一次或每2周一次给予。在所述方法的各种实施方案中,将所述抗体以每周约2mg/kg、每周约2.5mg/kg、每两周约2mg/kg、每两周约2.5mg/kg、每两周约3mg/kg和每两周约4mg/kg的剂量使用。在所述方法的各种实施方案中,将所述抗体以表1-5中列出的剂量使用。
[0038]
在所述方法的一些实施方案中,受试者具有小于30kg的体重。
[0039]
在所述方法的一些实施方案中,受试者具有至少约30kg的体重。在所述方法的一些实施方案中,受试者具有小于60kg的体重。
[0040]
在所述方法的一些实施方案中,受试者为1至17岁。
[0041]
在所述方法的一些实施方案中,受试者是人并且所述抗体是人抗体。
[0042]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中将所述抗体以每周或每两周约2mg/kg至约4mg/kg的剂量给予,其中受试者的体重大于或等于10kg且小于或等于60kg。
[0043]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周约2mg/kg至约3mg/kg的剂量或每周约2mg/kg的剂量给予。
[0044]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次约2mg/kg至约3mg/kg的剂量或每周一次约2mg/kg的剂量给予。
[0045]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周约2mg/kg或约3mg/kg的剂量给予。
[0046]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次约2mg/kg或约3mg/kg的剂量给予。
[0047]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周2mg/kg或3mg/kg或者每周2mg/kg的剂量给予。
[0048]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次2mg/kg或3mg/kg或者每周一次2mg/kg的剂量给予。
[0049]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周约2.5mg/kg至约4mg/kg的剂量或每周约2.5mg/kg的剂量给予。
[0050]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次约2.5mg/kg至约4mg/kg的剂量或每周一次约2.5mg/kg的剂量给予。
[0051]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周约2.5mg/kg或约4mg/kg的剂量给予。
[0052]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次约2.5mg/kg或约4mg/kg的剂量给予。
[0053]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周2.5mg/kg或4mg/kg或者每周2.5mg/kg的剂量给予。
[0054]
在所述方法的一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次2.5mg/kg或4mg/kg或者每周一次2.5mg/kg的剂量给予。
[0055]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于30kg且小于33kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的体重大于或等于33kg且小于37.5kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中受试者的体重大于或等于37.5kg且小于42kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中所述受试者的体重大于或等于42kg且小于46.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于46.5kg且小于50.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于50.5kg且小于55kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于55kg且小于59.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于59.5kg且小于64kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于64kg且小于68kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于68kg且小于72.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;或者其中受试者的体重大于或等于72.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予。
[0056]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于30kg且小于31kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于31kg且小于34kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于34kg且小于37kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中所述受试者的体重大于或等于37kg且小于39.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于39.5kg且小于42.5kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于42.5kg且小于45kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于45kg且小于48.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;其中受试者的体重大于或等于48.5kg且小于51.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予;其中受试者的体重大于或等于51.5kg且小于54.5kg,并且将所述抗体以每隔一周一次或每周一次157.5mg的剂量给予;其中受试者的体重大于或等于54.5kg且小于57kg,并且将所述抗体以每隔一周一次或每周一次166.25mg的剂量给予;其中受试者的体重大于或等于57kg且小于63kg,并且将所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于63kg,并且将所述抗体以每隔一周一次或每周一次192.5mg的剂量给予。
[0057]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于10kg且小于12.5kg,并且将所述抗体以每隔一周一次或每周一次26.25mg的剂量给予;其中受试者的体重大于或等于12.5kg且小于16kg,并且将所述抗体以每隔一周一次或每周一次35mg的剂量给予;其中受试者的体重大于或等于16kg且小于19.5kg,并且将所述抗体以每隔一周一次或每周一次43.75mg的剂量给予;其中受试者的体重大于或等于19.5kg且小于23kg,并且将所述抗体以每隔一周一次或每周一次52.5mg的剂量给予;其中受试者的体重大于或等于23kg且小于26.5kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的体重大于或等于26.5kg且小于30kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中受试者的体重大于或等于30kg且小于37.5kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中所述受试者的体重大于或等于37.5kg且小于42kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中受试者的体重大于或等于42kg且小于46.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于46.5kg且小于50.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于50.5kg且小于55kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于55kg且小于59.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于59.5kg且小于64kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于64kg且小于68kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于68kg且小于72.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;或者其中受试者的体重大于或等于72.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予。
[0058]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于10kg且小于12.5kg,并且将所述抗体以每隔一周一次或每周一次43.75mg的剂量给予;其中受试者的体重大于或等于12.5kg且小于14.5kg,并且将所述抗体以每隔一周一次或每周一次52.5mg的剂量给予;其中受试者的体重大于或等于14.5kg且小于16.5kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的体重大于或等于16.5kg且小于19kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中受试者的体重大于或等于19kg且小于21kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中受试者的体重大于或等于21kg且小于23.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于23.5kg且小于25.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中所述受试者的体重大于或等于25.5kg且小于27.5kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于27.5kg且小于30kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于30kg且小于39.5kg,并且
将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于39.5kg且小于42.5kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于42.5kg且小于45kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于45kg且小于48.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;其中受试者的体重大于或等于48.5kg且小于51.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予;其中受试者的体重大于或等于51.5kg且小于54.5kg,并且将所述抗体以每隔一周一次或每周一次157.5mg的剂量给予;其中受试者的体重大于或等于54.5kg且小于57kg,并且将所述抗体以每隔一周一次或每周一次166.25mg的剂量给予;其中受试者的体重大于或等于57kg且小于60.5kg,并且将所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于60.5kg且小于63kg,并且将所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于63kg,并且将所述抗体以每隔一周一次或每周一次192.5mg的剂量给予。
[0059]
本公开文本进一步提供了一种用于在有需要的受试者中治疗sjia的方法,其包括给予有效量的特异性结合il-6r的抗体,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中将所述抗体以每周约2mg/kg至约2.5mg/kg或每两周约3mg/kg至约4mg/kg的剂量给予,其中受试者的体重大于或等于30kg且小于或等于210kg。
[0060]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周约2mg/kg的剂量或每隔一周约3mg/kg的剂量给予。
[0061]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周一次约2mg/kg的剂量或每隔一周一次约3mg/kg的剂量给予。
[0062]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周2mg/kg的剂量或每隔一周3mg/kg的剂量给予。
[0063]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周一次2mg/kg的剂量或每隔一周一次3mg/kg的剂量给予。
[0064]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周约2.5mg/kg的剂量或每隔一周约4mg/kg的剂量给予。
[0065]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周一次约2.5mg/kg的剂量或每隔一周一次约4mg/kg的剂量给予。
[0066]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周2.5mg/kg的剂量或每隔一周4mg/kg的剂量给予。
[0067]
在所述方法的一些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周一次2.5mg/kg的剂量或每隔一周一次4mg/kg的剂量给予。
[0068]
本公开文本示出了一种用于在有需要的受试者中治疗jia的抗体,其中所述抗体特异性结合il-6r。
[0069]
在一个实施方案中,jia是sjia。
[0070]
在一个实施方案中,特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列。
[0071]
在各种实施方案中,所述抗体包含vh和vl,其中vh包含在seq id no:1序列中发现的三个cdr,并且其中vl包含在seq id no:2序列中发现的三个cdr。在各种实施方案中,抗il-6r抗体或其抗原结合片段包含三个hcdr(即hcdr1、hcdr2和hcdr3)和三个lcdr(即lcdr1、lcdr2和lcdr3),其中hcdr1包含seq id no:3的氨基酸序列;hcdr2包含seq id no:4的氨基酸序列;hcdr3包含seq id no:5的氨基酸序列;lcdr1包含seq id no:6的氨基酸序列;lcdr2包含seq id no:7的氨基酸序列;并且lcdr3包含seq id no:8的氨基酸序列。
[0072]
在一个实施方案中,所述抗体是萨瑞鲁单抗。
[0073]
在各种实施方案中,没有其他dmard与所述抗体一起给予。在一些实施方案中,将至少一种其他dmard给予至受试者。在一个实施方案中,将至少一种其他dmard与抗体一起或并行给予至受试者。
[0074]
在一些实施方案中,受试者患有sjia的至少一种症状,如至少1个关节的关节炎持续至少6周,伴有或之前有持续至少2周的发烧;主要发生在躯干和四肢上的短暂红斑疹;全身淋巴结病;肝肿大和/或脾肿大;多浆膜炎;体重减轻;疲劳;不适;发烧;外周血wbc计数升高(25 000至50 000/ml3);血小板计数增加(例如,》1x106),esr明显升高,为》100mm/h;贫血;和/或相对于健康受试者的高铁蛋白水平。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
[0075]
在各种实施方案中,受试者患有sjia的至少一种症状,如跛行;醒来时僵硬;受试者不愿使用手臂或腿;活动水平降低;每日高峰形热(例如,达到约40℃或至少40℃);关节肿胀;和/或精细运动活动困难。在所述抗体的一个实施方案中,在给予所述抗体后,受试者中sjia的至少一种症状得到改善。
[0076]
在各种实施方案中,所述抗体改善了至少一个得分或度量,如jia acr(例如,jia acr30、jia acr50、jia acr70、jia acr90和jia acr100)、jia acr组分(例如,患有活动性关节炎的关节的计数、具有有限运动范围的关节的计数、疾病活动性的医师全面评估、急性期反应物如红细胞沉降率或c反应蛋白、儿童健康评估问卷、患者(或父母)总体健康状况的全面评估)和/或退烧(例如,在首次给予所述抗体时发烧的受试者中)、皮质类固醇(例如,糖皮质激素)使用量自基线的减少和/或幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27)。在所述抗体的各种实施方案中,所述改善表征为至少一个得分或度量,如jia acr30、jia acr50、jia acr70、jia acr90和/或jia acr100。在所述抗体的各种实施方案中,所述改善表征为至少一个得分或度量,如疾病活动性得分的医师全面评估、总体健康状况的患者或父母评估、儿童健康评估问卷、患有活动性关节炎的关节的数量、活动受限的关节的数量、高敏c反应蛋白和/或退烧(例如,在首次给予所述抗体时发烧的受试者中)。在各种实施方案中,所述改善表征为至少一种生物标记物。
[0077]
在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如华莱士标准所定义。华莱士标准被定义为医师全面vas《1/10,无活动性关节炎,无活动性葡萄膜炎,以及crp《10mg/l。在所述方法的各种实施方案中,所述抗体引起临床上无活性的疾病,如由幼年关节炎疾病活动性得分-27-crp≤1所定义。在所述方法的各种实施方案中,所述抗体
引起临床上无活性的疾病或低疾病活性,如由幼年关节炎疾病活动性得分-27-crp≤3.8所定义。
[0078]
在所述用途的一个实施方案中,受试者对当前的治疗没有足够的响应,并且被认为是生物类改善疾病抗风湿药物的候选者。
[0079]
在所述用途的一个实施方案中,受试者年龄在约1岁与约17岁之间。在其他实施方案中,受试者的年龄在约4岁与约6岁之间,或者在约12岁与约18岁之间。
[0080]
在所述用途的一个实施方案中,受试者具有5个或更多个活动关节,或者2个活动关节和全身症状(例如发烧)。在所述抗体的一个实施方案中,尽管糖皮质激素的剂量稳定了至少3天,受试者在任何连续7天中的至少3天中具有5个或更多个活动关节或者2个或更多个活动关节,以及sjia发烧高于约37.5℃。
[0081]
在所述用途的一个实施方案中,jia是pcjia。在某些实施方案中,pcjia是扩展性少关节jia。在各种实施方案中,pcjia是rf阳性多关节jia。在一些实施方案中,pcjia是rf阴性多关节jia。
[0082]
在一个实施方案中,特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列。
[0083]
在各种实施方案中,所述抗体包含vh和vl,其中vh包含在seq id no:1序列中发现的三个cdr,并且其中vl包含在seq id no:2序列中发现的三个cdr。在各种实施方案中,抗il-6r抗体或其抗原结合片段包含三个hcdr(即hcdr1、hcdr2和hcdr3)和三个lcdr(即lcdr1、lcdr2和lcdr3),其中hcdr1包含seq id no:3的氨基酸序列;hcdr2包含seq id no:4的氨基酸序列;hcdr3包含seq id no:5的氨基酸序列;lcdr1包含seq id no:6的氨基酸序列;lcdr2包含seq id no:7的氨基酸序列;并且lcdr3包含seq id no:8的氨基酸序列。
[0084]
在一个实施方案中,所述抗体是萨瑞鲁单抗。
[0085]
在各种实施方案中,没有其他dmard与所述抗体一起给予。在一些实施方案中,将至少一种其他dmard给予至受试者。在一个实施方案中,将至少一种其他dmard与抗体一起或并行给予至受试者。
[0086]
在一些实施方案中,受试者患有rf阳性多关节jia的至少一种症状。在一些实施方案中,受试者患有rf阳性多关节jia的至少一种症状,如可发展为关节半脱位(例如,在手腕和/或拇指中)的变形性对称性多关节炎;关节挛缩(例如,近端和远端指间、近端指间的骨过度生长和手指畸形,如鹅颈或钮孔状畸形);慢性滑膜炎症;关节软骨丧失和关节旁骨侵蚀;正球性慢性贫血;esr和c反应蛋白升高;白细胞计数;无症状的颈椎关节炎;和/或小颌畸形。在各种实施方案中,受试者具有关节半脱位(例如,在手腕和/或拇指中)和/或关节挛缩(例如,近端和远端指间、近端指间的骨过度生长和手指畸形,如鹅颈或钮孔状畸形)。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
[0087]
在各种实施方案中,受试者患有rf阴性多关节jia的至少一种症状。在各种实施方案中,受试者患有rf阴性多关节jia的至少一种症状,如伴运动减少的对称性多关节炎、肌肉无力和/或身体机能降低。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
[0088]
在一个实施方案中,受试者患有扩展性少关节jia的至少一种症状,在一个实施方
案中,受试者患有扩展性少关节jia的至少一种症状,如在疾病的最初6个月后累及超过4个关节(例如,在较大的关节如膝盖、脚踝、手腕中)的无菌性炎性滑膜炎;跛行走动;慢性前葡萄膜炎;膝盖或脚踝慢性关节炎,导致该肢体过度生长,从而导致后续的腿部长度差异;肌肉萎缩(例如,当膝盖受到累及时,在伸肌如股外侧肌、股四头肌中);和/或膝盖或手腕的屈曲挛缩。在一个实施方案中,治疗受试者包括减轻、减缓或停止这些症状的进展,或以其他方式改善这些症状中的任何一种(或其任何组合)。
[0089]
在各种实施方案中,受试者患有pcjia的至少一种症状,如跛行;醒来时僵硬;受试者不愿使用手臂或腿;活动水平降低;关节肿胀;和/或精细运动活动困难。
[0090]
在所述抗体的一个实施方案中,在给予所述抗体后,受试者中pcjia的至少一种症状得到改善。在所述抗体的各种实施方案中,所述改善表征为至少一个得分或度量,如定义为以下核心组变量中的≥3/6有改善且不超过1/6恶化的jia acr30、jia acr50、jia acr70、jia acr90和jia acr 100:(i)疾病活动性的医师全面评估(例如,通过视觉模拟量表(vas)),(ii)总体健康状况的患者或父母评估,(iii)儿童健康评估问卷(例如,通过chaq-di)、(iv)患有活动性关节炎的关节数量、(v)活动受限的关节数量,和(vi)炎症指数(例如,高敏c反应蛋白)。在一个实施方案中,jia acr30响应是当≥3/6核心组变量自基线改善≥30%且不超过1/6的恶化≥30%时。
[0091]
在所述抗体的各种实施方案中,疾病活动性的改善表征为至少一个幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27,其包括4个测量:疾病活动性的医师全面评估、健康状况的父母/患者全面评估、患有活动性疾病的关节的计数和炎症指数(hs-crp或esr水平))。
[0092]
在所述抗体的各种实施方案中,所述改善表征为至少一个得分或度量,如疾病活动性的医师全面评估、总体健康状况的患者或父母评估、儿童健康评估问卷、患有活动性关节炎的关节的数量、活动受限的关节的数量、高敏c反应蛋白和/或幼年关节炎疾病活动性得分(如幼年关节炎疾病活动性得分-27)。
[0093]
在所述抗体的一个实施方案中,受试者年龄在约2岁与约17岁之间。
[0094]
在各种实施方案中,受试者已患有jia至少6个月。在一个实施方案中,受试者已患有关节炎,其在疾病的至少前6个月期间累及了多达4个关节,并且然后在至少前6个月之后发展为累及了5个或更多个关节。在一些实施方案中,受试者已患有关节炎,其在疾病的前6个月期间累及了多达4个关节,并且然后在前6个月之后发展为累及了5个或更多个关节。
[0095]
在所述抗体的一个实施方案中,受试者具有至少5个患有活动性关节炎的关节。在所述抗体的一个实施方案中,受试者患有在疾病的前6个月期间累及5个或更多个关节的关节炎。在所述抗体的一个实施方案中,关节炎是类风湿因子阴性的。可替代地,在一个实施方案中,关节炎是类风湿因子阳性的。
[0096]
在一个实施方案中,将所述抗体皮下给予。在各种实施方案中,将所述抗体每周或每两周给予。
[0097]
在一个实施方案中,将所述抗体以约2mg/kg至约4mg/kg的剂量给予。在一个实施方案中,将所述剂量每周一次或每2周一次给予。在各种实施方案中,将所述抗体以每周约2mg/kg、每周约2.5mg/kg、每两周约2mg/kg、每两周约2.5mg/kg、每两周约3mg/kg和每两周约4mg/kg的剂量使用。在各种实施方案中,将所述抗体以表1-5中列出的剂量使用。
[0098]
在一些实施方案中,受试者具有至少约10kg的体重。在一些实施方案中,受试者具有小于30kg的体重。
[0099]
在一些实施方案中,受试者具有至少约30kg的体重。
[0100]
在一些实施方案中,受试者具有小于60kg的体重。
[0101]
在各种实施方案中,受试者为1至17岁。
[0102]
在某些实施方案中,受试者是人并且所述抗体是人抗体。
[0103]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中将所述抗体以每周或每两周约2mg/kg至约4mg/kg的剂量给予,其中受试者的体重大于或等于10kg且小于或等于60kg。
[0104]
在一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周约2mg/kg至约3mg/kg的剂量或每周约2mg/kg的剂量给予。
[0105]
在某些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次约2mg/kg至约3mg/kg的剂量或每周一次约2mg/kg的剂量给予。
[0106]
在各种实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周约2mg/kg或约3mg/kg的剂量给予。
[0107]
在一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次约2mg/kg或约3mg/kg的剂量给予。
[0108]
在各种实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周2mg/kg或3mg/kg或者每周2mg/kg的剂量给予。
[0109]
在某些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每隔一周一次2mg/kg或3mg/kg或者每周一次2mg/kg的剂量给予。
[0110]
在一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周约2.5mg/kg至约4mg/kg的剂量或每周约2.5mg/kg的剂量给予。
[0111]
在各种实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次约2.5mg/kg至约4mg/kg的剂量或每周一次约2.5mg/kg的剂量给予。
[0112]
在某些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周约2.5mg/kg或约4mg/kg的剂量给予。
[0113]
在一些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次约2.5mg/kg或约4mg/kg的剂量给予。
[0114]
在各种实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周2.5mg/kg或4mg/kg或者每周2.5mg/kg的剂量给予。
[0115]
在某些实施方案中,受试者的体重大于或等于10kg且小于30kg,并且其中将所述抗体以每隔一周一次2.5mg/kg或4mg/kg或者每周一次2.5mg/kg的剂量给予。
[0116]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于30kg且小于33kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的体重大于或等于33kg且小于37.5kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其
中受试者的体重大于或等于37.5kg且小于42kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中所述受试者的体重大于或等于42kg且小于46.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于46.5kg且小于50.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于50.5kg且小于55kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于55kg且小于59.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于59.5kg且小于64kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于64kg且小于68kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于68kg且小于72.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;或者其中受试者的体重大于或等于72.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予。
[0117]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于30kg且小于31kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于31kg且小于34kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于34kg且小于37kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中所述受试者的体重大于或等于37kg且小于39.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于39.5kg且小于42.5kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于42.5kg且小于45kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于45kg且小于48.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;其中受试者的体重大于或等于48.5kg且小于51.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予;其中受试者的体重大于或等于51.5kg且小于54.5kg,并且将所述抗体以每隔一周一次或每周一次157.5mg的剂量给予;其中受试者的体重大于或等于54.5kg且小于57kg,并且将所述抗体以每隔一周一次或每周一次166.25mg的剂量给予;其中受试者的体重大于或等于57kg且小于63kg,并且将所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于63kg,并且将所述抗体以每隔一周一次或每周一次192.5mg的剂量给予。
[0118]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于10kg且小于12.5kg,并且将所述抗体以每隔一周一次或每周一次26.25mg的剂量给予;其中受试者的体重大于或等于12.5kg且小于16kg,并且将所述抗体以每隔一周一次或每周一次35mg的剂量给予;其中受试者的体重大于或等于16kg且小于19.5kg,并且将所述抗体以每隔一周一次或每周一次43.75mg的剂量给予;其中受试者的体重大于或等于19.5kg且小于23kg,并且将所述抗体以每隔一周一次或每周一次52.5mg的剂量给予;其中受试者的体重大于或等于23kg且小于26.5kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的
体重大于或等于26.5kg且小于30kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中受试者的体重大于或等于30kg且小于37.5kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中所述受试者的体重大于或等于37.5kg且小于42kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中受试者的体重大于或等于42kg且小于46.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于46.5kg且小于50.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中受试者的体重大于或等于50.5kg且小于55kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于55kg且小于59.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于59.5kg且小于64kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于64kg且小于68kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于68kg且小于72.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;或者其中受试者的体重大于或等于72.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予。
[0119]
本公开文本进一步提供了一种用于在有需要的受试者中治疗pcjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中受试者的体重大于或等于10kg且小于12.5kg,并且将所述抗体以每隔一周一次或每周一次43.75mg的剂量给予;其中受试者的体重大于或等于12.5kg且小于14.5kg,并且将所述抗体以每隔一周一次或每周一次52.5mg的剂量给予;其中受试者的体重大于或等于14.5kg且小于16.5kg,并且将所述抗体以每隔一周一次或每周一次61.25mg的剂量给予;其中受试者的体重大于或等于16.5kg且小于19kg,并且将所述抗体以每隔一周一次或每周一次70mg的剂量给予;其中受试者的体重大于或等于19kg且小于21kg,并且将所述抗体以每隔一周一次或每周一次78.75mg的剂量给予;其中受试者的体重大于或等于21kg且小于23.5kg,并且将所述抗体以每隔一周一次或每周一次87.5mg的剂量给予;其中受试者的体重大于或等于23.5kg且小于25.5kg,并且将所述抗体以每隔一周一次或每周一次96.25mg的剂量给予;其中所述受试者的体重大于或等于25.5kg且小于27.5kg,并且将所述抗体以每隔一周一次或每周一次105mg的剂量给予;其中受试者的体重大于或等于27.5kg且小于30kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于30kg且小于39.5kg,并且将所述抗体以每隔一周一次或每周一次113.75mg的剂量给予;其中受试者的体重大于或等于39.5kg且小于42.5kg,并且将所述抗体以每隔一周一次或每周一次122.5mg的剂量给予;其中受试者的体重大于或等于42.5kg且小于45kg,并且将所述抗体以每隔一周一次或每周一次131.25mg的剂量给予;其中受试者的体重大于或等于45kg且小于48.5kg,并且将所述抗体以每隔一周一次或每周一次140mg的剂量给予;其中受试者的体重大于或等于48.5kg且小于51.5kg,并且将所述抗体以每隔一周一次或每周一次148.75mg的剂量给予;其中受试者的体重大于或等于51.5kg且小于54.5kg,并且将所述抗体以每隔一周一次或每周一次157.5mg的剂量给予;其中受试者的体重大于或等于54.5kg且小于57kg,并且将所述抗体以每隔一周一次或每周一次166.25mg的剂量给予;其中受试者的体重大于或等于57kg且小于60.5kg,并且将
所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于60.5kg且小于63kg,并且将所述抗体以每隔一周一次或每周一次175mg的剂量给予;其中受试者的体重大于或等于63kg,并且将所述抗体以每隔一周一次或每周一次192.5mg的剂量给予。
[0120]
本公开文本进一步提供了一种用于在有需要的受试者中治疗sjia的抗体,其中所述抗体特异性结合il-6r,其中特异性结合il-6r的抗体包含序列seq id no:1的重链可变区和seq id no:2的轻链可变区序列,其中将所述抗体以每周约2mg/kg至约2.5mg/kg或每两周约3mg/kg至约4mg/kg的剂量给予,其中受试者的体重大于或等于30kg且小于或等于210kg。
[0121]
在一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周约2mg/kg的剂量或每隔一周约3mg/kg的剂量给予。
[0122]
在各种实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周一次约2mg/kg的剂量或每隔一周一次约3mg/kg的剂量给予。
[0123]
在某些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周2mg/kg的剂量或每隔一周3mg/kg的剂量给予。
[0124]
在一些实施方案中,受试者的体重大于或等于30kg且小于或等于60kg,并且其中将所述抗体以每周一次2mg/kg的剂量或每隔一周一次3mg/kg的剂量给予。
[0125]
在各种实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周约2.5mg/kg的剂量或每隔一周约4mg/kg的剂量给予。
[0126]
在某些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周一次约2.5mg/kg的剂量或每隔一周一次约4mg/kg的剂量给予。
[0127]
在一些实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周2.5mg/kg的剂量或每隔一周4mg/kg的剂量给予。
[0128]
在各种实施方案中,受试者的体重大于或等于30kg且小于或等于210kg,并且其中将所述抗体以每周一次2.5mg/kg的剂量或每隔一周一次4mg/kg的剂量给予。
[0129]
在某些实施方案中,所述抗体是药物组合物。在一些实施方案中,是体积小于约1ml的水性溶液。在各种实施方案中,是体积约0.5至1ml的水性溶液。在某些实施方案中,是体积约0.75至1ml的水性溶液。在一些实施方案中,是体积约0.5至0.75ml的水性溶液。在各种实施方案中,是体积约0.15至0.25ml的水性溶液。在某些实施方案中,是体积约0.25至0.5ml的水性溶液。
附图说明
[0130]
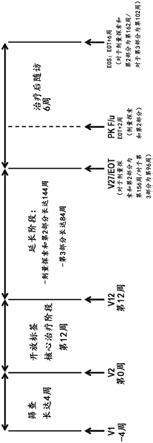
图1是在对当前疗法响应不足或不耐受或被认为是生物类dmard候选者的2至17岁的患有pcjia的儿童和青少年中进行的多国、多中心、开放标签、2阶段和2部分研究的两个剂量群组的研究流程图的图。所述研究的2个阶段是最初的12周核心治疗阶段,随后是144周延长阶段。缩写:eos=研究结束,eot=治疗结束,fda=食品和药物管理局,f/u=随访,imp=研究药物产品,pk=药代动力学,sil-6rα=可溶性白细胞介素6受体α亚基,v=访视,wk=周。
[0131]
注:所有患者必须在治疗完成时(剂量群组1和2在第154周最后一次注射imp,并且
剂量群组3在第155周最后一次注射imp)或在早期永久治疗中断时(无论治疗阶段如何)完成治疗结束(eot)访视(v27,第156周)。
[0132]
对于一名在12周核心治疗阶段期间过早中断研究治疗的患者,需要在eot访视后2周进行另外的pk访视,以采集血样(v88),并在eot访视时测量il-6和总sil-6rα。
[0133]
所有患者必须在eot访视后6周(即对于完成治疗的患者为第162周,并且对于过早中断治疗的患者为eot+6周)完成治疗后随访(v28)。然而,在核心治疗阶段期间过早中断治疗的患者应按照未进行治疗给药到12周而安排的方案继续返回到研究访视(根据fda关于缺失数据的指南)。
[0134]
图2是图1中描述的12周核心治疗阶段的图表研究设计。
[0135]
图3是示出pcjia患者中在首次皮下给予萨瑞鲁单抗后的一段时间内,血清中平均(具有标准偏差;sd)功能性萨瑞鲁单抗浓度的图。图中的缩写包括:q2w=每隔一周一次,qw=每周一次,sc=皮下,sd=标准偏差。低于lloq的治疗后浓度被lloq/2替代。
[0136]
图4是示出pcjia患者中在首次皮下给予萨瑞鲁单抗后的一段时间内,血清中(具有标准偏差;sd)观察到的平均萨瑞鲁单抗谷浓度的图。
[0137]
图5是示出在12周核心治疗阶段期间jia acr30(hs-crp)响应(如在治疗中观察到的)的发生率的图。
[0138]
图6是示出在12周核心治疗阶段期间jia acr30(hs-crp)响应(非响应归责方法)的发生率的图。
[0139]
图7是在对标准疗法响应不足或不耐受并且将接受q2w或qw给予的sc注射萨瑞鲁单抗的1至17岁(或国家规定的年龄要求)的患有sjia的儿童和青少年中进行的多国、多中心、开放标签、顺序的、2阶段研究的两个剂量群组的研究流程图的图。
[0140]
图8是示出患者处置的研究流程图的图。alt=丙氨酸转氨酶;qw=每周;q2w=每2周。
[0141]
图9a-9d是示出如在治疗中观察到的在jadas-27-crp中实现jia acr响应阈值的患者比例和从基线到第12周的平均变化的图。crp=c反应蛋白;jadas-27-crp=使用27-关节计数和crp的幼年关节炎疾病活动性得分;jia acr30/70/90=幼年特发性关节炎美国风湿病学会30/70/90%响应;se=标准误差。
[0142]
图10a-10b是如在治疗中观察到的在核心治疗阶段中的平均crp浓度和具有不可检测到的crp(《0.2mg/l)的患者比例的图。bl=基线;crp=c反应蛋白;se=标准误差。
[0143]
图11a-11c是通过非响应归责计算的实现jia acr应答的患者比例的图。crp=c反应蛋白。jia acr30/70/90=幼年特发性关节炎美国风湿病学会30/70/90%响应。
具体实施方式
[0144]
本公开文本提供了药物组合物和使用这些组合物治疗jia(例如,sjia和pcjia)以及改善所述障碍的至少一种症状的方法。这些组合物包括特异性结合人白细胞介素-6受体(hil-6r)的至少一种抗体。
[0145]
所述抗体治疗jia的功效通常是使用由临床医生和风湿病学家通常使用的本领域标准方法来测量,例如幼年特发性关节炎acr30/50/70/90/100、acr组分、糖皮质激素使用(用于sjia)的变化和幼年关节炎疾病活性得分-27(jadas)参见consolaro等人
development and validation of a composite disease activity score for juvenile idiopathic arthritis.arthritis&rheumatism.2009may;61(5):658-666),将其通过引用以其整体并入本文。jia acr是临床医生熟知的,并且可以由jia诊断和治疗领域的普通技术人员容易地确定。jia acr也称为儿科acr(并且与其是同义词)。与jia acr相关的非限制性描述在giannini等人(1994)“preliminary core of set of outcome variables for use in jra clinical trials”arthritis rheum.37增刊9:s428中提供,将其出于所有目的通过引用并入本文。
[0146]
如在本文的权利要求、发明内容和具体实施方式中所用,术语“约”在定量术语中是指它所修饰的值的加或减10%(如果所述值不可再分,如分子或核苷酸的数目,则上舍入到最接近的整数)。例如,短语“约100mg”将涵盖90mg至110mg,包含端值;短语“约2500mg”将涵盖2250mg至2750mg。当应用于百分比时,术语“约”是指相对于所述百分比的加或减10%。例如,短语“约20%”将涵盖18%-22%,并且“约80%”将涵盖72%-88%,包含端值。此外,在本文中结合定量术语使用“约”的情况下,应理解,除将值加或减10%以外,也涵盖和描述了定量术语的精确值。例如,术语“约23%”明确地考虑、描述并包括精确的23%。
[0147]
应注意,术语“一个/一种(a)”或“一个/一种(an)”实体是指一个/一种或多个/多种所述实体;例如,“一种症状”应理解为代表一种或多种症状。因此,术语“一个/一种(a)”(或“一个/一种(an)”)、“一个/一种或多个/多种”和“至少一个/至少一种”在本文中可以互换使用。
[0148]
此外,在本文中使用时将“和/或”视为对两个指定特征或组分中的每一个与或不与另一者一起的具体公开内容。因此,如在本文中以短语如“a和/或b”使用的术语“和/或”旨在包括“a和b”、“a或b”、“a”(单独)和“b”(单独)。同样地,如以短语如“a、b和/或c”使用的术语“和/或”旨在涵盖以下方面中的每一个:a、b和c;a、b或c;a或c;a或b;b或c;a和c;a和b;b和c;a(单独);b(单独);以及c(单独)。
[0149]
应理解,无论在本文中将多方面用语言“包含”来描述的情况如何,还提供以“由
……
组成”和/或“本质上由
……
组成”措辞描述的其他类似方面。
[0150]
jia
[0151]
关节炎是由医生观察的关节内肿胀,或关节活动范围受限,伴有关节疼痛或压痛,其持续至少6周,并且主要不是由于机械障碍。(petty re等人international league of associations for rheumatology classification of juvenile idiopathic arthritis:第二次修订,edmonton,2001.j rheumatol.2004年2月;31(2):390-2),将其通过引用以其整体并入本文)。
[0152]
jia是一种涉及关节炎症的自身免疫性障碍。这种障碍通常在患者16周岁前出现,并且可能持续到成年。每1000名儿童中有1名受到jia影响。典型症状包括跛行、醒来时僵硬、儿童不愿使用手臂或腿、活动水平降低、持续发烧、关节肿胀和精细运动活动困难。jia包括根据发病年龄、范围和发作后前6个月内的疾病特征分类的7个亚型(例如全身性jia rf阳性多关节jia和rf阴性多关节jia),如ilar在2001年所定义。
[0153]
在sjia中,关节炎症可能不在病患发作时出现,此时突出的体征是关节外的,从而延迟诊断,但最终在大多数患者中以多关节型发展。临床特征(由ilar分类定义)包括关节炎,伴有/之前有持续至少2周的每日发烧和以下中的一种或多种:短暂鲑鱼色红斑疹、全身
淋巴结病、肝/脾肿大和浆膜炎。在某些实施方案中,sjia的排除标准包括银屑病或一级亲属有银屑病;人白细胞抗原-b27(hla-b27)的存在;强直性脊柱炎、肌腱端炎相关关节炎、伴有慢性炎性肠病的骶髂关节炎、一级亲属有莱特尔病;以及至少相隔3个月至少2次出现类风湿因子(rf)。sjia占jia病例的10%,并且已知发生在儿童期和青春期期间的任何年龄。就结果而言,已知患有sjia的患者:50%在第1年减弱;25%患有严重的破坏性关节疾病;一般生长异常;和巨噬细胞活化综合征。
[0154]
sjia发烧典型地是每日峰形热,并且可能伴有以下中的一种或多种:短暂(非固定性)红斑疹、全身淋巴结肿大、肝肿大和/或脾肿大以及浆膜炎。(petty re等人international league of associations for rheumatology classification of juvenile idiopathic arthritis:第二次修订,edmonton,2001.j rheumatol.2004年2月;31(2):390-2)。sjia的发病在4至6岁的患者中具有一个高峰,并且在12至18岁的患者中偶尔可能发生在青春期。
[0155]
如本文所用,术语“多关节病程幼年特发性关节炎”或“pcjia”包括rf阳性多关节jia、rf阴性多关节jia和扩展性少关节jia。
[0156]
多关节jia是pcjia的一个亚型,累及至少5个关节。大关节和小关节两者均可能受累,并且经常呈对称的双侧分布,并经常累及手部承重关节和小关节。轻度发烧可能伴随关节炎。类风湿因子(rf)的存在区分了2种形式的多关节jia:rf阳性和rf阴性多关节jia:
[0157]
类风湿因子(rf)阳性多关节jia占患有jia的所有儿童和青少年的相对较小比例(3%-5%),但它是唯一类似于成人类风湿性关节炎(ra)的jia亚组,所述成人类风湿性关节炎是一种变形性对称性多关节炎,由慢性滑膜炎症、关节软骨丧失和关节旁骨侵蚀引起。rf阳性pcjia的特征是平均发病年龄为12至14岁、明显的女性性别优势(13:1的女性/男性比率)、小关节和大关节的对称受累、多克隆igm rf或抗ccp抗体的产生、遗传易感性(与hla-dr4等位基因相关)、以正球性慢性贫血(网状内皮阻滞)为特征的临床病程、和很少自发减弱的急性期蛋白(升高的esr和c反应蛋白),以及升高的白细胞计数。与坚固的、可移动的和非疼痛性类风湿结节形成的关联是可能的,但很少见。预期实验室的发现比与少关节炎相关的那些更严重。长期后遗症包括关节半脱位(手腕和拇指)、关节挛缩(近端和远端指间、近端指间的骨过度生长、和手指畸形(例如鹅颈或钮孔状畸形)。无症状的颈椎关节炎,伴有伸展性降低,可能导致半脱位(典型地c3上的c2椎骨的半脱位)和后椎骨元件的融合。颞下颌关节的关节炎也可能是无症状的,并导致小颌畸形(petty,r.e.等人,2004.j rheumatol.31(2):390-2)。
[0158]
rf阴性多关节jia比rf阳性多关节jia出现在更小的年龄(在儿童晚期,7至9岁),并且占患有jia的所有儿童和青少年的11%至28%。它可能不像rf阳性疾病那样具有破坏性和持久性,但根据定义,它确实累及5个或更多个关节。rf阴性疾病的放射学改变比rf阳性疾病发生得晚。严重的运动限制通常伴随着肌肉无力和身体机能下降。
[0159]
少关节jia(ojia),为pcjia的另一种亚型,是幼年关节炎最常见的亚型,占美国和西欧患有jia的所有患者的大约50%。发病范围为1至5年,并且高峰在2至3年。它被定义为无菌性炎性滑膜炎,其通常累及多达4个关节(通常是较大的关节,如膝盖、脚踝、手腕),并且与体质调查结果(如发烧、体重减轻、疲劳或全身炎症体征)不相关联。然而,如果在疾病的前6个月之后,大于4个关节受到累及,则被称为扩展性少关节炎,与在疾病的整个过程中
表征为仅最多4个关节的持续性ojia形成对比。尽管跛行走动,但孩子们通常看起来良好。ojia具有发展为慢性前葡萄膜炎的风险,尤其是当抗核抗体(ana)存在并且疾病在幼儿期发病时如此。它通常在发病时无症状,并且需要通过眼科裂隙灯检查进行筛查。膝盖或脚踝的慢性关节炎可能导致该肢体过度生长,从而导致后续的腿部长度差异。肌肉萎缩(通常是伸肌(例如,股外侧肌,当膝盖受到影响时是股四头肌)的肌肉萎缩),和/或膝盖屈曲挛缩,并且较少见的是手腕。
[0160]
当前的jia治疗包括类固醇,例如常规合成dmard和生物类dmard。能够选择性抑制单个分子和途径的新药的开发提供了提高缓解率的希望,同时最小化疾病损害和治疗相关副作用(giancane等人juvenile idiopathic arthritis:diagnosis and treatment.rheumatology and therapy.2016;3(2):187-207)。
[0161]
一种治疗jia的候选方案是白细胞介素-6(il6)抑制。il-6是一种重要的细胞因子,具有广泛的生物活性,包括调节免疫反应性、急性期响应、炎症、肿瘤发生和造血作用(kishimoto等人the cytokine handbook(london:academic出版社).2003,第281-304页)。已经发现il-6的过量产生在慢性炎性疾病中起病理作用。
[0162]
il-6直接与il-6rα亚基相互作用,并且il-6/il-6rα对与糖蛋白130(gp130)亚基形成高亲和力复合物。il-6rα也以可溶性形式存在,其参与反式信号传导,并使il-6r影响不表达il-6rα的细胞,包括关节滑膜细胞(rose-john等人,j leukoc biol.2006;80(2),227-36)。
[0163]
萨瑞鲁单抗,也称为sar153191或regn88,是一种针对il-6受体复合物(il-6rα)α亚基的全人序列的重组igg1κ单克隆抗体。萨瑞鲁单抗是一种有效且特异的il-6信号传导抑制剂。通过以高亲和力结合il-6rα,萨瑞鲁单抗阻断il-6的结合并中断细胞因子介导的信号传导级联。在某些实施方案中,白细胞介素-6是风湿性病症病因中的关键要素,并且抑制其信号传导是萨瑞鲁单抗作用机制的关键部分。在离体测定中,萨瑞鲁单抗并未在相关细胞类型上展现出抗体依赖性细胞毒性(adcc)或补体依赖性细胞毒性(cdc),其中通过荧光激活细胞分选仪(facs)分析验证了萨瑞鲁单抗结合(人类用药委员会,评估报告,2017年4月27日ema/292840/2017,可在https://www.ema.europa.eu/documents/assessment-report/kevzara-epar-public-assessment-report_en.pdf访问)。
[0164]
抗体
[0165]
本公开文本包括向受试者给予特异性结合hil-6r的抗体或其抗原结合片段的方法。如本文所用,术语“hil-6r”意指特异性结合人il-6的人细胞因子受体。在某些实施方案中,向患者给予的抗体特异性结合hil-6r的细胞外结构域。
[0166]
如本文所用,术语“抗体”是指包含通过二硫键相互连接的四条多肽链、两条重(h)链和两条轻(l)链的免疫球蛋白分子,以及其多聚体(例如,igm)。每条重链包含重链可变区(本文缩写为hcvr或vh)和重链恒定区。重链恒定区包含三个结构域,ch1、ch2和ch3。每条轻链包含轻链可变区(在本文中缩写为lcvr或vl)和轻链恒定区。轻链恒定区包含一个结构域(cl1)。vh和vl区可以进一步细分为具有高变性的区域,称为cdr,散布有更保守的区域,称为框架区(fr)。每个vh和vl由三个cdr和四个fr构成,按照以下顺序从氨基末端到羧基末端排列:fr1、cdr1、fr2、cdr2、fr3、cdr3、fr4。在一些实施方案中,抗体(或其抗原结合部分)的fr可以与人种系序列相同,或者可以是天然的或人工修饰的。可以基于两个或更多个cdr的
并排分析来定义氨基酸共有序列。
[0167]
如本文所用,术语“抗体”还包括完整抗体分子的抗原结合片段。如本文所用,术语抗体的“抗原结合部分”、抗体的“抗原结合片段”等包括特异性结合抗原以形成复合物的任何天然存在的、可酶促获得的、合成的或基因工程化的多肽或糖蛋白。抗体的抗原结合片段可以使用任何合适的标准技术,如蛋白水解消化或涉及操纵和表达编码抗体可变结构域和任选恒定结构域的dna的重组基因工程技术,例如从完整抗体分子衍生。这种dna是已知的和/或容易从例如商业来源、dna文库(包括例如噬菌体-抗体文库)获得,或可以合成。dna可以按化学方式或通过使用分子生物学技术进行测序和操纵,例如,以将一个或多个可变结构域和/或恒定结构域排列成合适的构型,或引入密码子,产生半胱氨酸残基,修饰、添加氨基酸或使之缺失等。
[0168]
抗原结合片段的非限制性例子包括:(i)fab片段;(ii)f(ab')2片段;(iii)fd片段;(iv)fv片段;(v)单链fv(scfv)分子;(vi)dab片段;和(vii)由模拟抗体高变区的氨基酸残基组成的最小识别单位(例如,分离的cdr,如cdr3肽),或受限fr3-cdr3-fr4肽。如本文所用,其他工程化分子,如结构域特异性抗体、单结构域抗体、结构域缺失抗体、嵌合抗体、cdr嫁接抗体、双抗体、三抗体、四抗体、微抗体、纳米抗体(例如单价纳米抗体和二价纳米抗体)、小的模块化免疫药物(smip)和鲨鱼可变ignar结构域也涵盖在表述“抗原结合片段”内。
[0169]
抗体的抗原结合片段通常将包含至少一个可变结构域。可变结构域可以具有任何大小或氨基酸组成,并且通常将包含与一个或多个框架序列相邻或同框的至少一个cdr。在其中vh结构域与vl结构域相缔合的抗原结合片段中,vh结构域和vl结构域可以按任何适合的排列相对彼此定位。例如,可变区可以是二聚体并含有vh-vh、vh-vl或vl-vl二聚体。可替代地,抗体的抗原结合片段可以含有单体vh或vl结构域。
[0170]
在某些实施方案中,抗体的抗原结合片段可以含有与至少一个恒定结构域共价连接的至少一个可变结构域。可以在抗体的抗原结合片段内发现的可变结构域和恒定结构域的非限制性示例性构型包括:(i)vh-ch1;(ii)vh-ch2;(iii)vh-ch3;(iv)vh-ch1-ch2;(v)vh-ch1-ch2-ch3;(vi)vh-ch2-ch3;(vii)vh-cl;(viii)vl-ch1;(ix)vl-ch2;(x)vl-ch3;(xi)vl-ch1-ch2;(xii)vl-ch1-ch2-ch3;(xiii)vl-ch2-ch3;和(xiv)vl-cl。在可变结构域和恒定结构域的任何构型中,包括上文列出的任何示例性构型,可变结构域和恒定结构域可以彼此直接连接或可以通过完整或部分铰链或接头区连接。在各种实施方案中,铰链区可以由至少2个(例如,5、10、15、20、40、60或更多个)氨基酸组成,其导致单个多肽分子中相邻可变结构域和/或恒定结构域之间的柔性或半柔性连接。此外,在各种实施方案中,抗体的抗原结合片段可以包含上文列出的任何可变结构域和恒定结构域构型的同型二聚体或异型二聚体(或其他多聚体),彼此非共价缔合和/或与一个或多个单体vh或vl结构域非共价缔合(例如,通过二硫键)。
[0171]
在具体实施方案中,用于本发明方法的抗体或抗体片段可以是多特异性抗体,其可以对一种靶多肽的不同表位具有特异性,或者可以含有对多于一种靶多肽的表位具有特异性的抗原结合结构域。可以用于本发明情形中的示例性双特异性抗体形式涉及使用第一免疫球蛋白(ig)c
h3
结构域和第二ig c
h3
结构域,其中所述第一ig c
h3
结构域和第二ig c
h3
结构域彼此相差至少一个氨基酸,并且其中至少一个氨基酸差异与缺乏所述氨基酸差异的双
特异性抗体相比降低了所述双特异性抗体与蛋白a的结合。在一个实施方案中,第一ig c
h3
结构域结合蛋白a并且第二ig c
h3
结构域含有减少或消除蛋白a结合的突变,如h95r修饰(根据imgt外显子编号;根据eu编号为h435r)。第二c
h3
可以进一步包含y96f修饰(根据imgt;根据eu为y436f)。在igg1抗体的情况下,可以在第二c
h3
中发现的其他修饰包括:d16e、l18m、n44s、k52n、v57m和v82i(根据imgt;根据eu为d356e、l358m、n384s、k392n、v397m和v422i);在igg2抗体的情况下,则为n44s、k52n和v82i(根据imgt;根据eu为n384s、k392n和v422i);并且在igg4抗体的情况下,则为q15r、n44s、k52n、v57m、r69k、e79q和v82i(根据imgt;根据eu为q355r、n384s、k392n、v397m、r409k、e419q和v422i)。上述双特异性抗体形式的变异被涵盖在本发明的范围内。在各种实施方案中,任何多特异性抗体形式,包括本文公开的示例性双特异性抗体形式,可以使用本领域中可利用的常规技术来改编,使其适用于抗il-6r抗体的抗原结合片段情形。
[0172]
如与对应种系序列相比,本文公开的全人抗il-6r抗体可以包含重链和轻链可变结构域的框架和/或cdr区中的一个或多个氨基酸取代、插入和/或缺失。通过将本文公开的氨基酸序列与可从例如公共抗体序列数据库获得的种系序列相比较,可以容易地确定此类突变。本发明包括来源于本文公开的任何氨基酸序列的抗体及其抗原结合片段,其中一个或多个框架和/或cdr区内的一个或多个氨基酸回复突变至一个或多个对应种系残基或者一个或多个对应种系残基的保守氨基酸取代(天然或非天然)(此类序列变化在本文中称为“种系回复突变”)。本领域普通技术人员可以容易地从本文公开的重链和轻链可变区序列开始产生包括一个或多个单独种系回复突变或其组合的许多抗体和抗原结合片段。在某些实施方案中,vh和/或vl结构域内的所有框架残基和/或cdr残基回复突变到种系序列。在其他实施方案中,仅将某些残基回复突变到种系序列,例如,仅在fr1的前8个氨基酸内或在fr4的最后8个氨基酸内发现的突变残基,或仅在cdr1、cdr2或cdr3中发现的突变残基。此外,本发明的抗体可以含有框架和/或cdr区内的两个或更多个种系回复突变的任何组合,即,其中某些单独残基回复突变到种系序列,同时保留不同于种系序列的某些其他残基。一旦获得,则可以容易地测试含有一个或多个种系回复突变的抗体和抗原结合片段的一种或多种所希望特性,如改善的结合特异性、增加的结合亲和力、改进或增强的拮抗或激动生物学特性(视情况而定)、降低的免疫原性等。以这种一般方式获得的抗体和抗原结合片段涵盖在本发明内。
[0173]
抗体的恒定区在抗体固定补体和介导细胞依赖性细胞毒性的能力上是重要的。因此,可以基于对于抗体来说介导细胞毒性是否是希望的来选择抗体的同种型。
[0174]
如本文所用,术语“人抗体”旨在包括具有来源于人种系免疫球蛋白序列的可变区和恒定区的抗体。尽管如此,在各种实施方案中,本公开文本中表征的人抗体(例如在cdr中,并且在一些实施方案中是在cdr3中)包括不由人种系免疫球蛋白序列编码的氨基酸残基(例如,通过体外随机或位点特异性诱变或通过体内体细胞突变引入的突变)。然而,如本文所用,术语“人抗体”不旨在包括其中来源于另一种哺乳动物物种(如小鼠)的种系的cdr序列已经被移植到人框架序列上的抗体。
[0175]
如本文所用,术语“重组人抗体”旨在包括通过重组方式制备、表达、产生或分离的所有人抗体,如使用转染到宿主细胞中的重组表达载体表达的抗体(下文进一步描述),从重组的组合人抗体文库(下文进一步描述)分离的抗体,从针对人免疫球蛋白基因为转基因
的动物(例如小鼠)分离的抗体(参见例如,taylor等人(1992)nucl.acids res.20:6287-6295,将其通过引用以其整体并入本文),或通过涉及将人免疫球蛋白基因序列剪接到其他dna序列的任何其他方式制备、表达、产生或分离的抗体。此类重组人抗体具有来源于人种系免疫球蛋白序列的可变区和恒定区。然而,在某些实施例中,对此类重组人类抗体进行体外诱变(或者,当使用针对人类ig序列转基因的动物时,进行体内体细胞诱变),从而尽管重组抗体的vh和vl区的氨基酸序列源自人类种系vh和vl序列且与其相关,但是重组抗体的vh和vl区的氨基酸序列为可能不是天然存在于体内人类抗体种系库内的序列。
[0176]
人抗体可以以与铰链异质性相关的两种形式存在。在一个实施方案中,免疫球蛋白分子包含大约150-160kda的稳定四链构建体,其中二聚体通过链间重链二硫键保持在一起。在另一个实施方案中,二聚体不经由链间二硫键连接,并且形成约75-80kda的分子,其由共价偶联的轻链和重链(半抗体)构成。这些实施方案/形式极难以分离,即使在亲和纯化后也是如此。
[0177]
在各完整igg同种型中第二种形式出现的频率是归因于但不限于与抗体的铰链区同种型相关的结构差异。人igg4铰链的铰链区中的单个氨基酸取代可以将第二种形式的出现率(angal等人,(1993)molecular immunology 30:105,将其通过引用以其整体并入)显著降低至通常使用人igg1铰链观察到的水平。在各种实施方案中,本公开文本涵盖在铰链区、ch2区或ch3区中具有一个或多个突变的抗体,所述突变例如在制造上可能是希望的,以改善所希望抗体形式的产率。
[0178]
如本文所用,“分离的抗体”意指已被鉴定并从它的天然环境的至少一种组分分离和/或回收的抗体。例如,已从生物体的至少一种组分或从其中天然存在或天然产生抗体的组织或细胞中分离或除去的抗体是“分离的抗体”。在各种实施方案中,分离的抗体还包括重组细胞内的原位抗体。在其他实施方案中,分离的抗体是已经受至少一个纯化或分离步骤的抗体。在各种实施方案中,分离的抗体可以基本上不含其他细胞材料和/或化学品。
[0179]
术语“特异性结合”等意指抗体或其抗原结合片段与抗原形成在生理条件下相对稳定的复合物。用于确定抗体是否特异性结合抗原的方法是本领域熟知的,并且包括例如平衡透析、表面等离子体共振等。例如,如本文所用,“特异性结合”il-6r的抗体包括结合il-6r的抗体或其部分,其kd小于约1000nm、小于约500nm、小于约300nm、小于约200nm、小于约100nm、小于约90nm、小于约80nm、小于约70nm、小于约60nm、小于约50nm、小于约40nm、小于约30nm、小于约20nm、小于约10nm、小于约5nm、小于约4nm、小于约3nm、小于约2nm、小于约1nm或约0.5nm,如在表面等离子体共振测定中所测量。特异性结合还可以表征为解离常数为至少约1x10-6 m或更小。在其他实施方案中,解离常数为至少约1x10-7 m、1x10-8 m或1x10-9 m。然而,特异性结合人il-6r的分离的抗体可以具有与其他抗原如来自其他(非人)物种的il-6r分子的交叉反应性。
[0180]
如本文所用,术语“表面等离子体共振”是指一种光学现象,它允许通过例如使用biacore系统(biacore life sciences division of ge healthcare,皮斯卡塔韦,新泽西州)检测生物传感器矩阵内蛋白质浓度的改变来分析实时相互作用。
[0181]
如本文所用,术语“kd”旨在是指抗体-抗原相互作用的平衡解离常数。
[0182]
术语“表位”是指与抗体分子可变区中称为互补位(paratope)的特异性抗原结合位点相互作用的抗原决定簇。单一抗原可以具有多于一个表位。因此,不同的抗体可以结合
抗原上的不同区域并且可以具有不同的生物效应。表位可以是构象的或线性的。通过来自线性多肽链的不同区段的空间并列氨基酸产生构象表位。线性表位是由多肽链中的相邻氨基酸残基产生的表位。在某些情况下,表位可包括抗原上的糖、磷酰基基团或磺酰基基团的部分。
[0183]
在各种实施方案中,与衍生所述抗体的对应种系序列相比,可用于本文所述方法的抗il-6r抗体可以在重链和轻链可变结构域的框架和/或cdr区中包含一个或多个氨基酸取代、插入和/或缺失。通过将本文公开的氨基酸序列与可从例如公共抗体序列数据库获得的种系序列相比较,可以容易地确定此类突变。在各种实施方案中,本公开文本包括涉及使用抗体及其抗原结合片段的方法,所述抗体及其抗原结合片段衍生自本文公开的任何氨基酸序列,其中一个或多个框架和/或cdr区内的一个或多个氨基酸突变为衍生所述抗体的种系序列的一个或多个对应残基,或另一个人种系序列的一个或多个对应残基,或所述一个或多个对应种系残基的保守氨基酸取代(此类序列变化在本文中统称为“种系突变”)。可以构建许多抗体和抗原结合片段,其包含一个或多个单独种系突变或其组合。在某些实施方案中,vh和/或vl结构域内的所有框架残基和/或cdr残基回复突变到衍生所述抗体的原始种系序列中发现的残基。在其他实施方案中,仅将某些残基回复突变到原始种系序列,例如,仅在fr1的前8个氨基酸内或在fr4的最后8个氨基酸内发现的突变残基,或仅在cdr1、cdr2或cdr3中发现的突变残基。在其他实施方案中,一个或多个框架和/或cdr残基中的一个或多个突变成不同种系序列(即与最初衍生所述抗体的种系序列不同的种系序列)的一个或多个对应残基。此外,所述抗体可以含有所述框架和/或cdr区内的两个或更多个种系突变的任何组合,例如,其中某些单独残基突变为某些种系序列的对应残基,同时保留不同于原始种系序列的某些其他残基或使之突变为不同种系序列的对应残基。一旦获得,则可以容易地测试含有一个或多个种系突变的抗体和抗原结合片段的一种或多种所希望特性,如改善的结合特异性、增加的结合亲和力、改进或增强的拮抗或激动生物学特性(视情况而定)、降低的免疫原性等。本公开文本涵盖使用以这种一般方式获得的抗体和抗原结合片段。
[0184]
本公开文本还包括涉及使用抗il-6r抗体的方法,所述抗il-6r抗体包含具有一个或多个保守取代的本文公开的任何hcvr、lcvr和/或cdr氨基酸序列的变体。例如,本公开文本包括使用具有hcvr、lcvr和/或cdr氨基酸序列的抗il-6r抗体,其具有例如相对于本文公开的任何hcvr、lcvr和/或cdr氨基酸序列的10个或更少、8个或更少、6个或更少、4个或更少等保守氨基酸取代。
[0185]
根据本公开文本,在各种实施方案中,抗il-6r抗体或其抗原结合片段包含hcvr、lcvr和/或cdr,所述hcvr、lcvr和/或cdr包含美国专利号7,582,298(将其通过引用以其整体并入本文)中所述的抗il-6r抗体的任何氨基酸序列。在某些实施方案中,抗il-6r抗体或其抗原结合片段包含含有seq id no:1的氨基酸序列的hcvr的hcdr和含有seq id no:2的氨基酸序列的lcvr的lcdr。根据某些实施方案,抗il-6r抗体或其抗原结合片段包含三个hcdr(即hcdr1、hcdr2和hcdr3)和三个lcdr(即lcdr1、lcdr2和lcdr3),其中hcdr1包含seq id no:3的氨基酸序列;hcdr2包含seq id no:4的氨基酸序列;hcdr3包含seq id no:5的氨基酸序列;lcdr1包含seq id no:6的氨基酸序列;lcdr2包含seq id no:7的氨基酸序列;并且lcdr3包含seq id no:8的氨基酸序列。在又其他实施方案中,抗il-6r抗体或其抗原结合
片段包含含有seq id no:1的氨基酸序列的hcvr和包含seq id no:2的氨基酸序列的lcvr。
[0186]
在另一个实施方案中,抗il-6r抗体或其抗原结合片段包含含有seq id no:9的氨基酸序列的重链和含有seq id no:10的氨基酸序列的轻链。在一些实施方案中,hil-6r的细胞外结构域包含seq id no:11的氨基酸序列。根据某些示例性实施方案,本公开文本的方法包括使用本领域中称作和已知为萨瑞鲁单抗的抗il-6r抗体或其生物等效物。
[0187]
seq id no:1的氨基酸序列是
[0188]
evqlvesggglvqpgrslrlscaasrftfddyamhwvrqapgkglewvsgiswnsgrigyadsvkgrftisrdnaenslflqmnglraedtalyycakgrdsfdiwgqgtmvtvss。
[0189]
seq id no:2的氨基酸序列是
[0190]
diqmtqspssvsasvgdrvtitcrasqgisswlawyqqkpgkapklliygasslesgvpsrfsgsgsgtdftltisslqpedfasyycqqansfpytfgqgtkleik。
[0191]
seq id no:3的氨基酸序列是rftfddya。
[0192]
seq id no:4的氨基酸序列是iswnsgri。
[0193]
seq id no:5的氨基酸序列是akgrdsfdi。
[0194]
seq id no:6的氨基酸序列是qgissw。
[0195]
seq id no:7的氨基酸序列是gas。
[0196]
seq id no:8的氨基酸序列是qqansfpyt。
[0197]
seq id no:9的氨基酸序列是
[0198]
evqlvesggglvqpgrslrlscaasrftfddyamhwvrqapgkglewvsgiswnsgrigyadsvkgrftisrdnaenslflqmnglraedtalyycakgrdsfdiwgqgtmvtvssastkgpsvfplapsskstsggtaalgclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslssvvtvpssslgtqtyicnvnhkpsntkvdkkvepkscdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspgk。
[0199]
seq id no:10的氨基酸序列是
[0200]
diqmtqspssvsasvgdrvtitcrasqgisswlawyqqkpgkapklliygasslesgvpsrfsgsgsgtdftltisslqpedfasyycqqansfpytfgqgtkleikrtvaapsvfifppsdeqlksgtasvvcllnnfypreakvqwkvdnalqsgnsqesvteqdskdstyslsstltlskadyekhkvyacevthqglsspvtksfnrgec。
[0201]
seq id no:11的序列是
[0202]
mvavgcallaallaapgaalaprrcpaqevargvltslpgdsvtltcpgvepednatvhwvlrkpaagshpsrwagmgrrlllrsvqlhdsgnyscyragrpagtvhllvdvppeepqlscfrksplsnvvcewgprstpslttkavllvrkfqnspaedfqepcqysqesqkfscqlavpegdssfyivsmcvassvgskfsktqtfqgcgilqpdppanitvtavarnprwlsvtwqdphswnssfyrlrfelryraersktfttwmvkdlqhhcvihdawsglrhvvqlraqeefgqgewsewspeamgtpwtesrsppaenevstpmqalttnkdddnilfrdsanatslpvqd。
[0203]
如本文所用的术语“生物等效物”是指在相同摩尔剂量和相似条件下(例如,相同的给予途径)给予后具有相似生物利用度(利用度的速率和程度),使得可以预期在功效和安全性两个方面的效果与对比分子本质上相同的分子。如果包含抗il-6r抗体的两种药物组合物在药学上等效,则它们是生物等效的,这意味着它们含有相同量的活性成分(例如il-6r抗体),以相同的剂型,用于相同的给予途径,并满足相同或相当的标准。生物等效性
可以例如通过比较两种组合物的药代动力学参数的体内研究来确定。生物等效性研究中常用的参数包括峰值血浆浓度(cmax)和血浆药物浓度时间曲线下面积(auc)。
[0204]
在某些实施方案中,本公开文本涉及包括向受试者给予抗体的方法,所述抗体包含含有序列seq id no:1的重链可变区和含有序列seq id no:2的轻链可变区。
[0205]
本公开文本提供了包含这种抗体的药物组合物,以及使用这些组合物的方法。
[0206]
在各种实施方案中,所述抗体包含含有序列seq id no:1的重链可变区和含有序列seq id no:2的轻链可变区,是特异性结合hil-6r的抗体。参见国际公开号wo 2007/143168,将其通过引用以其整体并入本文。在一个实施方案中,所述抗体包含含有序列seq id no:9的重链可变区和含有序列seq id no:10的轻链可变区。在各种实施方案中,所述抗体是萨瑞鲁单抗。
[0207]
dmard
[0208]
dmard是通过其在类风湿性关节炎和jia中使用来减慢疾病进展定义的药物。
[0209]
dmard已被分类为合成的(sdmard)和生物的(bdmard)。sdmard非穷尽地包括甲氨蝶呤、柳氮磺胺吡啶、来氟米特和羟氯喹。bdmard非穷尽地包括阿达木单抗、戈利木单抗、依那西普、阿巴西普、英利昔单抗、利妥昔单抗和托珠单抗。
[0210]
在一些实施方案中,没有其他dmard与所述抗体一起给予。在一些实施方案中,将至少一种其他dmard给予至受试者。在一个实施方案中,将至少一种其他dmard与抗体一起或并行给予至受试者。
[0211]
在一些实施方案中,受试者对当前的jia治疗没有足够的响应。在一些实施方案中,受试者对当前的jia治疗没有足够的响应,并且被认为是bdmard的候选者。在一些实施方案中,受试者的当前治疗是皮质类固醇、sdmard和/或bdmard。举例来说,但不作为限制,在一些实施方案中,皮质类固醇选自泼尼松、泼尼松龙和甲基泼尼松龙。
[0212]
给予方法和配制品
[0213]
本文所述的方法包括向受试者给予治疗有效量的抗il-6r抗体。如本文所用,“有效量”或“治疗有效量”是治疗剂的导致治疗sjia和/或pcjia的剂量。如本文所用,“治疗”是指引起与sjia和/或pcjia相关的一种或多种症状的可检测的改善,或引起与产生病症或者一种或多种症状的一个或多个潜在病理机制相关的生物效应(例如,特定生物标记物水平的降低)。例如,引起与幼年特发性关节炎相关的任何以下症状或病症改善的il-6r抗体剂量被视为是“治疗有效量”:跛行、醒来时僵硬、受试者不愿使用手臂或腿、活动水平降低、持续发烧、关节肿胀和精细运动活动困难。
[0214]
在各种实施方案中,jia相关症状的“改善”是指jia症状发生率的降低,这可能与一种或多种jia相关测试、得分或度量(如本文所述)的改善相关。例如,所述改善可能与以下相关:jia acr标准中的一者或多者从基线的增加,和/或皮质类固醇使用、幼年关节炎疾病活动性得分或发烧中的一者或多者从基线的降低。在一个实施方案中,改善可以包括僵硬(例如,活动受限的关节)从基线的降低。如本文所用,术语“基线”,关于jia相关参数,意指在给予本发明抗体之前或之时患者的jia相关参数的数值。还可以使用本文所述的至少一种测试、得分或度量来检测可检测的“改善”。在各种实施方案中,所述改善是使用选自以下的至少一种来检测:幼年特发性关节炎美国风湿病学会(acr)(例如,jia acr30、jia acr50、jia acr70、jia acr90和jia acr100)。在各种实施方案中,所述改善表征为至少一
个得分或度量,如疾病活动性得分的医师全面评估、总体健康状况的患者或父母评估、儿童健康评估问卷、患有活动性关节炎的关节的数量、活动受限的关节的数量和/或高敏c反应蛋白。在某些实施方案中,sjia症状的改善是退烧(例如,在首次给予所述抗体时发烧的受试者中)。在各种实施方案中,所述改善表征为至少一种生物标记物。在一些实施方案中,所述改善表征为至少一种生物标记物增加。在一些实施方案中,所述改善表征为至少一种生物标记物减少。
[0215]
在另一个例子中,当一定剂量的抗il-6r抗体没有引起与jia相关的一个或多个参数或症状的可检测的改善,或者没有引起与产生jia的病症或者一种或多种症状的一个或多个潜在病理机制相关的生物效应时,治疗没有效果。
[0216]
根据这些实施方案中的一些,将il-6r抗体皮下给予。根据这些实施方案中的一些,il-6r抗体是萨瑞鲁单抗。
[0217]
根据本发明的方法,向受试者给予的抗il-6r抗体的治疗有效量将根据受试者的年龄和大小(例如体重或体表面积)以及给予途径和本领域普通技术人员熟知的其他因素而变化。
[0218]
在某些实施方案中,所述抗体的剂量根据受试者的体重而变化。在各种实施方案中,如果受试者具有至少30kg(例如,高达60kg、70kg、80kg、90kg或100kg)的体重,则每2周一次向受试者给予2.0mg/kg的剂量。在其他实施方案中,如果受试者具有至少30kg且少于40、50、60、70、80、90或100kg的体重,则每2周一次向受试者给予2.0mg/kg的剂量。在一些实施方案中,如果受试者具有至少30kg的体重,则每2周一次向受试者给予3.0mg/kg的剂量。在一些实施方案中,如果受试者具有至少30kg的体重,则每2周一次向受试者给予4-6mg/kg的剂量。在其他实施方案中,如果受试者具有至少30kg且少于40、50、60、70、80、90或100kg的体重,则每2周一次向受试者给予3.0mg/kg的剂量。在某些实施方案中,如果受试者具有至少30kg的体重,则每周一次向受试者给予2.0mg/kg的剂量。在其他实施方案中,如果受试者具有至少30kg且少于40、50、60、70、80、90或100kg的体重,则每周一次向受试者给予2.0mg/kg的剂量。在各种实施方案中,如果受试者具有10-《30kg的体重,则每2周向受试者给予2.5mg/kg的剂量。在某些实施方案中,如果受试者具有10-《30kg的体重,则每2周向受试者给予4mg/kg的剂量。在一些实施方案中,如果受试者具有10-《30kg的体重,则每周向受试者给予2.5mg/kg的剂量。在各种实施方案中,如果受试者具有10-《30kg的体重,则每2周向受试者给予5-7mg/kg的剂量。在各种实施方案中,如果受试者具有10-《30kg的体重,则每2周向受试者给予4mg/kg的剂量。
[0219]
在某些实施方案中,向受试者给予的抗il-6r抗体的剂量是约10mg至约600mg。在一些实施方案中,向受试者给予的所述抗体的剂量是约25mg至约200mg。在各种实施方案中,向受试者给予的所述抗体的剂量是约60mg至约200mg。例如,本发明包括(但不限于)其中每周或每两周一次向患者给予约10mg、约15mg、约20mg、约25mg、约30mg、约35mg、约40mg、约45mg、约50mg、约55mg、约60mg、约65mg、约70mg、约75mg、约80mg、约85mg、约90mg、约95mg、约100mg、约105mg、约110mg、约115mg、约120mg、约125mg、约130mg、约135mg、约140mg、约145mg、约150mg、约155mg、约160mg、约165mg、约170mg、约175mg、约180mg、约185mg、约190mg、约195mg、约200、约205mg、约210mg、约215mg、约220mg、约225mg、约230mg、约235mg、约240mg、约245mg、约250mg、约255mg、约260mg、约265mg、约270mg、约275mg、约280mg、约
285mg、约290mg、约295mg、约300、约325mg、约350mg、约375mg、约400mg、约425mg、约450mg、约475mg、约500mg、约525mg、约550mg、约575mg、约600mg或更多抗il-6r抗体的方法。
[0220]
如本文所用,“给予约2.0至约4.0mg/kg”意指将物质以所述范围内的任何值(包括所述范围的端点)给予。例如,“向患者给予的抗il-6r抗体的剂量是2.0mg/kg至4.0mg/kg”,包括给予2.0mg/kg的抗il-6r抗体、4.0mg/kg的抗il-6r抗体以及之间的所有剂量。
[0221]
在各种实施方案中,将il-6r抗体以一周一次约25至150mg或每隔一周40mg至200mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次25至50mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次50至75mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次75至100mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次100至125mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次125至150mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次150至175mg的剂量给予。在一个实施方案中,将il-6r抗体以每周一次或每隔一周一次175至200mg的剂量给予。在一个实施方案中,将il-6r抗体以一周一次100mg的剂量给予。在一个实施方案中,将il-6r抗体以一周一次150mg的剂量给予。在一个实施方案中,将il-6r抗体以一周一次200mg的剂量给予。在一个实施方案中,将il-6r抗体以一周一次100至150mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次100至200mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次150至200mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次约100或约150mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次约100、150或200mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次100mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次150mg的剂量给予。在一个实施方案中,将il-6r抗体以每两周一次200mg的剂量给予。
[0222]
在所述方法的各种实施方案中,将所述抗体以表2-5中列出的剂量使用。
[0223]
向患者给予的抗il-6r抗体的量可以按照抗体毫克数/公斤患者体重(即mg/kg)表示。例如,本发明的方法包括以约0.01至约100mg/kg、约0.1至约50mg/kg、或约1至约10mg/kg患者体重的日剂量向患者给予抗il-6r抗体。在某些实施方案中,抗hil6r抗体是萨瑞鲁单抗,并且将其以约2mg/kg至约4mg/kg进行给予。在各种实施方案中,抗hil6r抗体是萨瑞鲁单抗,并且将其以约2mg/kg至约3mg/kg进行给予。在某些实施方案中,抗hil6r抗体是萨瑞鲁单抗,并且将其以约2.5mg/kg至约4mg/kg进行给予。在各种实施方案中,抗hil6r抗体是萨瑞鲁单抗,并且将其以一周一次(qw)或每两周一次(q2w)的频率从约2mg/kg至约3mg/kg进行给予。在一些实施方案中,将抗hil6r抗体以一周一次(qw)或每两周一次(q2w)的频率,以约2mg/kg至4mg/kg的剂量给予。在一些实施方案中,将抗hil6r抗体以一周一次(qw)或每两周一次(q2w)的频率,以约2.5mg/kg至4mg/kg的剂量给予。在各种实施方案中,将抗hil6r抗体以约1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5或5.0mg/kg的剂量给予。在一些实施方案中,抗hil6r抗体包含vh和vl,其中vh包含在seq id no:1序列中发现的三个cdr,并且其中vl包含在seq id no:2序列中发现的三个cdr。在其他实施方案中,抗hil6r抗体是萨瑞鲁单抗。
[0224]
本发明的方法包括在特定的时间过程内向患者给予多剂量的抗il-6r抗体。例如,可以将抗il-6r抗体每天给予约1至5次、每周给予约1至5次、每月给予约1至5次、或每年给
予约1至5次。在某些实施方案中,本发明的方法包括在第一时间点向患者给予第一剂量的抗il-6r抗体,随后在第二时间点向患者给予至少第二剂量的抗il-6r抗体。在某些实施方案中,第一和第二剂量可以含有相同量的抗il-6r抗体。例如,第一和第二剂量可以各自含有约10mg至约500mg、约20mg至约300mg、约100mg至约200mg或约100mg至约150mg所述抗体。第一和第二剂量之间的时间可以为约几小时到几周。例如,第二时间点(即,当给予第二剂量时的时间)可以在第一时间点(即,当给予第一剂量时的时间)之后约1小时至约7周。根据本发明的某些示例性实施方案,第二时间点可以在第一时间点之后约1小时、约4小时、约6小时、约8小时、约10小时、约12小时、约24小时、约2天、约3天、约4天、约5天、约6天、约7天、约2周、约4周、约6周、约8周、约10周、约12周、约14周或更长。在某些实施方案中,第二时间点是约1周或约2周。在患者的整个治疗过程中,可以类似地给予第三剂量和随后的剂量。本发明提供了使用治疗组合物的方法,所述治疗组合物包含抗il-6r抗体或其抗原结合片段,以及任选的一种或多种另外的治疗剂。本发明的治疗组合物将与合适的载体、赋形剂和掺入配制品中以提供改进传递、递送、耐受性等的其他药剂一起给予。在所有药物化学家都知道的配方中可以找到许多适当的配制品:remington's pharmaceutical sciences,mack publishing company,伊斯顿,宾夕法尼亚州,将其通过引用以其整体并入本文。这些配制品包括例如粉末、糊剂、软膏、胶状物、蜡、油、脂质、含有脂质(阳离子或阴离子)的囊泡(如lipofectin)、dna缀合物、无水吸收糊剂、水包油和油包水乳液、乳液卡波蜡(carbowax)(具有不同分子量的聚乙二醇)、半固体凝胶和含有卡波蜡的半固体混合物。还参见powell等人“compendium of excipients for parenteral formulations”pda(1998)j pharm sci technol 52:238-311,将其通过引用以其整体并入本文。
[0225]
各种递送系统是已知的并可以用于给予本发明的药物组合物,例如在脂质体中包封、微粒、微胶囊、受体介导的胞吞作用(参见例如,wu等人(1987)j.biol.chem.262:4429-4432,将其通过引用以其整体并入本文)。引入的方法包括但不限于皮内、肌肉内、腹膜内、静脉内、皮下、鼻内、硬膜外和口服途径。可以将组合物通过任何方便的途径给予,例如通过输注或推注,通过上皮或粘膜皮肤内层(例如,口腔粘膜、直肠和肠粘膜等)吸收,并且可以与其他生物活性剂一起给予。给予可以是全身性或局部的。可以将il-6r抗体皮下给予。
[0226]
还可以将药物组合物在囊泡如脂质体中递送(参见langer(1990)science 249:1527-1533,将其通过引用以其整体并入本文)。在某些情况下,可以将药物组合物在受控释放系统中递送,例如,使用泵或聚合材料。在另一个实施方案中,受控释放系统可以被放置成接近组合物的靶,因此只需要全身剂量的一部分。
[0227]
可注射制剂可以包括用于静脉内、皮下、皮内和肌肉内注射、局部注射、点滴输注等的剂型。这些可注射制剂可以通过公共已知的方法来制备。例如,可以通过例如将上述抗体或其盐溶解、悬浮或乳化在常规用于注射的无菌水性介质或油性介质中来制备可注射制剂。作为用于注射的水性介质,例如有生理盐水、含葡萄糖的等渗溶液和其他辅助剂等,其可以与以下组合使用:适当的增溶剂,如醇(例如乙醇);多元醇(例如丙二醇、聚乙二醇);非离子表面活性剂[例如,聚山梨醇酯80、hco-50(氢化蓖麻油的聚氧乙烯(50mol)加合物)]等。作为油性介质,采用例如芝麻油、大豆油等,其可以与如苯甲酸苄酯、苄醇等的增溶剂组合使用。可以将由此所制备的注射剂填充在适当的安瓿中。
[0228]
所述抗体通常是如本文和国际公开号wo 2011/085158(将其通过引用以其整体并
入本文)中所述进行配制的。
[0229]
在各种实施方案中,所述抗体被作为约ph 6.0的水性缓冲溶液给予,其含有
[0230]-约21mm组氨酸,
[0231]-约45mm精氨酸,
[0232]-约0.2%(w/v)聚山梨醇酯20,
[0233]-约5%(w/v)蔗糖,以及
[0234]-约100mg/ml与约200mg/ml之间的所述抗体。
[0235]
在另一个实施方案中,所述抗体被作为约ph 6.0的水性缓冲溶液给予,其含有
[0236]-约21mm组氨酸,
[0237]-约45mm精氨酸,
[0238]-约0.2%(w/v)聚山梨醇酯20,
[0239]-约5%(w/v)蔗糖,以及
[0240]-至少约130mg/ml的所述抗体。
[0241]
在另一个实施方案中,所述抗体被作为约ph 6.0的水性缓冲溶液给予,其含有
[0242]-约21mm组氨酸,
[0243]-约45mm精氨酸,
[0244]-约0.2%(w/v)聚山梨醇酯20,
[0245]-约5%(w/v)蔗糖,以及
[0246]-约131.6mg/ml的所述抗体。
[0247]
在另一个实施方案中,所述抗体被作为约ph 6.0的水性缓冲溶液给予,其含有
[0248]-约21mm组氨酸,
[0249]-约45mm精氨酸,
[0250]-约0.2%(w/v)聚山梨醇酯20,
[0251]-约5%(w/v)蔗糖;以及
[0252]-约175mg/ml的所述抗体。
[0253]
在其他实施方案中,所述抗体被作为ph 6.0的水性缓冲溶液给予,其含有
[0254]-21mm组氨酸,
[0255]-45mm精氨酸,
[0256]-0.2%(w/v)聚山梨醇酯20,
[0257]-5%(w/v)蔗糖,以及
[0258]-100mg/ml与200mg/ml之间的所述抗体。
[0259]
在另一个实施方案中,所述抗体被作为ph 6.0的水性缓冲溶液给予,其含有
[0260]-21mm组氨酸,
[0261]-45mm精氨酸,
[0262]-0.2%(w/v)聚山梨醇酯20,
[0263]-5%(w/v)蔗糖,以及
[0264]-至少130mg/ml的所述抗体。
[0265]
在另一个实施方案中,所述抗体被作为ph 6.0的水性缓冲溶液给予,其含有
[0266]-21mm组氨酸,
[0267]-45mm精氨酸,
[0268]-0.2%(w/v)聚山梨醇酯20,
[0269]-5%(w/v)蔗糖,以及
[0270]-131.6mg/ml的所述抗体。
[0271]
在另一个实施方案中,所述抗体被作为ph 6.0的水性缓冲溶液给予,其含有
[0272]-21mm组氨酸,
[0273]-45mm精氨酸,
[0274]-0.2%(w/v)聚山梨醇酯20,
[0275]-5%(w/v)蔗糖;以及
[0276]-175mg/ml的所述抗体。
[0277]
有利地,将上述的用于口服或肠胃外使用的药物组合物制备成适于配合活性成分剂量的单位剂量的剂型。单位剂量的此类剂型包括例如片剂、丸剂、胶囊、注射剂(安瓿)、栓剂等。
[0278]
根据本文公开的方法,可以使用任何可接受的装置或结构向患者给予抗il-6r抗体(或包含所述抗体的药物配制品)。例如,所述给予可以使用注射筒和针或者使用可重复使用的笔和/或自动注射器递送装置来完成。本发明的方法包括使用许多可重复使用的笔和/或自动注射器递送装置来给予抗il-6r抗体(或包含所述抗体的药物配制品)。此类装置的例子包括但不限于autopen(owen mumford,inc.,woodstock,英国),disetronic笔(disetronic medical systems,波道夫,瑞士),humalog mix 75/25笔,humalog笔,humalin 70/30笔(eli lilly and co.,印第安纳波利斯,印第安纳州),novopen i、ii和iii(novo nordisk,哥本哈根,丹麦),novopen junior(novo nordisk,哥本哈根,丹麦),bd笔(becton dickinson,富兰克林湖,新泽西州),optipen,optipen pro,optipen starlet,以及opticlik(sanofi-aventis,法兰克福,德国)。用于皮下递送本发明药物组合物的一次性笔和/或自动注射器递送装置的例子包括但不限于solostar笔(sanofi-aventis)、flexpen(novo nordisk)和kwikpen(eli lilly)、sureclick自动注射器(amgen,千橡市,加利福尼亚州)、penlet(haselmeier,stuttgart,德国)、epipen(dey,l.p.)以及humira笔(abbvie inc.,北芝加哥,伊利诺伊州),仅举几例。
[0279]
在一个实施方案中,将所述抗体用预填充注射筒给予。在另一个实施方案中,将所述抗体用含有安全系统的预填充注射筒给予。例如,安全系统防止意外针刺伤害。在各种实施方案中,将所述抗体用含有安全系统(west pharmaceutical services inc.)的预填充注射筒给予。还参见美国专利号5,215,534和9,248,242,将其通过引用以其整体并入本文。
[0280]
在另一个实施方案中,将所述抗体用自动注射器给予。在各种实施方案中,将所述抗体用特征为pushclick技术的自动注射器(shl group)给予。在各种实施方案中,自动注射器是包括注射筒的装置,所述注射筒允许向受试者给予一定剂量的组合物和/或抗体。还参见美国专利号9,427,531和9,566,395,将其通过引用以其整体并入本文。
[0281]
本文还考虑使用微融合器向患者递送抗il-6r抗体(或包含所述抗体的药物配制品)。如本文所用,术语“微融合器”意指皮下递送装置,其被设计成在延长的时间段(例如,约10、15、20、25、30或更多分钟)内缓慢给予大体积(例如,高达约2.5ml或更多)的治疗配制
品。参见例如,u.s.6,629,949;us 6,659,982;以及meehan等人,j.controlled release 46:107-116(1996),将其通过引用以其整体并入本文。微融合器特别适用于递送包含在高浓度(例如,约100、125、150、175、200mg/ml或更高)和/或粘稠溶液中的大剂量治疗性蛋白质。
[0282]
在各种实施方案中,将根据本公开文本的抗体给予至患有一类型jia(例如全身型幼年特发性关节炎)的受试者。在某些实施方案中,本文公开的方法包括选择患有一类型jia(例如全身型幼年特发性关节炎)的受试者的步骤。全身型幼年特发性关节炎的诊断是根据ilar 2001jia分类标准(petty re等人international league of associations for rheumatology classification of juvenile idiopathic arthritis:第二次修订,edmonton,2001.j rheumatol.2004年2月;31(2):390-2)。在一些实施方案中,患者年龄在约1岁与约17岁之间。在各种实施方案中,患者年龄在约4岁与约6岁之间、在约12岁与约18岁之间、在约12岁与约14岁之间,或者在约7岁与约9岁之间。在各种实施方案中,受试者展现出5个活动关节的关节炎。在一些实施方案中,尽管以稳定剂量使用糖皮质激素至少3天,受试者在任何连续7天中的至少3天中,展现出2个活动关节的关节炎伴全身型幼年特发性关节炎发烧(体温高于37.5℃)。在各种实施方案中,受试者对当前的治疗没有足够的响应,并且被认为是bdmard的候选者。
[0283]
在各种实施方案中,将根据本公开文本的抗体给予至患有pcjia的受试者。在某些实施方案中,本文公开的方法包括选择患有一类型jia(例如pc jia)的受试者的步骤。类风湿因子阴性或rf阳性多关节幼年特发性关节炎亚型或少关节扩展幼年特发性关节炎亚型的诊断是根据ilar 2001jia分类标准(petty re等人international league of associations for rheumatol ogy classification of juvenile idiopathic arthritis:第二次修订,edmonton,2001.j rheumatol.2004年2月;31(2):390-2)。在一些实施方案中,患者年龄在约2岁与约17岁之间。在各种实施方案中,患者年龄在约4岁与约6岁之间、在约12岁与约18岁之间、在约12岁与约14岁之间,或者在约7岁与约9岁之间。在各种实施方案中,根据活动性关节炎的美国风湿病学会定义,受试者展现出至少5个活动关节的关节炎。在各种实施方案中,受试者对当前的治疗没有足够的响应,并且被认为是bdmard的候选者。
[0284]
在某些实施方案中,对先前治疗的不充分响应是指在以最大耐受典型剂量接受先前治疗(例如,糖皮质激素关节注射和/或甲氨蝶呤)后,其jia未得到良好控制的受试者。在一个实施方案中,对先前治疗的不充分响应是指尽管有先前治疗但仍具有中度或高度疾病活动性和不良预后特征的受试者。在各种实施方案中,对先前治疗的不充分响应是指尽管有先前治疗但具有未改善或已经恶化的jia症状(例如,本文列出的任何症状)的受试者。
[0285]
sjia和pcjia的各种治疗方法的有效性可以使用本文所述的任何方法例如幼年关节炎疾病活动性得分-27(jadas)进行评估。(consolaro等人development and validation of a composite disease activity score for juvenile idiopathic arthritis.arthritis&rheumatism.2009年5月;61(5):658-666)。
[0286]
在本文中提到的所有出版物出于所有目的通过引用以其整体并入本文。
[0287]
缩写列表
[0288]
acr:美国风湿病学会
[0289]
ada:抗药物抗体
[0290]
ae:不良事件
[0291]
aesi:特别关注的一个或多个不良事件
[0292]
alp:碱性磷酸酶
[0293]
alt:丙氨酸转氨酶
[0294]
ana:抗核抗体
[0295]
anc:绝对嗜中性粒细胞计数
[0296]
抗dsdna:抗双链dna
[0297]
ast:天冬氨酸转氨酶
[0298]
bp:血压
[0299]
bun:血尿素氮
[0300]
chaq:儿童健康评估问卷
[0301]
cox-2:环氧合酶-2抑制剂
[0302]
crf:病历报告表
[0303]
crp:c反应蛋白
[0304]
csr:临床研究报告
[0305]
cyp:细胞色素
[0306]
dec:剂量递增委员会
[0307]
dmard:改善疾病抗风湿药物
[0308]
dmc:数据监测委员会
[0309]
dna:脱氧核糖核酸
[0310]
ebv:爱泼斯坦-巴尔病毒
[0311]
e-crf:电子病历报告表
[0312]
eos:研究结束
[0313]
eot:治疗结束
[0314]
esr:红细胞沉降率
[0315]
gcp:良好临床规范
[0316]
haq-di:健康评估问卷残疾指数
[0317]
hbc-ab:乙型肝炎核心抗体
[0318]
hiv:人类免疫缺陷病毒
[0319]
hlgt:高层群组术语
[0320]
hr:心率
[0321]
hs-crp:高敏c反应蛋白
[0322]
icf:知情同意书
[0323]
ich:国际协调理事会
[0324]
iec:独立伦理委员会
[0325]
igg:免疫球蛋白g
[0326]
il-6:白细胞介素6
[0327]
il-6r:il-6受体
[0328]
ilar:国际风湿病学协会联盟
[0329]
imp:研究药物产品
[0330]
irb:机构审查委员会
[0331]
ivrs:交互式语音响应系统
[0332]
iwrs:交互式网络响应系统
[0333]
jadas:幼年关节炎疾病活动性得分
[0334]
jak:janus激酶
[0335]
jia:幼年特发性关节炎
[0336]
jia acr:幼年特发性关节炎美国风湿病学会
[0337]
ldh:乳酸脱氢酶
[0338]
ldl:低密度脂蛋白
[0339]
lft:肝功能测试,肝功能测试
[0340]
lloq:定量下限
[0341]
mab:单克隆抗体
[0342]
mas:巨噬细胞活化综合征
[0343]
mtss:修正的总sharp得分
[0344]
mtx:甲氨蝶呤
[0345]
nimp:非研究药物产品
[0346]
nsaid:非类固醇抗炎药
[0347]
ojia:少关节幼年特发性关节炎
[0348]
pcjia:幼年特发性关节炎的多关节病程
[0349]
pcsa:潜在的临床显著异常
[0350]
pd:药效学
[0351]
pk:药代动力学
[0352]
poppk:群体药代动力学
[0353]
ppd:纯化蛋白衍生物
[0354]
pt:首选术语
[0355]
q2w:每隔一周一次
[0356]
qw:一周一次
[0357]
ra:类风湿性关节炎
[0358]
rf:类风湿因子
[0359]
sae:严重不良事件
[0360]
sc:皮下
[0361]
sd:标准偏差
[0362]
sjia:全身型幼年特发性关节炎
[0363]
soc:系统器官类别
[0364]
susar:疑似意外严重不良反应
[0365]
tb:结核病
[0366]
teae:治疗突发性不良事件
[0367]
uln:正常上限
[0368]
vas:视觉模拟量表
[0369]
wbc:白细胞
[0370]
实施例
[0371]
提出以下实施例以向本领域普通技术人员提供关于如何制备和使用本发明中表征的方法和组合物的完整公开内容和描述,并且不旨在限制诸位发明人视为其发明的范围。已经努力确保关于所使用的数字(例如,量、温度等)的准确性,但是应该考虑一些实验误差和偏差。
[0372]
实施例1:在患有pcjia的2至17岁的儿童和青少年中进行的用皮下(sc)注射给予的萨瑞鲁单抗(随后是延长阶段)的开放标签、顺序、递增、重复剂量探索研究。(研究编号dri13925,研究标题:在患有pcjia的儿童和青少年中进行的萨瑞鲁单抗的开放标签、递增、重复剂量探索研究)
[0373]
开始了阶段2受控试验(编号nct02776735),以测试在治疗pcjia中皮下给予的人igg1抗il-6rα单克隆抗体萨瑞鲁单抗。本研究是在对当前疗法响应不足或不耐受或被认为是生物类改善疾病抗风湿药物(dmard)候选者的2至17岁(或国家规定的年龄要求)的患有pcjia的儿童和青少年中进行的多国、多中心、开放标签、2阶段和2部分研究。所述研究的2个阶段是最初的12周核心治疗阶段,随后是144周延长阶段(参加图1和图2)。
[0374]
整体研究设计和计划:
[0375]
12周核心治疗阶段
[0376]
这项多国、多中心、开放标签的研究纳入了根据ilar 2001jia分类标准(1)诊断为rf阳性或rf阴性pjia或eojia的2-17岁患者。患者具有≥5个活动关节,并且根据研究者的判断必须被考虑作为bdmard的候选者。
[0377]
患者经由皮下(sc)注射接受萨瑞鲁单抗,由调查场所的专业照护者提供。在2个体重组(a组,≥30-60kg和b组,10-《30kg)中以阶梯式方法测试剂量方案,从较高体重组(a组)中的最低萨瑞鲁单抗剂量方案开始(图2)。对于剂量方案1、2和3,a/b组中的剂量方案分别为2.0/2.5mg/kg q2w、3.0/4.0mg/kg q2w和2.0/2.5mg/kg qw。在至少4周的治疗后,在审查了纳入a组中相同剂量方案中的前3名患者的数据后,允许在b组中开始剂量方案1。一旦纳入当前体重和剂量组中的前3名患者完成了6周的治疗,在审查了安全性、功效、药代动力学(pk)和药效学(pd)数据后,允许在每个体重组内递增到更高剂量方案。在审查了来自所有其他体重和剂量组的数据后,允许纳入剂量3,b组。剂量递增决定由剂量递增委员会(dec)做出。如果纳入所述剂量和体重组中的前3名患者在第6周未达到jia acr30响应,则dec可以宣布剂量无效,停止所述剂量和体重组的纳入,并中断所述方案中的患者得到研究者批准的护理标准。在整个研究过程中,由包括3名儿科风湿病学家的独立数据监测委员会正式监测了萨瑞鲁单抗的安全性。
[0378]
本研究的主要目的是研究被给予12周的萨瑞鲁单抗的3种剂量方案的pk特征,以便选择一种方案进行进一步研究。基于建模和模拟来选择剂量方案,以提供与成人ra中评价的剂量方案相似的暴露。剂量方案1对应于每2周150mg(q2w),最低批准的成人剂量;剂量方案2对应于200mg q2w,建议批准的成人剂量;并且剂量方案3对应于每周150mg(qw),在成人中测试的最高剂量,其证明以可接受的安全性比安慰剂更有效(20)。
[0379]
12周核心治疗阶段(如图2所展示)分为3个部分。第一个顺序的递增剂量探索部分,其中从a组和剂量群组1开始,在每剂量和体重组的6个可评价患者(总共大约36名患者)中,在以下2个体重组中的每一组中研究了3种剂量方案(表1):患者≥30kg且≤60kg(a组)和患者《30kg且≥10kg(b组)。接下来的第二部分,其中将大约24名另外的患者(每个体重组12名,≥30kg(a组)和《30kg且≥10kg(组)直接纳入所选择剂量方案,以在此所选择剂量方案下每体重组总共获得18名可评价患者。第三部分,其中大约28名另外的患者(每个体重组上限为70%,患者≥30kg[a组]和患者《30kg且≥10kg[b组])将被直接纳入所选择剂量方案,以在整个研究中总共获得大约100名治疗的患者。在12周核心治疗阶段期间,这些患者将经历与在剂量探索和第二部分中招募的患者相同的现场访视;然而,在基线与第2周之间,第三部分中的患者将不进行萨瑞鲁单抗pk取样访视。
[0380]
12周核心治疗阶段是从第2次访视(基线-第0周)到第12次访视给予研究药物产品(imp)的时间。在12周核心治疗阶段期间,将较低体重组(b组:《30kg且≥10kg)中的患者随机分配到以下萨瑞鲁单抗pk取样时间表1或2,以便最小化抽血量和访视次数,同时保持对主要终点的评价:
[0381]
■
时间表1:基线、第3天、第8天、第2周、第4周、第8周和第12周
[0382]
■
时间表2:基线、第5天、第12天、第2周、第4周、第8周和第12周。
[0383]
评估。两个体重组的3种剂量方案的数据均已提供,并从研究基线到第12周进行了报告。
[0384]
主要终点是pk暴露,包括最大血清浓度、在给药间隔期间使用梯形法计算的血清浓度对时间曲线下的面积、以及在重复给药期间在治疗给予前观察到的血清浓度。次要终点是pd,包括高敏感性c反应蛋白(crp)浓度、安全性和临床功效。功效由第12周,实现jia acr30/70/90响应的患者比例、jia acr响应组分自基线的变化、以及使用27-关节计数和crp评估得到的幼年关节炎疾病活动性得分(jadas-27-crp)的平均变化来确定。
[0385]
此外,在完成剂量探索和第二部分的纳入后,按照卫生当局的建议,在第三部分中大约28名另外的患者将被直接纳入所选择剂量方案,以在整个研究中总共获得大约100名治疗的患者。在12周核心治疗阶段期间,这些患者将经历与在剂量探索和第二部分中招募的患者相同的现场访视;然而,在基线与第2周之间,第三部分中的患者将不进行萨瑞鲁单抗pk取样访视。
[0386]
延长阶段中的另外的治疗期将允许在pcjia患者中收集萨瑞鲁单抗的长期安全性和临床响应数据。此延长阶段对于纳入剂量探索和第二部分中的患者,长达144周,并且对于纳入第三部分中的患者,长达84周。纳入剂量探索部分中的患者(在选择剂量方案时已经处于延长阶段的患者,以及没有接受所选择剂量方案的患者)将会将他们的剂量方案调整为所选择剂量方案。
[0387]
为了最小化采集的血液量,剂量探索和第二部分中的b组患者(《30kg且≥10kg)将被随机分配到2个不同的萨瑞鲁单抗pk样品采集时间表中的1个。pk分析将整合这些取样时间表,以描述萨瑞鲁单抗的pk特征。在基线与第2周之间,没有计划为纳入第三部分的患者进行pk样品采集。在4周的时间内,用于研究的总抽血量将不超过总血量的3%,并且在任何单个时间都不超过总血量的1%(39)。此外,计划的频繁评估控制了研究参与者的风险。
[0388]
最后,考虑到在成人ra程序中观察到的pk特征,在针对teae、萨瑞鲁单抗pk和pd评
估的最后一次治疗访视后6周的治疗后随访期是适当的。
[0389]
统计分析。
[0390]
使用非线性混合效应建模建立了混合数据的群体pk模型,以描述萨瑞鲁单抗的pk特征。仅使用描述性统计分析了pd、安全性和功效数据。功效终点是基于如在治疗中观察(方法)得到的数据进行分析的(没有缺失数据的归责)。此外,jia acr30/70/90结果也是使用非响应归责方法(非响应状态自动分配给数据不足或中断治疗后的患者)得出的。
[0391]
144周/84周延长阶段:
[0392]
第12次访视的imp被认为是延长阶段的第一个imp。仅在第12次访视(第12周)时达到jia acr30响应的患者被允许继续进行延长阶段。患者继续接受他们在本研究的12周核心治疗阶段中被分配接受的萨瑞鲁单抗的相同剂量方案,直到所选择剂量方案被确定。一旦选择了剂量方案,尚未采用此剂量方案的患者将其剂量方案调整至所选择剂量方案,并遵循新的访视时间表,其中与未调整剂量方案的患者相比,在前12周进行更频繁的监测(pk、安全性和功效)。
[0393]
在第27次访视时,使用eot程序评估过早中断研究治疗的患者。这些患者被要求在eot访视后6周(eot+6周)返回进行研究结束(eos)评估。对于在12周核心治疗阶段期间中断研究治疗的患者,在eot访视后2周(eot+2)进行另外的萨瑞鲁单抗pk评估,并在eot访视时测量il-6和sil6r。这些患者被要求执行除了萨瑞鲁单抗给予之外的所有方案安排的访视和评估,直到第12次访视。
[0394]
为纳入第三部分中的患者安排了84周的延长阶段。第12次访视的imp被认为是延长阶段的第一个imp。如表32所示,将对采用所选择剂量方案纳入第三部分中的患者进行随访。
[0395]
测试的剂量方案
[0396]
对于每个体重组,待测试的3个顺序递增剂量方案是基于pk建模定义的,其基本原理如下:
[0397]
·
剂量群组1:类似于萨瑞鲁单抗150mg q2w的靶向pk暴露的剂量,是患有ra的成年患者中的最低有效剂量方案。
[0398]
·
剂量群组2:类似于萨瑞鲁单抗200mg q2w的靶向pk暴露的剂量,是患有ra的成年患者中的推荐剂量方案。
[0399]
·
剂量群组3:类似于萨瑞鲁单抗150mg qw的靶向pk暴露的剂量,其在患有ra的成年患者中在长期给药研究中,产生最高暴露。
[0400]
表1.按体重和剂量群组的剂量
[0401][0402][0403]
缩写:qw=每周一次,q2w=每隔一周一次
[0404]
在基线处计算向患者给予的剂量(mg)。无论患者体重如何变化,在试验的12周核
心治疗阶段的整个过程中,药物产品的剂量和对应体积都保持相同。在延长阶段,每次访视时测量患者的体重,并且仅在计算显示需要增加剂量时,才根据体重的增加调整剂量。分别地,上限剂量对于剂量群组1和3为150mg,并且对于剂量群组2为200mg。对于剂量群组1至3,待注射的萨瑞鲁单抗体积在下表2-5中呈现。
[0405]
表2和表3示出了高体重组(a组)中患者的剂量信息。高体重患者是对于剂量探索部分体重≥30kg且≤60kg的那些患者,以及第二部分中体重≥30kg的另外的患者。表4和表5示出了低体重组(b组):《30kg且≥10kg中患者的剂量信息。
[0406]
表2:高体重组(a组)群组1和3的剂量信息
[0407][0408]
表3:高体重组(a组)群组2的剂量信息
[0409][0410][0411]
表3和表4示出了低体重组(b组):《30kg且≥10kg中患者的剂量信息。
[0412]
表4.低体重组(b组)群组1和3的剂量信息
[0413][0414]
表5.低体重组(b组)群组2的剂量信息
[0415]
[0416][0417]
在12周核心治疗阶段期间,每次注射的待给予体积是基于基线(第2次访视)体重来计算,并在所有12周核心治疗阶段期间保持不变。如果体重增加,并且计算显示需要增加剂量,则在延长治疗阶段期间待给予的体积可以通过每次访视时测量的体重进行调整。如果所选择剂量方案是3个预定义剂量群组中的一个,则随后纳入或改为所选择剂量方案的患者使用表2-表5中的对应剂量。
[0418]
每位患者参与研究的持续时间
[0419]
研究的总持续时间(每位患者)对于纳入剂量探索和第二部分中的患者,长达166周,并且对于纳入第三部分中的患者,长达106周:
[0420]
■
长达4周+3天的筛查(长达31天)
[0421]
■
12周核心治疗阶段
[0422]
■
对于纳入剂量探索和第二部分中的患者,长达144周延长阶段,并且对于纳入第三部分中的患者,长达84周延长阶段
[0423]
■
治疗后6周随访。
[0424]
对于所有访视,使用第1天作为参考,剂量群组1和2的
±
3天以及剂量群组3的
±
1天的时间框架是可接受的,除了
[0425]
■
对于第3次访视(第3天)、第4次访视(第5天)和第5次访视(第8天),在进行pk取样时不允许有访视窗口。对于纳入第三部分中的患者,pk取样访视,即访视3、4、5和6,在核心治疗阶段期间不适用。
[0426]
■
对于所有3个剂量群组,第6次访视(第12天)
±
1天
[0427]
■
对于所有3个剂量群组,第7次访视(第2周)
±
1天
[0428]
■
对于剂量群组1和2,第8次访视(第4周)
±
2天,以及对于剂量群组3,为
±
1天。
[0429]
纳入标准
[0430]
本研究的纳入标准如下所示:
[0431]
1.筛查访视时年龄≥2且≤17岁(或国家规定的年龄要求)的男性和女性患者
[0432]
2.根据ilar 2001jia分类标准(2),诊断为rf阴性或rf阳性多关节jia亚型或ojia亚型,在筛查时根据acr定义的“活动性关节炎”具有至少5个活动关节(表6)
[0433]
3.根据研究者的判断,对当前的治疗没有足够的响应并且被认为是bdmard的候选者的患者
[0434]
4.达到法定同意年龄的患者,或者父母一方或双方或者一个或多个法定监护人签署伦理委员会批准的书面知情同意书并注明日期。应基于当地指南以及患者的成熟度和理解研究相关信息的智力能力获得患者的允许。在涉及具有充分决策能力的独立或成熟未成年人的情况下,或者在法律允许的其他情况下,将直接从父母一方或双方或者一个或多个法定监护人处获得签署的知情同意书。
[0435]
排除标准
[0436]
患者因以下原因被排除在本研究之外:体重《10kg或》60kg;除了rf阳性、rf阴性或eojia之外的jia亚型;先前用抗il-6或il-6r拮抗剂治疗;在首次萨瑞鲁单抗剂量的5个半衰期内用任何其他生物制剂进行治疗;在研究基线之前4周内使用肠胃外或关节内糖皮质激素注射;机会性感染(包括结核病)的活跃或既往病史;基线之前4周内接种减毒活疫苗;或将对患者参与本研究产生不利影响的任何情况。如果非类固醇抗炎药(nsaid)或口服糖皮质激素的剂量在基线之前2周内不稳定或者如果口服糖皮质激素剂量超过0.5mg/kg/天(或30mg/天)的等效泼尼松剂量,如果常规合成改善疾病抗风湿药物(csdmard)的剂量在基线之前6周内不稳定或者如果csdmard剂量超过当地标签的推荐剂量,则接受nsaid、口服糖皮质激素或csdmard治疗的患者被排除在外。基于当地法规指南,需要获得所有患者/监护人的书面知情同意书。
[0437]
表6.类风湿因子(rf)阴性、类风湿因子(rf)阳性多关节和(ojia)的亚分类
[0438][0439][0440]
a.患者或一级亲属有银屑病或银屑病病史。
[0441]
b.hla b27阳性男性在6岁后开始患关节炎;
[0442]
c.强直性脊柱炎、肌腱端炎相关关节炎、伴有炎性肠病的骶髂关节炎、莱特尔综合征、急性前葡萄膜炎或一级亲属中这些障碍之一的病史;
[0443]
d.间隔至少3个月至少2次出现igm类风湿因子;
[0444]
e.患者存在全身型jia。
[0445]
研究治疗
[0446]
研究药物产品(imp)
[0447]
萨瑞鲁单抗,为抗il-6r mab(抗白细胞介素-6受体α亚基单克隆抗体)。
[0448]
配制品
[0449]
在ph 6.0的水性缓冲媒介物中,以175mg/ml提供萨瑞鲁单抗药物产品。将其装在填充有2.7ml萨瑞鲁单抗的5ml小瓶中,其中可抽出体积为2.0ml。
[0450]
一个或多个给予途径:
[0451]
在自我注射时在腹部或大腿上或者另外由专业或非专业照护者在上臂(外侧)上进行皮下给予萨瑞鲁单抗。优选的是,sc注射部位在腹部(除了肚脐或腰部区)或大腿(前部和侧部)的4个象限之间交替。
[0452]
对于接受剂量方案1或2(q2w)的患者,在本研究的12周核心治疗阶段期间,由专业照护者在现场进行注射。对于接受剂量方案3(每周注射)的患者,已安排合格的现场人员或家庭护士针对未计划在研究现场给出的剂量给予imp。对于本研究的延长阶段,如果患者或者父母一方或双方或者一个或多个法定监护人或者一个或多个照护者愿意并能够进行注射,则允许家庭注射。在这些情况下,在核心治疗阶段,从第10次访视(第8周)开始,需要并提供培训来准备和给予imp。将此培训记录在患者的研究文件中。在允许在延长治疗阶段中的家庭注射之前,允许患者或者父母一方或双方或者一个或多个法定监护人或者一个或多个照护者在一个或多个研究者或者一个或多个代表人的观察/监督下在第10次访视(第8周)、第11次访视(第10周)和第12次访视(第12周)时给予注射。
[0453]
在患者进行研究访视的日子里,遵循临床程序和血液采集给予imp。提供日记来记录与这些注射有关的信息;这些日记作为源数据保存在患者的研究文件中。如果照护者不能或不愿意给予imp,则安排合格的现场人员针对未计划在研究现场给出的剂量给予imp。
[0454]
剂量方案
[0455]
将萨瑞鲁单抗按q2w或qw给予。然而,根据方案,允许萨瑞鲁单抗给予窗口对于剂量群组1和2为
±
3天并且对于剂量群组3为
±
1天,以适应特殊情况,例如实验室测试结果待定、持续不良事件(ae)、除了第5次访视(第8天)之外的患者时间安排困难。第5次访视(第8天)时没有给予窗口。在第7次访视(第2周)时,剂量群组1、2和3患者仅有
±
1天的给予窗口,并且在第8次访视(第4周)时,剂量群组3仅有
±
1天的给予窗口或者剂量群组1和2仅有
±
2天的给予窗口。
[0456]
请注意,对于q2w给予在少于11天并且对于每周给予在少于6天的时间间隔内,imp的过量(意外或故意)被定义为所述剂量的至少两倍
[0457]
q2w或qw给予以下萨瑞鲁单抗剂量:
[0458]
剂量群组1:
[0459]
·
a组(≥30kg且≤60kg):2mg/kg q2w
[0460]
·
b组(《30kg且≥10kg):2.5mg/kg q2w。
[0461]
剂量群组2:
[0462]
·
a组(≥30kg且≤60kg):3mg/kg q2w
[0463]
·
b组(《30kg且≥10kg):4mg/kg q2w。
[0464]
剂量群组3:
[0465]
·
a组(≥30kg且≤60kg):2mg/kg qw
[0466]
·
b组(《30kg且≥10kg):2.5mg/kg qw。
[0467]
剂量修改
[0468]
在基线处计算向患者给予的剂量(mg)。无论患者体重如何变化,在试验的12周核心治疗阶段的整个过程中,药物产品的剂量和对应体积都保持相同。在延长阶段,每次访视时测量患者的体重,并且仅在计算显示需要增加剂量时,才根据体重的增加调整剂量。分别地,上限剂量对于剂量群组1和3为150mg,并且对于剂量群组2为200mg。表2-表5进一步详述了根据患者体重注射的体积。
[0469]
将患者分配到治疗组的方法
[0470]
集中生成随机治疗试剂盒编号的列表。根据此列表包装imp。通过交互式语音响应系统(ivrs)或交互式网络响应系统(iwrs)为患者分配治疗试剂盒。
[0471]
符合入选标准的患者将被包括在内,并将在2个体重组(a组:针对在剂量探索部分期间纳入的患者,≥30kg且≤60kg,并且针对在第二和第三部分期间纳入的患者,≥30kg;b组:针对全部3个部分,《30kg且≥10kg)内分配治疗试剂盒。
[0472]
·
对于剂量探索和第二部分,2个体重组的计划纳入比率为1:1。
[0473]
对于第三部分,2个体重组没有预先规定的纳入比率,但每个体重组的上限为70%(即20名患者)。因此,按体重分组最不平衡的纳入可能将是第三部分中的20名患者对8名患者。
[0474]
包装和加标签
[0475]
imp在含有加标签小瓶的患者治疗试剂盒中提供。
[0476]
储存条件
[0477]
将研究药物在现场或家中保持冷藏在2℃与8℃之间。
[0478]
药代动力学、功效和安全性评估
[0479]
根据以下流程表,患者计划接受12周核心治疗阶段评估(表7)。
[0480]
药代动力学测量和定时
[0481]
12周核心治疗阶段中的血液采集取样时间表可以见于表7。如果患者出现sae,则在可能的情况下,在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度。对于在核心治疗阶段期间(在第12次访视时或之前)过早中断研究治疗的患者,在计划的eot评估后2周(对于剂量群组3患者为在最后一次imp注射后3周,或者[对于剂量群组1和2患者]为在最后一次imp注射后4周)收集另外的萨瑞鲁单抗pk评估。这些患者被要求在eot评估后6周返回进行研究结束(eos)评估。样品采集的日期应已被记录。对于早期中断治疗的患者,在可能的情况下,应在eos访视时已采集血液样品。使用经验证的酶联免疫吸附测定(elisa)法分析血清样品的功能性萨瑞鲁单抗浓度。
[0482]
[0483]
[0484]
[0485]
[0486][0487]
缩写:ada=抗药物抗体,ana=抗核抗体,bp=血压,dna=脱氧核糖核酸,d=天,ebv=爱泼斯坦-巴尔病毒,ec=伦理委员会,eot=治疗结束,esr=红细胞沉降率,hba1c=
糖化血红蛋白,hiv=人类免疫缺陷病毒,hs-crp=高敏c反应蛋白,ig=免疫球蛋白,ivrs=交互式语音响应系统,il=白细胞介素,imp=研究药物产品,jadas=幼年关节炎疾病活动性得分,jia acr=幼年特发性关节炎美国风湿病学会,pk=药代动力学,ppd=纯化蛋白衍生物,sae=严重不良事件,sil-6r=可溶性白细胞介素-6受体,tb=结核病,v=访视,wk=周,yrs=岁。
[0488]
a.对于在12周核心治疗阶段(在第12次访视时或之前)期间中断治疗的患者,将在eot访视后2周(eot++2周)进行另外的萨瑞鲁单抗pk评估。对于纳入第三部分中的患者,第88次访视不适用。
[0489]
b.在第2次访视与第7次访视之间的萨瑞鲁单抗pk取样在患者之间有所不同:对于a组(≥30kg),第3次访视(第3天)、第4次访视(第5天)、第5次访视(第8天)、第6次访视(第12天);对于b组(《30kg)时间表1,在第3次访视(第3天)、第5次访视(第8天)的访视和天数;对于b组(《30kg)时间表2,在第4次访视(第5天)、第6次访视(第12天)的访视和访视天数。萨瑞鲁单抗pk取样可以在家里进行。将提供pk日记,以记录样品采集的日期和时间。对于第3次访视(第3天)、第4次访视(第5天)和第5次访视(第8天)不允许样品采集窗口。对于第6次访视(第12天)允许有
±
1天的窗口。对于纳入第三部分中的患者,第3、4、5和6次访视不适用。然而,在第二次萨瑞鲁单抗给予之前必须进行另外的血液学测试(详情参见脚注“o”)。
[0490]
c.根据研究者的判定,排除标准e
24
中提到的实验室测试可以在筛查访视与首次imp给予之间通过中心实验室再测试来重复,以确保患者符合关于排除标准e
24
的资格。将由需要吉尔伯特综合征基因测试的患者签署一份当地批准的具体同意书(在进行此评估之前必须获得同意书/允许书,并且应遵守当地法规)。
[0491]
d.在所有筛查评估之前,患者(如果他/她已达到基于当地法规的法定同意年龄)、父母一方或双方或者一个或多个法定监护人必须签署ec批准的书面知情同意书并注明日期。患者、父母一方或双方和一个或多个法定监护人将从研究者处接受关于一个或多个研究目标和程序的信息。应获得父母一方或双方或者一个或多个法定监护人允许其子女参加用于药物基因组研究的任选唾液样品采集、并允许主办方保留其剩余/未使用的血液样品用于未来研究的单独书面同意书
[0492]
e..根据当地法规,将由需要吉尔伯特遗传疾病测试的任何患者完成一份单独的(当地批准的)知情同意书。应基于当地法规和患者对研究信息的理解成熟度,从患者处获得签署的允许书。
[0493]
f.关于imp给予的家庭日记要在家中针对给予的imp来完成。
[0494]
g.完整的身体检查将在第1次访视(第-28天至第-1天,最多31天)、第2次访视(第1天,第0周)、第8次访视(第4周)、第9次访视(第6周)和第12次访视(第12周)时进行,包括皮肤、鼻腔、眼睛、耳朵、呼吸系统、心血管系统、胃肠系统、神经系统、淋巴系统和肌肉骨骼系统。
[0495]
h.任选的:基于研究者的判断,可以在筛查时进行爱泼斯坦-巴尔病毒(ebv)滴度检测(包括igg和igm);基于患者的家族史和病史以及研究者的判断,可以在筛查时进行乙型肝炎表面抗原(hbs-ag)、乙型肝炎表面抗体(hbs-ab)、总乙型肝炎核心抗体(hbc-ab)和丙型肝炎抗体(hcv-ab)检测。hiv血清学检查将仅基于研究者对那些疑似hiv患者的评估进行。仅对于阿根廷和德国:必须在筛查访视时对所有患者进行乙型和丙型肝炎以及hiv的血
清学测试,以便筛查对应的排除标准e
14
。
[0496]
i.应在基线第2次访视(第1天,第0周)之前对≤5岁的患者进行纯化蛋白衍生物(ppd)皮肤测试。患者应在放置ppd皮肤测试后48至72小时内接受评估。对于在72小时内未接受评估的患者,应重复皮肤测试。在确定皮肤测试结果时,应考虑对风险(tb接触和/或最近从tb高发国家移居)疫苗接种史水平的评估。对于所有细节,请参考排除标准e
13
。将对》5岁的患者进行干扰素-γ(ifn-r)释放测定即quantiferon-tb测试。(1)在初始tb筛查后,如果ppd或quantiferon结果为阴性,但对tb的临床怀疑高于中度,应基于研究者的评估,在研究期间随时对患者进行tb再测试。基于当地ppd可获得性、tb筛查的当地法规和研究者的判断,将在年轻患者组(≤5岁)中考虑quantiferon-tb测试。
[0497]
j.对在基线第2次访视(第1天,第0周)前3个月内没有进行过胸部x光检查的jia患者开始进行生物疗法之前,仅在基于研究者的判断或根据当地tb筛查指南认为有必要的情况下,才可以对患者进行胸部x光检查。
[0498]
k.剂量方案1和2群组中的患者每隔一周一次,并且剂量方案3群组中的患者每周一次给予研究药物产品。将安排家庭护士在中期访视时对剂量方案3群组患者给予imp(如果可能的话,家庭给予)。在imp给予后,应对患者进行至少30分钟的监测,以发现过敏反应的任何体征或症状。仅对于德国:在首次imp给予后,必须对患者进行至少1小时的监测,以评估局部耐受性。在剂量探索和第二部分中,在12周核心治疗期结束时未实现jia美国风湿病学会(acr)30的患者将中断研究治疗,以根据研究者的临床判断接受护理标准。仅在第12次访视后,主办方将向研究者提供幼年特发性关节炎acr响应(包括jia acr30响应),以评价此标准。对于在第12次访视(第12周)时完成核心治疗阶段,但由于除了缺乏jia acr30响应之外的原因而没有继续到延长阶段的患者,在第12次访视时将不进行imp注射。
[0499]
l.在核心治疗阶段期间,研究药物产品将被分配给处于剂量方案3群组的患者。
[0500]
m.在研究期间,将使用测距仪在现场测量身高。
[0501]
n.幼年特发性关节炎acr核心集包括:医师对疾病严重程度的全面评估、患者或者父母一方或双方/一个或多个监护人对总体健康状况的全面评估、患有活动性关节炎(定义为不是由于畸形导致的肿胀或者伴随疼痛或压痛或两者的活动受限)的关节数量、活动受限的关节数量、儿童健康评估问卷(chaq)、hs-crp(2,3,4)。在筛查时,将仅评估患有活动性关节炎的关节数量和活动受限的关节数量。
[0502]
o.本文解释了幼年关节炎疾病活动性得分评分。。
[0503]
p.血液学(在药物给予之前应抽血):血红蛋白、血细胞比容、红细胞(rbc)计数和形态学(如果rbc计数异常)、白细胞(wbc)分类(嗜中性粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞)、血小板计数、绝对嗜中性粒细胞计数(anc)。另外的血液学测试必须在第6次访视(第12
±
1天)对剂量方案1和2群组进行,以便在第二剂量的萨瑞鲁单抗给予(第7次访视:第2周,第15天)之前获得结果,或者在第5次访视(第8天)之前或之时对剂量方案3群组(不在第4次访视[第5天]之前)进行,以便在第二剂量的萨瑞鲁单抗给予之前获得结果。对于纳入剂量探索和第二部分中的患者,可以在第二剂量的萨瑞鲁单抗给予之前,在中心实验室或当地实验室进行另外的血液学测试,以确认嗜中性粒细胞计数和血小板计数。对于纳入第三部分中但未进行现场第6次访视的患者,将在第二剂量的萨瑞鲁单抗给予之前但不早于第12天,在当地实验室进行另外的血液学测试。如果使用当地实验室,仍应在
如对于剂量方案1和2群组所安排的第7次访视(第15天,第2周)以及对于剂量方案3群组为第5次访视(第8天)时给予imp前抽取用于血液学检查的中心实验室样品。对于所有患者,在对于剂量方案3群组患者的第5次访视(第8天)或对于剂量方案1和2群组患者的第7次访视(第2周,第15天)时给予第二剂量的萨瑞鲁单抗之前,必须审查第2次访视(第1天)和另外的血液学实验室评估。
[0504]
q.化学(在药物给予前应抽血):整个化学检查将仅在筛查访视、基线第2次访视(第1天,第0周)和第12次访视(第12周)或eot时进行:钠、钾、氯、碳酸氢盐、血尿素氮、肌酐和肌酐清除率、肾小球滤过率(使用修改的schwartz公式)、钙、磷酸盐、总蛋白、白蛋白、丙氨酸转氨酶(alt)、天冬氨酸转氨酶(ast)、碱性磷酸酶(alp)、总胆红素、结合胆红素和未结合胆红素。在所有其他访视时:将仅测试alt、ast、alp、总胆红素、结合胆红素、未结合胆红素和白蛋白。
[0505]
r.空腹血脂(在药物给予前应抽血):甘油三酯(tg)、总胆固醇、高密度脂蛋白(hdl)胆固醇、低密度脂蛋白(ldl)胆固醇。需要患者在测试前禁食至少8小时。
[0506]
s.任选的:将仅基于患者的病史和研究者的判断来测量hba1c水平。
[0507]
t.尿分析试纸:比重、ph、葡萄糖、血液、蛋白质、硝酸盐、白细胞酯酶、胆红素。如果试纸上的任何参数异常,则应将尿样送到中心实验室进行测试。如果蛋白质呈阳性,则由中心实验室进行显微镜分析。
[0508]
u.对于已经开始月经来潮的女性,在筛查访视时必须进行血清妊娠测试。
[0509]
v.对于已经开始月经来潮的女性,应在第2次访视(第0周,第1天)、第8次访视(第4周)、第10次访视(第8周)和第12次访视(第12周)时进行尿液妊娠当地测试。可以在当地进行尿液妊娠测试。在暴露于imp和eot之前,应通过尿液妊娠测试检查妊娠状态。
[0510]
w.在治疗期期间的给药日,将在imp给予之前采集血液样品。如果患者出现sae,则在可能的情况下,应在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度和抗药物抗体(ada)评估。在剂量探索和第二部分中,如果患者在12周核心治疗阶段期间过早中断研究治疗,则需要在eot访视后2周进行另外的pk访视,以用于血液采样(第88次访视)。
[0511]
x.对于在核心治疗阶段期间过早中断研究治疗的患者,将在eot评估时测量il-6和总sil-6r。
[0512]
y.父母一方或双方或者一个或多个法定监护人必须在用于药物基因组研究的任选唾液样品采集之前签署一份单独的知情同意书。优选将样品在基线第2次访视(第1天,第0周)时收集,但可以在任何访视时收集。患者还可以基于他/她的年龄、当地法规以及他/她对研究信息的理解成熟度签署知情同意书。如果患者或者其父母一方或双方或者其一个或多个法定监护人不希望患者参与唾液样品采集,则患者仍有资格纳入本研究。
[0513]
药代动力学变量
[0514]
萨瑞鲁单抗pk变量包括在首次剂量后观察到的最大血清浓度(cmax)和在剂量间隔(auc0-τ)期间使用梯形法计算的血清浓度对时间曲线下面积(auc),从基线到第12周重复给药期间在治疗给予前观察到的浓度(c谷)。
[0515]
使用非线性混合效应建模建立了混合数据的群体pk(poppk)模型,以描述萨瑞鲁单抗的pk特征。使用血清中功能性萨瑞鲁单抗的poppk模型计算了以下pk参数(poppk分析
将在另函中报告)。pk参数包括但不限于表8中列出的那些参数。
[0516]
表8.-药代动力学参数和定义列表
[0517]
药代动力学参数定义
[0518][0519]
功效评估
[0520]
jia acr响应
[0521]
在本研究中使用了用于评估体征和症状的jia acr响应水平(参考表7)。jia acr 30/50/70/90/100(jia acr30/50/70/90/100)响应被定义为6个核心集变量中的3个变量自基线改善至少30%/50%/70%/90%/100%同时剩余变量中不多于1个变量恶化多于30%的患者。
[0522]
jia acr核心集包括6个变量:
[0523]
1.疾病活动性的医师全面评估
[0524]
2.总体健康状况的患者/父母评估
[0525]
3.由儿童健康评估问卷(chaq)确定的功能能力
[0526]
4.患有活动性关节炎的关节数量(0至71个关节)
[0527]
5.活动受限的关节数量(0至67个关节)
[0528]
6.高敏c反应蛋白。
[0529]
在12周核心治疗阶段期间,除了第3次访视(第3天)、第4次访视(第5天)、第5次访视(第8天)和第6次访视(第12天)之外,将在每次现场访视时评估jia acr疾病核心集。在延长阶段期间(对于仍保持当前剂量(所选择剂量方案)的患者),将在第24、48、72、96周(第三部分的eot)、120周(不适用于第三部分)、144周(不适用于第三部分)和eot(对于剂量探索和第二部分为第156周/对于第三部分为第96周)时定期地评估jia acr核心集。对于在延长阶段期间变为所选择剂量方案的患者。
[0530]
疾病活动性的医师全面评估
[0531]
研究者被要求在锚定的100毫米(mm)水平视觉模拟量表(vas)上评级患者的疾病活动性,其中0被认为是最佳疾病活动性,并且100是最差的。总体健康状况的患者/父母评估
[0532]
在100mm水平vas上测量总体健康状况的患者/父母评估。要求患者或同一个家长或监护人完成表格,以确保一致性。
[0533]
儿童健康评估问卷(chaq)
[0534]
chaq是一种针对≥8岁儿童和管理小于8岁儿童的家长/代理人的访谈或自我管理工具。chaq是对1至19岁儿童健康状况的一种通用测量。评估总共包括43个项目,并且需要大约10分钟来完成。chaq问卷在imp给药前和随后的时间点完成(根据由卫生经济和结果研究部门提供的指导)。回忆期为1周(singh,g.等人1994arthritis rheum.1994年12月;37(12):1761-9)。
[0535]
在chaq发展中报告的对应于轻度、轻度至中度和中度残疾的chaq得分中值分别是
0.13、0.63和1.75(dempster,h.等人,2001arthritis rheum.44(8):1768-74)。
[0536]
为了消除可能由生长和发育带来的差异,
[0537]
父母被要求仅注意由于病患造成的那些困难(例如,如果儿童因为太小而不能做某项活动,则响应被标记为“不适用”)。评估难度的响应选项基于5分李克特量表
[0538]
(0=没有任何困难,1=有一些困难,2=有很多困难,3=不能做,以及4=不适用)。
[0539]
将chaq分为3个部分:
[0540]
1.残疾指数:评估日常生活运行能力的41个项目,进一步分为8个分量表/域:着装和梳洗;起床;吃饭;行走;卫生;到达;抓握;和活动。对于每个域,评估了:日常功能难以执行的程度评级,需要使用特殊辅助工具或设备,和需要来自他人的帮助。
[0541]
为了计算chaq-di,首先计算每个域得分:
[0542]
■
具有最高响应的问题决定了所述功能领域的得分
[0543]
■
如果使用了辅助工具或设备或需要帮助来完成某个领域的任务,则记录对应功能领域的最低得分为2
[0544]
■
对8个分量表/域进行平均化,以计算平均得分,即残疾指数(范围为0至3)。
[0545]
较低的残疾指数得分表示比健康状况更好/更健康的相关生活质量/较少的体征和症状,而较高的残疾指数得分表示较差的健康状况/较差的hrql/较多的体征和症状(singh,g.等人1994arthritis rheum.1994年12月;37(12):1761-90;klepper s.2003arthritis rheum.49(3):435-43)。chaq残疾指数中的最小临床重要改善是得分降低了0.13。chaq残疾指数中的最小临床重要恶化是变化得分中值为0.75(dempster,h.等人,2001arthritis rheum.44(8):1768-74)。
[0546]
2.不适指数:由过去一周内疼痛的严重程度决定,在vas上评级(锚点为“0无疼痛”和“100非常严重疼痛”)。为了计算chaq不适指数,测量从项目67中vas左端到响应者标记的距离,并乘以0.2。范围是0至3。
[0547]
3.健康状况:病患的患者或父母全面评估的量度。
[0548]
为了计算chaq健康状况得分,测量从项目69中vas左端到响应者标记的距离,并乘以0.2。范围是0至3。
[0549]
患有活动性关节炎的关节数量和活动受限的关节数量
[0550]
活动关节被定义为具有不是由于畸形导致的关节内肿胀,或者具有伴随疼痛或压痛的活动受限的关节。通过计数活动关节的数量,评估了七十一个关节的活动性疾病(bazso,a.等人,2009j rheumatol.36(1):183-90;beukelman,t.等人,2011arthritis care res(hoboken).63(4):465-82)。
[0551]
71个关节包括颈椎(计为1个关节)、颞下颌(2个关节,右侧和左侧)、胸锁(2个关节)、肩锁(2个关节)、肩(2个关节)、肘(2个关节)、腕(2个关节)、掌指(总共10个关节,每侧5个)、近端指间(总共10个关节,每侧5个)、远端指间(总共8个关节,每侧4个)、髋(2个关节)、膝(2个关节)、踝(2个关节)、距下(2个关节)、跗跖(2个关节)、跖趾(10个关节,每侧5个)和脚趾间(10个关节,每侧5个)。
[0552]
除了胸锁关节(n=2)和肩锁关节(n=2)之外,针对活动受限检查的67个关节与针对活动性疾病检查的关节相同。由培训过的评估员进行关节的正式计数。关节压痛被定义
为由评估者的拇指和食指施加的关节压力引起的疼痛。
[0553]
高敏c反应蛋白(hs-crp)
[0554]
在每次访视时,均评估hs-crp。参见表7。高敏c反应蛋白水平与il-6r活性直接相关。预期主动剂量方案将对c反应蛋白(crp)水平具有显著的降低作用。
[0555]
幼年关节炎疾病活动性评分
[0556]
在本研究中使用了用于评估疾病活动性的jadas得分(参考表7)。jadas包括4项测量:
[0557]
1.疾病活动性的医师全面评估,在10厘米(cm)vas上测量,其中0=无活动性且10=最大活动性
[0558]
2.健康状况的父母/患者全面评估,在10cm vas上测量,其中0=非常好且10=非常差
[0559]
3.患有活动性疾病的关节计数
[0560]
4.炎症指数:hs-crp或esr水平:
[0561]
■
根据以下公式将红细胞沉降率归一化为0至10级:(esr[mm/小时]-20)/10,其中在进行计算之前,将《20mm/小时的esr值转换为0,并且将》120mm/小时的esr值转换为120。
[0562]
■
或者,根据以下公式将hs-crp归一化为0至10级:(crp[mg/l]-10)/10,其中在进行计算之前,将《10mg/l的crp值转换为0,并且将》110mg/l的crp值转换为110。
[0563]
jadas被计算为其4个组分得分的简单线性和。参见consolaro,a.等人2009arthritis rheum.61(5):658-66。jadas-27包括以下关节:颈椎、肘、腕、掌指关节(从第一至第三)、近端指间关节、髋、膝和脚踝。jadas被认为是评估jia中疾病活动性的有效工具,并有可能应用于标准临床护理、观察性研究和临床试验(同上)。
[0564]
药效学测量和定时
[0565]
通过测量以下生物标记物来评估萨瑞鲁单抗的药效作用:hs-crp、il-6和总sil-6rα:
[0566]
■
由covance cls使用lloq为0.20mg/l的比浊法(hs)测定测量n高敏crp浓度
[0567]
■
由covance cls使用lloq为3.12pg/ml的经验证的定量夹心酶免疫测定法确定血清il-6浓度
[0568]
■
由covance使用lloq为15ng/ml的经验证的elisa法(doh1163)测量血清总sil-6rα浓度。
[0569]
血液采集的取样时间表可见于表7中的研究图表。
[0570]
临床实验室评价
[0571]
所有实验室变量(中心实验室值和自基线的变化)的汇总统计数据(包括数量、平均值、中值、sd、最小值和最大值)是针对每次访视或研究评估(基线、每个基线后时间点、终点)按剂量群组计算的并且按生物学功能组织。通过生物学功能和剂量群组汇总了teae时期期间任何时候pcsa的发生率和异常实验室值。对于脂质,基线使用ncep aptiii分类。按最高级别汇总了嗜中性粒细胞减少症、血小板减少症和淋巴细胞减少症的发生率。
[0572]
药代动力学数据分析
[0573]
使用描述性统计学汇总了血清中功能性萨瑞鲁单抗的浓度。使用pk群体进行所有pk分析。这由安全群体中具有至少1个剂量后非缺失萨瑞鲁单抗浓度值的所有患者组成。使
用描述性统计学(包括数量、算术和几何平均值、sd、平均值的se(sem)、cv、最小值、中值和最大值)按剂量、总体和体重组对每次访视时血清中功能性萨瑞鲁单抗的谷浓度进行汇总。如果前一次给药时间在每隔一周方案的取样时间前《11天或》17天;在每周方案的取样时间前《5天或》9天,则样品被认为不符合这些分析的条件。在给药前(第0周),样品的低于lloq的浓度被设置为零。低于lloq的其他浓度被lloq/2替代。使用非线性混合效应建模建立了poppk模型,以描述萨瑞鲁单抗的pk特征。poppk模型在独立于csr的文件中呈现。
[0574]
药效学数据分析
[0575]
从基线到第12周,使用描述性统计学(包括数量、算术和几何平均值、sd、sem、cv、最小值、中值和最大值)按各自的剂量和体重组对血清中的il-6、总sil-6rα和hs-crp的浓度进行汇总。
[0576]
药代动力学-药效学数据分析
[0577]
进行探索性分析以阐明与关键功效终点和/或生物标记物的暴露-响应关系。与关键功效终点的暴露-响应关系的结果在独立于csr的文件中呈现。
[0578]
人口统计和其他基线特征
[0579]
本文的结果涉及本研究12周核心阶段的剂量探索部分。
[0580]
人口统计学
[0581]
在表9中按治疗组描述了所有治疗群体的人口统计学和患者特征。总的来说,患者主要是女性(64.3%)和白种人(78.6%),其中在体重和剂量组中的分布相似。分别地,平均体重和年龄在a组中为45.0kg和13.0岁并且在b组中为19.7kg和5.2岁。九名(21.4%)患者是在南美国家招募,并且33名(78.6%)患者是在西部地区和世界其他地区纳入。这些人口统计学特征在每个体重组内的三个剂量群组中具有可比性。详细基线特征在表9中呈现。
[0582]
病史
[0583]
剂量群组1、2和3中分别有总共9名[69.2%]、11名[78.6%]和9名[60.0%]患者有任何病史。最常见的原发性soc是在总共11名患者中发现的感染和侵染,其中超过半数在剂量群组2中(6名[42.9%]患者)。来自a组的剂量群组2中的一名患者在进入本研究前3.5年因外科手术而具有肠道问题史,其符合排除标准21,并且随后导致永久中断治疗。
[0584]
在基线时,患者具有高疾病活动性,其平均jadas-27-crp为20.6,平均活动关节计数为13.9,并且平均crp为9.5mg/l。伴服药物和既往药物使用在体重组之间是平衡的(表21)。
[0585]
[0586]
[0587]
[0588][0589]
百分比是使用被评估为分母的患者数量来计算的。
[0590]
基线时的疾病特征
[0591]
在表10中按治疗组描述了所有治疗群体的基线疾病特征。基线时的疾病特征在每个体重组内的三个剂量群组中具有可比性。患有rf阴性pcjia、rf阳性pcjia和e-ojia亚型
的患者分布分别是64.3%、21.4%和14.3%。在2个体重组中,rf阴性pcjia的分布相似;而与b组相比,a组中患有rf阳性pcjia的患者数量是约两倍,并且患有e-ojia的患者数量是一半。
[0592]
正如预期的那样并且由于剂量群组的年龄,在剂量群组中,自诊断以来,a组的jia持续时间中值(2.6年[范围:0.3至15.1年])高于b组(1.1年[范围:0.1至7.2年])。活动关节和活动受限关节的数量中值分别是10.0(范围:5.0至45.0)和8.0(范围:1.0至46.0),其中分成细目:a组中为15.5(范围:5.0至45.0)和8.5(范围:1.0至46.0)并且b组中为8.5(5.0至32.0)和7.0(范围:1.0至34.0)。基线jadas-27-crp中值是19.3(范围:8.5至35.5)(在a组和b组中分别是23.2[范围:9.2至33.8]和16.8[范围:8.5至35.5]),反映了具有高度活动性疾病的群体。a组和b组中的基线hs-crp中值分别是1.1mg/l(范围:0.1至86.5)(1.0mg/l[范围:0.1至45.9]和1.6mg/l[范围:0.1至86.5]。在体重和剂量组中,anc的分布相似。
[0593]
总的来说,进入本研究的76.2%患者接受伴随的csdmard治疗并且31.0%接受伴随的口服糖皮质激素;28.6%患者接受了≥1次在本研究之前停止的bdmard治疗。其他患者和疾病特征在体重组之间具有可比性。
[0594]
[0595]
[0596]
[0597][0598]
治疗依从性的测量
[0599]
如果患者没有按照方案的要求接受计划剂量的imp,则认为给定的给予是非依从
的。患者的依从性百分比被定义为患者依从的给予次数除以患者被安排在最后一次给药日期之时或之前接受的总给予次数。
[0600]
总体依从性是97.6%。对imp摄入的依从性是96.4%。在剂量体重组中,依从率是相似的。由于嗜中性粒细胞减少症导致的剂量中断,b组剂量群组2中的一名患者的依从性《80%。没有患者接受了错误的剂量或过量。
[0601]
在纳入的42名患者中(a组=20,b组=22),34名完成了12周核心治疗阶段(a组=17;b组=17),并且8名患者中断:5名因治疗突发性不良事件(ae),2名因研究者判断缺乏功效,并且1名因依从性差(图8)。
[0602]
药代动力学和药效学评价
[0603]
血清中功能性萨瑞鲁单抗的浓度
[0604]
血清中功能性萨瑞鲁单抗的所有给药前浓度均低于lloq(0.3125mg/l)。对于每个治疗组,用描述性统计学分析12周核心治疗阶段中血清中的单独功能浓度。首次sc给予后观察到的萨瑞鲁单抗的平均(sd)浓度在图3中示出。在时间曲线上观察到的萨瑞鲁单抗的平均(sd)谷浓度在图4中示出。
[0605]
药代动力学参数
[0606]
从群体pk模型(poh0516)估算的在首次剂量后萨瑞鲁单抗pk参数的描述性统计在表11a中示出,并且在重复给药后的描述性统计在表11b中示出。在患有pcjia的患者中,萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk(图3)。auc0-τ是对于q2w方案具有2周剂量间隔或对于qw方案具有一周剂量间隔的auc。在第10周至第12周重复萨瑞鲁单抗的sc剂量后,使用auc0-14天来比较不同q2w和qw方案的暴露(对于qw方案,auc0-14天=2
×
auc0-τ,以及对于q2w方案,auc0-14天=auc0-τ)。
[0607]
表11a.-患有pcjia的儿科患者中,首次sc给予后单独萨瑞鲁单抗暴露的平均值(cv%)[中值]
[0608][0609][0610]
表11b.-患有pcjia的儿科患者中,在第10周至第12周或者第11周至第12周重复sc
[0611]
给予后单独萨瑞鲁单抗暴露的平均值(cv%)[中值]
[0612][0613]
缩写:auc0τ:在2周(q2w方案)或一周(qw方案)的剂量间隔τ期间血清浓度对时间曲线下面积,cmax=观察到的最大血清浓度,c谷=重复给药期间在治疗给予前观察到的浓度,cv=变异系数,pcjia=多关节病程幼年特发性关节炎,q2w=每隔一周一次,qw=每周一次,sc=皮下。
[0614]
群体pk模型衍生的贝叶斯暴露
[0615]
在第10至第12周重复q2w sc和qw给予后,萨瑞鲁单抗暴露以大于剂量比例的方式增加,其中从剂量群组1到剂量群组2对于萨瑞鲁单抗剂量的1.5至1.6倍增加,auc0-14天增加2.4至2.6倍,并且从剂量群组1到剂量群组3对于萨瑞鲁单抗剂量的2倍增加,auc0-14天增加3.4至4.4倍。
[0616]
与首次剂量相比,在第12周,剂量群组1和2的累积为1.9至2.2倍,并且剂量群组3的累积为4.1至4.5倍。(表22)在2个体重组中测试的剂量方案在每个剂量群组中实现了相似的暴露。在患有pcjia的患者中,重复sc剂量后,cmax和auc0-τ的变异性在13.6%至36.6%的范围内。
[0617]
对12周剂量探索研究的数据进行了分析。对按体重分为2组的患者的数据进行分析:a组(30-60kg)和b组(10-《30kg)。如上所述,患者接受顺序递增剂量的萨瑞鲁单抗:剂量1(a/b组):2.0/2.5mg/kg q2w;剂量2(a/b组):3/4mg/kg q2w;剂量3(a/b组):2.0/2.5mg/kg qw。pcjia剂量的目标是在成人中实现类似于约150mg q2w、200mg q2w和150mg qw的暴露。如上所述,主要结果是药代动力学(pk),并且次要结果是萨瑞鲁单抗的安全性、pd和功效。
[0618]
数据显示42名患者被纳入(20/22,在a/b组中),并且平均年龄是13.0/5.2岁。在基线时,在a/b组中,平均pcjia持续时间、活动关节数量和jadas27-crp分别是4.6/1.7年、17.2/11.0和22.2/19.1。与成人患者一样,萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk。在重复sc给予后,暴露以大于剂量比例的方式增加,并在12周内累积了1.9-4.5倍。在两个体重组中,对于每个剂量,萨瑞鲁单抗暴露是相似的(表12),并且与对应的成人剂量是相当的。在36/42(85.7%)的患者中报告了治疗突发性不良事件(剂量和体重组间相
当);感染(28/42,66.7%)是最常报告的ae。鉴定了12例3/4级嗜中性粒细胞减少症,主要在剂量3(n=6)和b组(n=8)中。无一例与感染相关;几天内全部消退。总的来说,4名患者由于嗜中性粒细胞减少症而中断治疗,并且1名患者由于丙氨酸转氨酶升高而中断治疗。无严重ae,无胃肠道穿孔病例,并且无死亡。到第12周,如在治疗中观察到的:所有患者达到jia acr30;50%、62%和100%的患者分别以剂量1、2和3达到jia acr70;在剂量1、2和3中,jadas27-crp自基线的平均变化分别是-74.6%、-73.1%和-87.9%。
[0619][0620]
药代动力学结论
[0621]
在患有pcjia的患者中,萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk。
在重复sc给予后,萨瑞鲁单抗暴露以大于剂量比例的方式增加,并在12周内累积了1.9至4.5倍。对于每个剂量群组,在2个体重组中测试的剂量方案实现了相似的暴露。对于另外的患者a组(30-60kg)和患者b组(10-《30kg)的数据分析,观察到萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk。此外,所有剂量方案都证明对降低疾病活动性有效。安全性与类效应一致;使用剂量3时并且在体重为10-《30kg的患者中,观察到嗜中性粒细胞减少症的较高发生率。
[0622]
crp、esr、il-6和sil-6r浓度
[0623]
从基线到第12周,对每个治疗组分析hs-crp、esr、il-6和总sil-6rα浓度的描述性统计。对于每个群组均获得了随时间轮廓变化的平均(se)crp、esr、il-6和sil-6rα浓度。在sc给予萨瑞鲁单抗后,与剂量群组1相比,剂量群组2和3中对crp的平均抑制和sil-6rα的增加更大。在剂量群组2和3中,平均crp浓度在2至4周内降低,其中与剂量群组1相比,在剂量群组2和3中观察到的降低更大且更快(图10a)。从基线到第12周,使用剂量1、2和3,平均crp浓度的变化分别是7.4至5.8mg/l、14.6至0.5mg/l和6.5至0.1mg/l。在第2周与第6周之间,所有患者使用剂量2和3实现了正常的crp浓度。在第4周时,与使用剂量1时为25.0%相比,大多数患者使用剂量2和3实现了低于检测限(《0.2mg/l)的crp水平(分别是71.4%和83.3%,如在治疗中观察到的)。这种效应持续一段时间,其中与使用剂量1时为30.0%相比,在第12周时,分别有66.7%和90.9%的患者使用剂量2和3实现crp《0.2mg/l(图10b)。
[0624]
a组剂量群组1中的高平均crp值主要由一名患者驱动,其在12周核心治疗阶段期间的crp保持升高(在40至110mg/l之间)。
[0625]
第12周时的红细胞沉降率与对于crp观察到的一致。一般来说,在第12周时,平均红细胞沉降率(esr)浓度降低,并且平均il-6和sil-6rα浓度升高。与crp水平一样,观察到剂量群组2和3的降低更多,其中剂量群组1、2和3从基线到第12周的平均变化分别是22.2至18.2mm/h、21.4至6.9mm/h和21.9至4.4mm/h。
[0626]
生物标记物的药效学药代动力学关系
[0627]
进行探索性分析以阐明与pd生物标记物的暴露-响应关系。在第12周时,将单独的crp、esr、il-6和sil-6rα浓度相对于血清谷浓度进行绘图。在单独的il-6水平与萨瑞鲁单抗谷浓度之间没有明显的关系。数据显示,在患有pcjia的单独患者中,在重复给药后,总sil-6rα随着萨瑞鲁单抗谷浓度的渐增而增加。与萨瑞鲁单抗谷浓度≤1mg/l的患者相比,萨瑞鲁单抗谷浓度》1mg/l的单独患者中的c反应蛋白水平较低(≤1mg/l)。与剂量群组1的患者相比,剂量群组2和3中对crp的抑制更大。与萨瑞鲁单抗谷浓度≤1mg/l的患者相比,萨瑞鲁单抗谷浓度》1mg/l的单独患者中的红细胞沉降率较低(≤10mm/小时)。与剂量群组1的患者相比,剂量群组2和3中的esr抑制作用更大。
[0628]
在sc给予萨瑞鲁单抗后,与剂量群组1相比,剂量群组2和3中对crp的抑制和sil-6rα的增加更大。一般来说,在第12周时,相对于基线,平均esr浓度降低,并且平均il-6和sil-6rα浓度升高。
[0629]
安全性2周核心治疗阶段。总暴露(患者-治疗年数)在剂量组与体重组之间是相似的,其中在a组和b组中分别是4.6年和5.0患者-年。
[0630]
没有严重ae和死亡(表23)。最常观察到的ae是感染和嗜中性粒细胞减少症(分别是n=28[66.7%]和n=11[26.2%])。剂量和体重组中的感染率是相似的,并且主要是由上
呼吸道感染引起的(n=9[21.4%];表25)。未报告严重感染。未鉴定出肺炎或结核病病例。发生了一例口腔念珠菌病(剂量群组3,a组),其在不进行纠正性治疗的情况下自行消退,并且萨瑞鲁单抗治疗没有中断。没有患者因任何感染而中断治疗。
[0631]
预先指定的实验室监测鉴定了12名具有暂时性3/4级嗜中性粒细胞减少症的患者(28.6%)。在这些患者中,3人接受了剂量1(a组2人,b组1人),3人接受了剂量2(a组1人,b组2人),并且6人接受了剂量3(a组1人,b组5人;表24)。所有患者均无症状,但3名患者需要方案规定的治疗中断(以下中各1名:剂量2,b组;剂量3,a组和b组)。没有嗜中性粒细胞减少症的事件与感染相关,并且在几天内全部消退了。实验室监测,包括红细胞计数、血小板计数、脂质水平和肝功能评估,未揭示任何其他潜在的临床显著异常。具体地,没有如由hy定律定义的由药物引起的肝损害病例(21)。
[0632]
5名患者(11.9%)由于ae发生了方案规定的永久性中断治疗。其中,1例是在第2周时剂量1,a组中的3级嗜中性粒细胞减少症;3例是剂量3,b组中的4级嗜中性粒细胞减少症;并且1例是剂量1,a组中的丙氨酸转氨酶(alt)增加(在第4周时》5
×
正常的上限[uln])。
[0633]
七名患者报告了注射后的局部反应(a组4名和b组3名)。这些反应主要是红斑和疼痛;强度均为轻度至中度;没有导致治疗中断。在第12周时,两名患者的抗药物抗体测试呈阳性:剂量1,a组中的一例(中和抗体阳性;由于在第4周时alt》5
×
uln而中断治疗);剂量3,b组中的一例(中和抗体非阳性;由于在首次注射后的4级嗜中性粒细胞减少症而中断治疗)。
[0634]
功效评价
[0635]
jia acr响应率
[0636]
在这项研究中,使用jia acr响应水平来评估体征和症状。jia acr30/50/70/90/100响应被定义为6个核心集变量中的3个变量自基线改善至少30%/50%/70%/90%/100%同时剩余变量中不多于一个变量恶化多于30%的患者。
[0637]
对于正在接受治疗的患者通过直接观察的方法(“如在治疗中观察到的”)并且对于中断治疗的患者通过归责(“非响应归责方法”,其中中断治疗的患者在中断治疗后的随访中自动被视为非响应者)来计算jia acr响应率。在第12周时所有剂量和体重组中的jia acr响应率在表13中汇总。jia acr响应率的精度估计值在表33中示出。
[0638]
如在治疗中观察到的,在第12周时,所有剂量和体重组中的jia acr30响应率是100%。在剂量群组1、2和3中,jia acr70响应率分别是50.0%、61.5%和100%。如由非响应归责方法所计算,在第12周时,剂量群组1、2和3中的jia acr30率分别是76.9%、92.9%和73.3%。在剂量群组1、2和3中,jia acr70响应率分别是38.5%、57.1%和73.3%。
[0639]
总的来说,使用如在治疗中观察的方法,在第2周的首次评估中,在所有剂量群组和体重组中都看到了jia acr30响应的发生。对于剂量群组2(a组和b组),分别在第4周和第8周实现趋于100%的稳定水平,并且对于剂量群组3(a组和b组),分别在第6周和第8周实现趋于100%的稳定水平,而剂量群组1中的响应率波动直到第12周(图5)。非响应归责方法的结果在图6中绘出。
[0640]
jia acr组分
[0641]
在12周核心治疗阶段期间,对jia acr组分的数据进行了汇编。第12周时的jia acr组分的汇总在表14中示出。在第12周时,所有剂量群组和体重组的所有jia acr组分均
显示出自基线的下降,其与剂量群组1相比,通常在剂量群组2和3中更明显地出现。在整个12周核心治疗阶段,两个体重组中所有剂量群组的患者患有活动性关节炎的关节数量都减少了。在第12周时,观察到剂量群组3,a组中患者最大的平均下降(-16.5)。在整个12周核心治疗阶段,两个体重组所有剂量群组中患者患有活动受限的关节数量都减少了。在第12周时,对于剂量群组3,b组中的患者,观察到最大的平均下降(-11.3)。在整个12周核心治疗阶段,两个体重组所有剂量群组中患者的医师全面vas都降低了。在第12周时,对于剂量群组3,b组中的患者,观察到最大的平均下降(-5.0)。在整个12周核心治疗阶段,两个体重组所有剂量群组中患者的患者全面vas都降低了。在第12周时,对于剂量群组3,b组中的患者,观察到最大的平均下降(-5.7)。总的来说,在整个12周核心治疗阶段,两个体重组所有剂量群组中患者的chaq-di得分都降低了。在第12周时,对于b组,剂量群组2中的患者,观察到最大的平均下降(-1.0)。在第12周时,两个体重组的所有剂量群组中的平均hs-crp浓度均下降。剂量群组2和3中的平均crp浓度在2至4周内降低。剂量群组1,a组中的高平均crp值主要由一名患者引起,所述患者在12周核心治疗阶段期间的crp保持升高(在40至110mg/l之间)。在第12周时,对于b组,剂量群组2中的患者,观察到最大的平均下降(-15.2)。
[0642]
幼年关节炎疾病活动性评分
[0643]
疾病活动性通过jadas-27 crp得分进行评估。分析每个剂量群组和体重组中随时间的jadas-27 crp得分。表15中提供了12周核心治疗阶段期间jadas-27(esr)的汇总。研究者报告的teae在表30中示出。数据显示,与基线相比,从第2周开始,所有剂量群组和体重组都显示出jadas-27 crp得分的下降。在第12周时,剂量群组1、2和3中自基线的平均百分比变化分别是-74.6%、-73.1%和-87.9%。
[0644][0645]
表14.-第12周时按剂量群组和体重组汇总的jia acr组分-功效群体
[0646][0647][0648]
表14.续
[0649][0650]
缩写:chaq-d1=儿童健康评估问卷残疾指数,hs-crp=高敏c反应蛋白,vas=视觉模拟量表。
[0651]
表15-在12周核心治疗阶段期间,按剂量群组
[0652]
和体重组对jadas-27 crp的汇总-功效群体
[0653]
[0654]
[0655][0656]
功效结论
[0657]
从第2周开始,在所有剂量群组和体重组中均看到如通过jia acr响应评估的体征和症状的改善,无论通过在治疗中观察到的还是非响应归责方法都如此。没有剂量群组由于缺乏功效而过早地停止。如由研究者所判断,两名患者由于功效不足而中断治疗:1名在第10周时的剂量群组1,b组中;并且另一名在第4周时的剂量群组3,b组中。使用如在治疗中观察的方法,在第12周时,在所有剂量和体重组中,jia acr30响应率是100%(34/34),其中与剂量群组1相比,剂量群组2和3产生更快速和持续的jia acr30响应(图9a)。在第12周时,如通过非响应归责方法所计算,77%、93%和73%的患者实现了jia acr30(分别为剂量群组1、2和3;图11)。大部分患者还达到了较高的响应阈值,其中如在治疗中观察到的,在第12周时,剂量群组1、2和3分别有50%、62%和100%的患者实现了jia acr70并且40%、31%和64%的患者实现了jia acr90(图9b、9c;根据图11b、11c所示的非响应归责方法的结果)。3种萨瑞鲁单抗剂量方案中的每一种都减少了所有jia acr组分(表26)。
[0658]
与剂量群组1相比,在剂量群组2和3中有更快速的jia acr响应的趋势。使用如在治疗中观察的方法,剂量群组2和3(a和b组)在萨瑞鲁单抗治疗的前2个月内,jia acr30响应发生率实现趋于100%的稳定水平,而剂量群组1中的响应率波动直到第12周。
[0659]
在第12周时,在所有剂量群组和体重组的患者中看到,通过jadas-27 crp评估的疾病活动性降低,其中从第2周开始还看到,剂量群组1、2和3中自基线的平均百分比变化分别是-74.6%、-73.1%和-87.9%(图9d)。在事后分析中,在第12周时,20名患者实现了低疾病活动性(lda;jadas-27-crp≤3.8):剂量群组1、2和3中分别有6、5和9名患者。如由jadas-27-crp≤1定义的临床非活动性疾病(cid)由3名患者(剂量群组2中1名和剂量群组3中2名)实现。如由华莱士标准定义的cid由9名患者(剂量1、2和3中分别有3、2和4名患者)实现(22)。十三名患者在第12周时没有活动关节,其中剂量群组1和2中各有4名,并且剂量群组3中有5名(表27)。
[0660]
讨论
[0661]
在本研究中测试的萨瑞鲁单抗的实验剂量方案是基于来自成人ra患者的萨瑞鲁单抗pk数据的建模和模拟而选择的。儿科患者的体重范围、身体组分和药物代谢与成人不同,并且对治疗的响应和副作用可能因年龄而异。因此,不建议在儿科临床试验中不经过测试而直接从成人剂量推断儿科剂量(23,24)。单克隆抗体的清除率经常以相对于渐增体重而言的非线性方式增加(25),因此考虑到pcjia群体中体重和年龄的范围,体重调整剂量被认为适合于本研究。基于群体pk模拟,选择在患有pcjia的患者中测试的剂量方案,以在两个体重组中实现类似的暴露,这将与在患有ra的成年患者中研究的剂量方案暴露相当。研究结果支持基于体重的原始模型设计的准确性。萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk。体重调整剂量方案在两个体重组中产生了相当的暴露,其中所实现的暴露与在患有ra的成人中测试的固定剂量方案相当。
[0662]
在这项研究中,萨瑞鲁单抗治疗与平均crp值的降低和esr的降低相关,证明了患
有pcjia的患者中il-6的功能抑制(20)。具体地,已知crp水平的降低是il-6中和的直接结果,并表明il-6中和在患有ra的患者中的有效性(26-28)。剂量群组2和3的平均crp降低更明显,这些患者中有更多的人实现并维持crp水平低于本研究中利用的高灵敏度方法的检测限(《0.2mg/l)。
[0663]
从第2周开始,在萨瑞鲁单抗的所有剂量方案中均看到jia acr响应。基于jia acr70,与剂量群组1相比,剂量群组2和3的疾病体征和症状的减少显现更显著和持续。如由jadas-27-crp所评估,与剂量群组1或2相比,剂量群组3中更多的患者实现cid或lda。这些功效结果与在pd结果中观察到的明确趋势(即crp浓度)平行,进一步支持了il-6中和与治疗响应之间的关系(20,29)。总体而言,剂量群组1中对crp的持续影响较小,这些结果支持了将剂量群组1排除在进一步研究之外。
[0664]
在本研究中观察到的治疗响应率与用il-6r阻断剂托珠单抗治疗的患pcjia患者的更大群组已发布数据相比是有利的(18)。当前结果表明,用萨瑞鲁单抗治疗可符合由国际工作组最近发布的指南的目标,其将治疗的最终目标建立为疾病缓解,并建议在3个月内达到在疾病活动性方面≥50%的改善,并且一旦实现则应维持治疗目标(11)。本文所述的研究纳入了已确立疾病的患者,其中三分之一的患者已用生物剂治疗失败≥1次。然而,在接受剂量群组2或3的24名患者中,在3个月时,23名和19名分别实现了jia acr50和jia acr70,14名实现了jadas-27-crp lda,并且9名没有活动关节。在研究的延长阶段期间进行长期随访将提供对萨瑞鲁单抗满足长期治疗目标以及一旦实现则维持持久临床缓解的能力的更深入了解。
[0665]
研究中采用萨瑞鲁单抗观察到的ae类型与在成人患者萨瑞鲁单抗治疗中观察到的一致,并且与儿科患者中抗il-6类效应观察到的一致(18,30-32)。很少有局部注射部位反应的报告,并且强度都是轻度到中度的,展示出良好的局部耐受性;这在儿科患者中很重要,因为害怕用注射筒注射引起的疼痛是常见的。最常报告的ae是感染和嗜中性粒细胞减少症。感染率主要是由上呼吸道感染引起的,这在儿科群体中很常见,并且在体重和剂量组之间是平衡的。未报告严重感染、导致中断治疗的感染、和机会性感染。
[0666]
根据研究方案,在整个治疗中的时期(包括第一次萨瑞鲁单抗给药后和第二次给予前)定期地评估绝对嗜中性粒细胞计数(anc)。这种彻底的血液学监测,其中在q2w方案的第12天和qw方案的第5-8天进行首次剂量后评估,导致检测到12例3/4级嗜中性粒细胞减少症。在用萨瑞鲁单抗或托珠单抗治疗的成年患者中,首次治疗给予后几天(第7天前)出现anc最低点,此后anc值恢复,与类效应一致(33)。基于这些来自成人研究的数据,本研究中的评估定时有助于解释剂量群组3(n=6,包括第5天发现的4例病例)与其他2种剂量方案(n=3,在剂量1和剂量2中)相比的事件数量失衡。
[0667]
在12例3/4级嗜中性粒细胞减少症中,8例发生在b组中。还在用托珠单抗治疗的pcjia患者中看到,更年轻、较低体重患者的嗜中性粒细胞减少症有明显增加的趋势(34,35)。没有明确的机制来解释为什么更年轻患者比更年长的儿科患者或成人更容易出现嗜中性粒细胞减少症,但总的来说,已报告儿童中的嗜中性粒细胞减少症的患病率更高(36)。在本研究中,嗜中性粒细胞减少症是导致大部分ae引起的中断治疗的原因(n=4/5):3例是4级嗜中性粒细胞减少症,根据方案定义的标准导致中断治疗,并且1例是患有3级嗜中性粒细胞减少症的患者,其基线anc低于2,000/mm3。重要的是,在这项研究期间鉴定的3/4级嗜
中性粒细胞减少症病例中没有一例与感染相关,并且大多数病例在几天内得到消退。这一观察结果与从用托珠单抗或萨瑞鲁单抗治疗的儿童和成人获得的数据一致,增加了进一步的数据以支持由il-6抑制引起的嗜中性粒细胞减少症不会引起感染风险增加的结论(18,32-34,37)。尽管所有可用数据都支持由il-6抑制引起的嗜中性粒细胞减少症与感染风险增加无关的结论,但研究中获得的嗜中性粒细胞减少症数据,以及在托珠单抗或萨瑞鲁单抗给予后在成人中anc的动态变化,反对选择剂量群组3(32,33,37)。这种谨慎的选择降低了嗜中性粒细胞减少症的风险,尤其是在风险较高的幼儿中。此外,在剂量2中使用2周的给药方案允许更长的anc恢复时间。
[0668]
在对来自本研究12周剂量探索部分的所有数据进行审查后,选择剂量群组2作为在本研究核心治疗阶段的第二部分中纳入另外的患者的最佳萨瑞鲁单抗方案,并用于已经纳入剂量群组1或3并持续到第二部分的患者。与群组1和3相比,剂量群组2方案显示了功效与安全性之间的有利平衡。此剂量方案实现了与等效成人剂量方案200mg q2w(是患有ra的成人中的推荐批准剂量)相似的暴露,并反映了其功效特征(20,30,31)。剂量群组2还遵循了2周的给药方案(与剂量群组3的每周方案相比),其减少了注射压力,这在儿童中尤其有意义,并且如上所述,增加了anc恢复时间。
[0669]
将通过纳入将接受所选择剂量方案并将在研究的延长阶段中被跟踪的另外的患者来提供在更大的患者群组中关于萨瑞鲁单抗在患有pcjia的患者中随时间推移的安全性和功效的另外的数据。如所述,这项研究不是盲法的,并且不包括安慰剂或活性对照。这是由于考虑到所述疾病潜在的慢性和严重性质,以及在患有关节炎的成人和儿童中存在关于il-6抑制的可用数据,使儿童暴露于无效治疗的伦理关怀(18,38)。这些问题反映在以下中:研究设计(采用测试剂量的顺序方法和频繁监测),使用独立数据监测委员会在整个研究中正式评估萨瑞鲁单抗的安全性,使用dec监测患者对治疗的响应,以及dec中断患者接受任何视为无效的剂量并将其返回研究者批准的标准治疗的能力。
[0670]
总之,在患有pcjia的患者中,萨瑞鲁单抗展现出具有靶标介导的药物处置的非线性pk,显示出与抗il-6类效应一致的安全性,并降低了疾病活动性。本研究核心治疗阶段的此12周剂量探索部分显示,剂量群组2(30-60kg患者中为3.0mg/kg q2w,并且10-《30kg患者中为4.0mg/kg q2w)实现了与患有ra的成人中200mg q2w的批准剂量相似的暴露。剂量群组2在患有pcjia的患者的安全性/耐受性与功效之间提供了良好的平衡,并将作为pcjia中的最佳萨瑞鲁单抗剂量方案被进一步研究。
[0671]
实施例2:在患有全身型幼年特发性关节炎(sjia)的1至17岁的儿童和青少年中进行的用皮下(sc)注射给予的萨瑞鲁单抗(随后是延长阶段)的开放标签、顺序、递增重复剂量探索研究。(研究编号dri13926,研究标题:在患有全身型幼年特发性关节炎的儿童和青少年中进行的萨瑞鲁单抗的重复剂量探索研究)
[0672]
计划了阶段2受控试验[nct02991469],以测试皮下给予用于治疗sjia的人igg1抗il-6rα单克隆抗体萨瑞鲁单抗。这项研究针对的是1至17岁患有sjia的儿童和青少年,sjia是类似于成人发作的still病的jia子集,是一种相对罕见的炎性疾病(估计全球男性和女性发病率分别为每10万人0.22和0.34;bennett,a.n.等人,adult onset still's disease and collapsing glomerulopathy:successful treatment with intravenous immunoglobulins and mycophenolate mofetil.rheumatology(oxford).2004;43(6):
795-9)。在这一儿科群体中,计划测试萨瑞鲁单抗的3种剂量方案。
[0673]
研究设计
[0674]
方案的描述
[0675]
研究dri13926是在对标准疗法响应不足或不耐受并且将接受q2w或qw给予的萨瑞鲁单抗sc注射的1至17岁(或国家规定的年龄要求)患有sjia的儿童和青少年中进行的多国、多中心、开放标签、顺序的、2阶段研究。2个阶段如下:
[0676]
1.12周核心治疗阶段,分为2个部分:
[0677]
·
第一个顺序、递增剂量群组、剂量探索部分,其中将在2个体重组中研究至少2个剂量方案:≥30kg且≤60kg的患者(a组)和《30kg且≥10kg的患者(b组),每个剂量方案体重组有6名可评价的患者(总共至少24名患者)。患者纳入将按体重组和剂量方案错开,从a组(≥30kg且≤60kg)和剂量群组2开始。
[0678]
·
随后的部分,其中大约24名另外的患者(每个体重组中12名:≥30kg[a组]和《30kg且≥10kg的患者[b组])将被直接纳入所选择剂量方案(基于来自纳入第一部分中的患者的综合数据而鉴定),以在此所选择选剂量方案下实现每个体重组总共18名可评估患者。
[0679]
12周核心治疗阶段是从第2次访视(基线-第0周)到第12次访视(第12周)给予研究药物产品(imp)的时间。在12周核心治疗阶段期间,为了最小化抽血量和访视次数,同时保持对主要终点的评价,b组(《30kg和210kg)的患者将被随机分配到以下萨瑞鲁单抗pk取样时间表1或2中:
[0680]
·
时间表1:基线、第3天、第8天、第2周、第4周、第8周和第12周
[0681]
·
时间表2:基线、第5天、第12天、第2周、第4周、第8周和第12周
[0682]
2.144周延长阶段:
[0683]
第12次访视的imp被认为是延长阶段的第一个imp。
[0684]
仅在第12次访视(第12周)时达到jia acr30(在没有发烧的情况下)响应的患者将被允许继续进行延长阶段。患者将继续接受他们在本研究的12周核心治疗阶段中被分配接受的萨瑞鲁单抗的相同剂量方案,直到所选择剂量方案被确定。一旦选择了剂量方案,尚未采用此剂量方案的患者将其剂量方案调整至所选择剂量方案,并将遵循新的访视时间表,其中与未调整剂量方案的患者相比,在前12周进行更频繁的监测(pk、安全性和功效)。
[0685]
在第27次访视时,将使用eot程序评估过早中断研究治疗的患者。这些患者将被要求在eot访视后6周(eot+6周)返回进行研究结束(eos)评估。对于在12周核心治疗阶段期间中断研究治疗的患者,在eot访视后2周(eot+2)将进行另外的萨瑞鲁单抗pk评估,并在eot访视时将测量il-6和sil-6r。这些患者将被要求执行除了萨瑞鲁单抗给予之外的所有方案安排的访视和评估,直到第12次访视。
[0686]
测试的剂量方案
[0687]
对于每个体重组,最初计划测试的3个顺序递增剂量方案是基于pk建模定义的,其基本原理如下:
[0688]
■
剂量群组1:类似于萨瑞鲁单抗150mg q2w的靶向pk暴露的剂量,是患有ra的成年患者中的最低有效剂量
[0689]
■
剂量群组2:类似于萨瑞鲁单抗200mg q2w的靶向pk暴露的剂量,是患有ra的成年患者中的推荐剂量方案
[0690]
■
剂量群组3:类似于萨瑞鲁单抗150mg qw的靶向pk暴露的剂量,其在患有ra的成年患者中在长期给药研究中,产生最高暴露。关于本研究待利用的按体重和剂量群组的剂量的信息,参见表1。还参见图7。
[0691]
将在基线时计算有待向患者给予的剂量(mg)。无论患者体重如何变化,在试验的12周核心治疗阶段的整个过程中,药物产品的剂量和对应体积都将保持相同。在延长阶段,每次访视时将测量患者的体重,并且将仅在计算显示需要增加剂量时,才根据体重的增加调整剂量。分别地,上限剂量对于剂量群组2将为200mg,并且对于剂量群组3将为150mg。剂量群组待注射的萨瑞鲁单抗体积在表2-表5中呈现。
[0692]
即使12周核心治疗阶段从第2次访视(基线-第0周)开始,直到第12次访视(第12周)(包含端值),第12次访视时的imp仅被给予至即将进入延长治疗阶段并且在第12次访视后完成了评估(包括功效、安全性和pk/pd)的患者。
[0693]
每位患者参与研究的持续时间
[0694]
研究的总持续时间(每位患者)预期长达166周:
[0695]
■
长达4周+3天的筛查(长达31天)
[0696]
■
12周核心治疗期
[0697]
■
长达144周延长阶段
[0698]
■
治疗后6周随访
[0699]
对于所有访视,使用第1天作为参考,剂量群组2(包括任何潜在的q2w中间剂量)的
±
3天以及剂量群组3(包括任何潜在的qw中间剂量)的
±
1天的时间框架是可接受的,除了
[0700]
■
第3次访视(第3天)、第4次访视(第5天)和第5次访视(第8天),在进行pk取样时不允许有访视窗口。
[0701]
■
对于所有剂量群组,第6次访视(第12天)
±
1天
[0702]
■
对于所有剂量群组,第7次访视(第2周)
±
1天
[0703]
■
对于剂量群组2,第8次访视(第4周)
±
2天,以及对于剂量群组3,为
±
1天
[0704]
sjia的3/4级嗜中性粒细胞减少症的停止规则
[0705]
在无感染体征的4级嗜中性粒细胞减少症中,建议暂时终止萨瑞鲁单抗治疗,一旦anc大于1000/mm3,研究者可以基于对益处和风险的全面评估恢复所述治疗。anc的减少是一种在用抗il-6抗体治疗的成人和儿科患者中看到的药效学抗白细胞介素-6(il-6)类效应。在患有类风湿性关节炎(ra)的成人的长期研究中,反复给药萨瑞鲁单抗后,anc的降低通常是短暂的,并且与感染风险的增加无关。在儿科患者中,尽管当前公开的数据有限,但在患有3/4级嗜中性粒细胞减少症的那些患者中,抗il-6的使用与感染风险的增加无关。表31显示了按照研究中所记录的最低值嗜中性粒细胞计数,3级和4级嗜中性粒细胞减少症的发生。
[0706]
患者的选择
[0707]
纳入标准
[0708]
1.筛查访视时年龄≥1且≤17岁的男性和女性患者
[0709]
2.根据ilar 2001jia分类标准诊断sjia亚型(petty,r.e.等人2001.j rheumatol.2004;31(2):390-2),其中在筛查时具有以下特征
[0710]
■
≥5个活动关节或
[0711]
■
筛查时≥2个活动关节,其中在基线前的3天中或在筛查期间任何连续7天中持续至少3天出现全身型jia发烧》37.5℃(尽管糖皮质激素以稳定剂量持续了至少3天)
[0712]
3.根据研究者的判断,对当前的治疗没有足够的响应并且被认为是bdmard的候选者的患者
[0713]
4.达到法定同意年龄的患者,或者父母一方或双方或者一个或多个法定监护人签署伦理委员会(ec)批准的书面知情同意书并注明日期。应基于当地指南以及患者的成熟度和理解研究相关信息的智力能力获得患者的允许。在涉及具有充分决策能力的独立或成熟未成年人的情况下,或者在法律允许的其他情况下,将直接从父母一方或双方或者一个或多个法定监护人处获得签署的知情同意书。
[0714]
研究治疗
[0715]
研究药物产品
[0716]
萨瑞鲁单抗,其为抗il-6r mab。
[0717]
配制品
[0718]
将在ph 6.0的水性缓冲媒介物中,以175mg/ml提供萨瑞鲁单抗药物产品。将使药物产品在填充有2.7ml萨瑞鲁单抗的5ml小瓶中供应,其中可抽出体积为2.0ml。
[0719]
一个或多个给予途径:
[0720]
在自我注射时将在腹部或大腿上或者另外由专业或非专业照护者在上臂(外侧)上进行皮下给予萨瑞鲁单抗。优选的是,sc注射部位在腹部(除了肚脐或腰部区)或大腿(前部和侧部)的4个象限之间交替。
[0721]
对于接受剂量方案2(q2w)的患者,在本研究的12周核心治疗阶段期间,将由专业照护者在现场进行注射。参见图1。对于接受剂量方案3(每周注射)的患者,必须安排合格的现场人员或家庭护士针对未计划在研究现场给出的剂量给予imp。对于本研究的延长阶段,如果患者或者父母一方或双方或者一个或多个法定监护人或者一个或多个照护者愿意并能够进行注射,则将允许家庭注射。在这些情况下,在核心治疗阶段,从第10次访视(第8周)开始,将需要并提供培训来准备和给予imp。此培训必须记录在患者的研究文件中。在允许在延长治疗阶段中的家庭注射之前,允许患者或者父母一方或双方或者一个或多个法定监护人或者一个或多个照护者在一个或多个研究者或者一个或多个代表人的观察/监督下在第10次访视(第8周)、第11次访视(第10周)和第12次访视(第12周)时给予注射。
[0722]
在患者进行研究访视的日子里,将遵循临床程序和血液采集给予imp。将提供日记来记录与这些注射有关的信息;这些日记将作为源数据保存在患者的研究文件中。如果照护者不能或不愿意给予imp,则必须安排合格的现场人员针对未计划在研究现场给出的剂量给予imp。
[0723]
剂量方案
[0724]
萨瑞鲁单抗应按q2w或qw给予。然而,根据方案,允许萨瑞鲁单抗给予窗口对于剂量群组2(包括任何潜在的q2w中间剂量)为
±
3天并且对于剂量群组3(包括任何潜在的qw中间剂量)为
±
1天,以适应特殊情况,例如实验室测试结果待定、持续不良事件(ae)、除了第5次访视(第8天)之外的患者时间安排困难。第5次访视(第8天)时没有给予窗口。在第7次访视(第2周)时,剂量群组2和3患者将仅有
±
1天的给予窗口,并且在第8次访视(第4周)时,剂量群组3将仅有
±
1天的给予窗口或者剂量群组2将仅有
±
2天的给予窗口。请注意,对于q2w
给予在少于11天并且对于每周给予在少于6天的时间间隔内,imp的过量(意外或故意)被定义为所述剂量的至少两倍。
[0725]
将以q2w或qw给予以下萨瑞鲁单抗剂量。
[0726]
剂量群组2:
[0727]
■
a组(》30kg且≤60kg):3mg/kg q2w
[0728]
■
b组(《30kg和210kg):4mg/kg q2w。
[0729]
剂量群组3:
[0730]
■
a组(》30kg且≤60kg):2mg/kg qw
[0731]
■
b组(《30kg和210kg):2.5mg/kg qw。
[0732]
还对剂量群组3作为两周一次的方案进行测试,但保持相同的pk暴露。因此,范围如下:
[0733]-小于30kg的患者:5至7mg/kg q2w(剂量2是4mg/kg q2w)
[0734]-30kg或更重的患者:4至6mg/kg q2w(剂量2是3mg/kg q2w)
[0735]
剂量修改
[0736]
将在基线时计算有待向患者给予的剂量(mg)。无论患者体重如何变化,在试验的12周核心治疗阶段的整个过程中,药物产品的剂量和对应体积都将保持相同。在延长阶段,每次访视时将测量患者的体重,并且将仅在计算显示需要增加剂量时,才根据体重的增加调整剂量。分别地,上限剂量对于剂量群组2将为200mg,并且对于剂量群组3将为150mg。本文描述了有待根据患者体重注射的对于剂量群组2至3的体积。参见表2-表5。
[0737]
包装和加标签
[0738]
imp将在含有加标签小瓶的患者治疗试剂盒中提供。加标签的内容符合当地监管规范和要求。一个试剂盒含有一个小瓶。
[0739]
储存条件和保质期
[0740]
应将研究药物在现场或家中保持冷藏在2℃与8℃之间。
[0741]
[0742]
[0743]
[0744]
[0745][0746]
i对在基线第2次访视(d1,第0周)前3个月内没有进行过胸部x光检查的jia患者开始进行生物疗法之前,仅在基于研究者的判断或根据当地tb筛查指南认为有必要的情况
下,才可以对患者进行胸部x光检查。
[0747]
j对剂量群组2中的患者以q2w,以及对剂量群组3中的患者每周一次给予研究药物产品。将安排家庭护士在中期访视时对剂量群组3患者给予imp(如果可能的话,家庭给予)。在imp给予后,应对患者进行至少30分钟的监测,以发现过敏反应的任何体征或症状。在12周核心治疗期结束时未实现幼年特发性关节炎美国风湿病学会(jia acr)30的患者将中断研究治疗,以根据研究者的临床判断接受护理标准。仅在v12后,主办方将向研究者提供jia acr响应(包括jia acr30响应),以评价此标准。对于在v12时完成核心治疗阶段,但由于除了缺乏jia acr30响应之外的原因而没有继续延长阶段的患者,在第12次访视时将不进行imp注射。
[0748]
仅对于德国:在首次imp给予后,必须对患者进行至少1小时的监测,以评估局部耐受性。
[0749]
k在核心治疗阶段期间,研究药物产品将被分配给处于剂量群组3的患者。
[0750]
l患者体温/皮疹日记将包括使用acr jia功效核心集评估,在筛查与基线访视之间的每一天以及在每次随后的安排访视之前持续至少7天,在家中记录鼓膜温度并报告皮疹(如果发生的话)。每天应在固定的时间点(在起床时和就寝前)以及怀疑发烧的任何时候至少两次测量体温。
[0751]
m收集基线前最接近1年测量的另外的身高。在研究期间,将使用测距仪在现场测量身高。
[0752]
n幼年特发性关节炎acr核心集包括:医师对疾病严重程度的全面评估、患者或者父母一方或双方/一个或多个法定监护人对总体健康状况的全面评估、患有活动性关节炎(定义为不是由于畸形导致的关节内肿胀,或者伴随疼痛或压痛或两者的活动受限)的关节数量、活动受限的关节数量、儿童健康评估问卷(chaq)、hs-crp和发烧(petty,r.e.等人,international league of associations for rheumatology classification of juvenile idiopathic arthritis:second revision,edmonton,2001.j rheumatol.2004;31(2):390-2;giannini,e.h.等人preliminary definition of improvement in juvenile arthritis.arthritis rheum.1997;40(7):1202-9;4.beukelman,t.等人,2011american college of rheumatology recommendations for the treatment of juvenile idiopathic arthritis:initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features.arthritis care res(hoboken).2011;63(4):465-82。在筛查时,将仅评估发烧、患有活动性关节炎的关节数量和活动受限的关节数量。本文中解释了jadas评分。
[0753]
o sjia的全身性特征包括筛查、基线和第12次访视时的发烧、短暂鲑鱼色红斑疹、全身淋巴结肿大、肝肿大、脾肿大和浆膜炎以及其他并发症。将在第2周、第4周、第6周、第8周和第10周仅评估发烧和短暂鲑鱼色红斑疹。
[0754]
p血液学(在药物给予之前应抽血):血红蛋白、血细胞比容、红细胞(rbc)计数和形态学(如果rbc计数异常)、白细胞(wbc)、白细胞分类(嗜中性粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞)、血小板计数、绝对嗜中性粒细胞计数(anc)。在第6次访视(第12
±
1天)时,必须对群组2的患者进行另外的血液学测试,以便在第7次访视(第15天,第2周),即第二剂量萨瑞鲁单抗注射之前获得结果。对于剂量群组3的患者,应在第5次访视
(第8天)之前或之时但不早于第4次访视(第5天)进行另外的血液学测试,并且结果需要在第5次访视(第8天)时第二次萨瑞鲁单抗注射之前进行审查。可以在中心实验室或当地实验室完成另外的血液学测试,以确认anc和血小板计数是否不在研究药物暂时或永久中断的由方案限定的限度内。如果使用当地实验室,仍应在如对于群组2所安排的第7次访视(第15天,第2周)以及对于群组3为第5次访视(第8天)时给予imp前抽取用于血液学检查的中心实验室样品。对于所有患者,在对于剂量群组3患者的第5次访视(第8天)或对于剂量群组2患者的第7次访视(第2周,第15天)时给予第二剂量的萨瑞鲁单抗之前,必须审查第2次访视(第1天)血液学实验室评估。
[0755]
q化学(在药物给予之前应抽血):整个化学检查将仅在筛查访视、基线第2次访视(第1天,第0周)和第12次访视(第12周)或eot时进行:钠、钾、氯、碳酸氢盐、血尿素氮、肌酐和肌酐清除率、肾小球滤过率、钙、磷酸盐、总蛋白、白蛋白、丙氨酸转氨酶(alt)、天冬氨酸转氨酶(ast)、碱性磷酸酶(alp)、乳酸脱氢酶(ldh)、总胆红素、结合胆红素和未结合胆红素。在所有其他访视时:将仅测试alt、ast、alp、ldh、总胆红素、结合胆红素、未结合胆红素和白蛋白。
[0756]
r空腹血脂(在药物给予之前应抽血):甘油三酯(tg)、总胆固醇、高密度脂蛋白(hdl)胆固醇、低密度脂蛋白(ldl)胆固醇。需要患者在测试前禁食至少8小时。
[0757]
s任选的:将仅基于患者的病史和研究者的判断来测量hba1c水平。
[0758]
t对于≥30kg的患者(a组),将在第3天和第8天测量hs-crp。对于pk时间表1组中《30kg的患者(b组),将在第3天和第8天测量hs-crp。对于pk时间表2组中《30kg的患者(b组),将在第5天和第12天测量hs-crp。
[0759]
u当怀疑巨噬细胞活化综合征(mas)时,基于研究者的判断,如有必要,需要进行铁蛋白、血细胞计数(红细胞、白细胞、血小板和血红蛋白)、ast/alt、甘油三酯和纤维蛋白原测试。本文描述了用于mas诊断的临床和实验室特征。
[0760]
v尿分析试纸:比重、ph、葡萄糖、血液、蛋白质、亚硝酸盐、白细胞酯酶、胆红素。如果试纸上的任何参数异常,则应将尿样送到中心实验室进行测试。如果蛋白质呈阳性,则由中心实验室进行显微镜分析。
[0761]
w对于已经开始月经来潮的女性,在筛查访视时必须进行血清妊娠测试。
[0762]
x对于已经开始月经来潮的女性,应在筛查时进行血清妊娠测试,并且应在第2次访视(第0周,第1天)、第8次访视(第4周)、第10次访视(第8周)和第12次访视(第12周)时进行尿液妊娠测试。可在当地进行尿液妊娠测试。在暴露于imp和eot之前,应通过尿液妊娠测试检查妊娠状态。
[0763]
y在治疗期期间的给药日,将在imp给予之前采集血液样品。如果患者出现sae,则在可能/*98+-的情况下,应在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度和抗药物抗体(ada)评估。
[0764]
z对于在核心治疗阶段期间过早中断研究治疗的患者,将在eot评估时测量il-6和总sil-6r。
[0765]
aa父母一方或双方或者一个或多个法定监护人必须在用于药物基因组研究的唾液样品采集之前签署一份单独的书面受试者信息(wsi)。优选将样品在基线第2次访视(d1,第0周)时收集,但可以在任何访视时收集。患者还可以基于他/她的年龄、当地法规以及他/
她对研究信息的理解成熟度签署wsi。如果患者或者其父母一方或双方或者其一个或多个法定监护人不希望患者参与唾液样品采集,则患者仍有资格纳入本研究。
[0766]
[0767]
[0768][0769]
物产品,ivrs=交互式语音响应系统,il=白细胞介素,jadas=幼年关节炎疾病活动性得分,jia acr=幼年特发性关节炎美国风湿病学会,pk=药物动力学,ppd=纯化蛋
白衍生物,q2w=每隔一周一次,sae=严重不良事件,sjia=全身型幼年特发性关节炎;tb=结核病,v=访视,wk=周。
[0770]
a第27次访视是研究计划的治疗结束(eot)访视。如果患者过早中断研究治疗,则将在剂量群组3的最后一次imp注射后1周和剂量群组2的最后一次imp注射后2周发生eot。
[0771]
b第28次访视是研究计划的研究结束(eos)访视。如果患者过早中断研究治疗,则这些患者将被要求在eot访视后6周(eot+6周)返回进行eos评估。此eos(eot+6周)访视将适用于核心治疗阶段和延长阶段两者中的所有患者。
[0772]
c关于imp给予的患者日记要在家中针对给予imp来完成。
[0773]
d完整的身体检查将在第15次访视(第24周)、第18次访视(第48周)、第20次访视(第72周)、第22次访视(第96周)、第24次访视(第120周)、第26次访视(第144周)、第27次访视eot和第28次访视eos时进行,包括皮肤、鼻腔、眼睛、耳朵、呼吸系统、心血管系统、胃肠系统、神经系统、淋巴系统和肌肉骨骼系统。
[0774]
e将imp对于剂量群组2中的患者以q2w给予,并且对于剂量群组3中的患者每周给予。对于完成延长阶段的患者,如果所选择剂量方案是双周给药(q2w),则最后一次imp注射将在第154周发生;如果所选择剂量方案是每周给药(qw),则最后一次imp注射将在第155周发生。患者将在第27次访视(第156周)时接受eot评估。对于在延长阶段期间过早中断研究治疗的患者,将优选在对剂量群组2患者的最后一次imp注射后2周和对剂量群组3患者的最后一次imp注射后1周进行eot评估。在患者不能满足计划的时间表的情况下,在最后一次imp注射后的由下一次方案限定的访视时,将使用eot访视进行评估。患者将被要求在eot访视后6周返回现场进行eos评估。在imp给予后,应对患者进行至少30分钟的监测,以发现过敏反应的任何体征或症状。在第27次访视(第156周)时将没有imp注射。
[0775]
f患者体温/皮疹日记将包括使用acr jia功效核心集评估,在每次随后的安排访视之前持续至少7天,在家中记录鼓膜温度并报告皮疹(如果发生的话)。每天应在固定的时间点(在起床时和就寝前)以及怀疑发烧的任何时候,至少两次测量体温。
[0776]
g收集基线前最接近1年测量的另外的身高。在研究期间,将使用测距仪在现场测量身高。
[0777]
h jia acr核心集包括:医师对疾病严重程度的全面评估、患者或父母/法定监护人对总体健康状况的全面评估、患有活动性关节炎(定义为不是由于畸形导致的关节内肿胀,或者伴随疼痛或压痛或两者的活动受限)的关节数量、活动受限的关节数量、儿童健康评估问卷(chaq)、hs-crp和发烧。本文中解释了jadas评分。
[0778]
i sjia的全身性特征包括发烧、短暂鲑鱼色红斑疹、全身淋巴结肿大、肝肿大、脾肿大、浆膜炎以及第156周(eot访视)时的其他并发症。将在第24周、第48周、第72周、第96周、第120周、第144周和第162周(eos访视)时仅评估发烧和短暂鲑鱼色红斑疹。
[0779]
j血液学(在药物给予之前应抽血):血红蛋白、血细胞比容、红细胞(rbc)计数和形态学(如果rbc计数异常)、白细胞(wbc)分类(嗜中性粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞)、血小板计数、绝对嗜中性粒细胞计数(anc)。
[0780]
k化学(在药物给予前应抽血):将测试alt、ast、alp、乳酸脱氢酶(ldh)、总胆红素、结合胆红素、未结合胆红素和白蛋白。完整的化学检查应在第27次访视eot时完成。
[0781]
l血脂(在药物给予前应抽血):甘油三酯(tg)、总胆固醇、高密度脂蛋白(hdl)胆固
醇、低密度脂蛋白(ldl)胆固醇。需要患者在测试前禁食至少8小时。
[0782]
m当怀疑mas时,基于研究者的判断,如有必要,需要进行铁蛋白、血细胞计数(红细胞、白细胞、血小板和血红蛋白)、ast/alt、甘油三酯和纤维蛋白原测试。参考本文所述的临床和实验室mas诊断特征。
[0783]
n对于已经开始月经来潮的女性,应在每次安排的访视时暴露于imp注射之前和eot时进行尿液妊娠测试。可在当地进行尿液妊娠测试。
[0784]
o在治疗期期间的给药日,将在imp给予之前采集血液样品。如果患者出现sae,则在可能的情况下,应在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度和抗药物抗体(ada)评估。
[0785]
[0786]
[0787]
[0788][0789]
b第28次访视是研究计划的研究结束(eos)访视。如果患者过早中断研究治疗,则这些患者将被要求在eot访视后6周(eot+6周)返回进行eos评估。此eos(eot+6周)访视将适
用于核心治疗阶段和延长阶段两者中的所有患者。
[0790]
c关于imp给予的患者日记要在家中针对给予imp来完成。
[0791]
d完整的身体检查将包括皮肤、鼻腔、眼睛、耳朵、呼吸系统、心血管系统、胃肠系统、神经系统、淋巴系统和肌肉骨骼系统。
[0792]
e对剂量群组2中的患者以q2w,以及对剂量群组3中的患者每周给予研究药物产品。对于完成延长阶段的患者,如果所选择剂量方案是双周给药(q2w),则最后一次imp注射将在eot前2周发生;如果所选择剂量方案是每周给药(qw),则最后一次imp注射将在eot前1周发生。患者将在第27次访视(第156周)时接受eot评估。对于在延长阶段期间过早中断研究的患者,将优选在对剂量群组2患者的最后一次imp注射后2周和对剂量群组3患者的最后一次imp注射后1周进行eot评估。在患者不能满足计划的时间表的情况下,在最后一次imp注射后的由下一次方案限定的访视时,将使用eot访视进行评估。患者将被要求在eot访视后6周返回现场进行eos评估。在imp给予后,应对患者进行至少30分钟的监测,以发现过敏反应的任何体征或症状。在第27次访视(第156周)时将没有imp注射。
[0793]
f患者体温/皮疹日记将包括使用acr jia功效核心集评估,在每次随后的安排访视之前持续至少7天,在家中记录鼓膜温度并报告皮疹(如果发生的话)。每天应在固定的时间点(在起床时和就寝前)以及怀疑发烧的任何时候,至少两次测量体温。
[0794]
g收集基线前最接近1年的另外的身高。在研究期间,将使用测距仪在现场测量身高。
[0795]
h幼年特发性关节炎acr核心集包括:医师对疾病严重程度的全面评估、患者或父母/法定监护人对总体健康状况的全面评估、患有活动性关节炎(定义为不是由于畸形导致的关节内肿胀,或者伴随疼痛或压痛或两者的活动受限)的关节数量、活动受限的关节数量、儿童健康评估问卷(chaq)、hs-crp和发烧。本文中解释了jadas评分。
[0796]
i将仅评估发烧和短暂鲑鱼色红斑疹。
[0797]
j血液学(在药物给予之前应抽血):血红蛋白、血细胞比容、红细胞(rbc)计数和形态学(如果rbc计数异常)、白细胞(wbc)分类(嗜中性粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞)、血小板计数、绝对嗜中性粒细胞计数(anc)。
[0798]
k化学(在药物给予前应抽血):将测试alt、ast、alp、乳酸脱氢酶(ldh)、总胆红素、结合胆红素、未结合胆红素和白蛋白。完整的化学检查应在第27次访视eot时完成。
[0799]
l血脂(在药物给予前应抽血):甘油三酯(tg)、总胆固醇、高密度脂蛋白(hdl)胆固醇、低密度脂蛋白(ldl)胆固醇。需要患者在测试前禁食至少8小时。
[0800]
m当怀疑mas时,基于研究者的判断,如有必要,需要进行铁蛋白、血细胞计数(红细胞、白细胞、血小板和血红蛋白)、ast/alt、甘油三酯和纤维蛋白原测试。
[0801]
n对于已经开始月经来潮的女性,应在前述流程表中提及的所安排访视时在暴露于imp注射之前进行尿液妊娠测试。可在当地进行尿液妊娠测试。
[0802]
o在治疗期期间的给药日,将在imp给予之前采集血液样品。如果患者出现sae,则在可能的情况下,应在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度和抗药物抗体(ada)评估。
[0803]
药代动力学处理程序
[0804]
尽可能接近方案规定的日期收集所有血液样品是极其重要的(取样时间表参见表
17的研究流程表)。任何遗漏或丢失血液样品的原因都应记录在案。可以利用描述用于收集、存储和运送血清的特殊程序。参见表20。
[0805]
表20.-针对萨瑞鲁单抗和抗萨瑞鲁单抗抗体的样品处理程序
[0806][0807]
a对于进行pk和抗萨瑞鲁单抗抗体样品的血液采集的研究访视(例如,基线第2次访视),建议抽取1ml血液。血清将被平均分成2个等分试样,以获得一个pk血清等分试样和一个抗萨瑞鲁单抗抗体血清等分试样。
[0808]
生物分析方法
[0809]
将使用经验证的生物分析方法来测定功能性萨瑞鲁单抗和抗萨瑞鲁单抗抗体的血清水平。
[0810]
药代动力学参数
[0811]
将使用非线性混合效应建模开发poppk模型,以描述萨瑞鲁单抗的pk特征。将使用血清中功能性萨瑞鲁单抗的poppk模型来计算以下pk参数。pk参数将包括但不限于以下:c
max
、c
谷
、t
max
和auc0-τ。
[0812]
表21-研究基线时的患者和疾病特征*
[0813]
[0814]
[0815][0816]
*anc=绝对嗜中性粒细胞计数;bdmard=生物类dmard;chaq-di=儿童健康评估问卷-残疾指数;crp=c反应蛋白;csdmard=常规合成dmard;dmard=改善疾病抗风湿药物;esr=红细胞沉降率;gc=糖皮质激素;jadas-27crp=幼年关节炎疾病活动性得分=使用c反应蛋白的27-关节计数得分;jia=幼年特发性关节炎;n=治疗期间至少一次满足标准的患者总数;qw=每周;q2w=每2周;rf=类风湿因子;sd=标准偏差;vas=视觉模拟量表;yrs=岁/年。
[0817]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。
[0818]
剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0819]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0820]
表22.首次给予时和重复给予后第12周时的萨瑞鲁单抗pk数据*
[0821][0822]
*auc
0-τ
=在2周(q2w方案)或1周(qw方案)的剂量间隔τ期间,血清浓度对时间曲线
下面积;c
max
=观察到的最大血清浓度;c
谷
=重复给药期间在治疗给予前观察到的浓度;cv%=变异系数;n=治疗期间至少一次满足标准的患者总数;qw=每周;q2w=每2周;sc=皮下。
[0823]
在第10-12周时测试的q2w剂量方案;在第11-12周时测试的qw剂量方案。
[0824]
表23.研究者报告的ae*
[0825]
[0826][0827]
*ae=治疗突发性不良事件;alt=丙氨酸转氨酶;n=治疗期间至少一次满足标准的患者总数;ne=事件数量;py=患者-年;qw=每周;q2w=每2周;sae=严重ae。来自meddra 21.0的所有术语。
[0828]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0829]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0830]
表24.研究中记录的最低值的绝对嗜中性粒细胞计数*
[0831][0832]
*n=治疗期间至少一次满足标准的患者总数;qw=每周;q2w=每2周。
[0833]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0834]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0835]
表25.所有研究者报告的感染*
[0836]
[0837][0838]
*ne=事件数量;py=患者-年;qw=每周;q2w=每2周。来自meddra 21.0的所有术语。
[0839]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。
[0840]
剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0841]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0842]
表26.在第12周时,jia acr组分自基线的平均变化*
[0843][0844][0845]
*acr=美国风湿病学会;bl=基线;chaq-di=儿童健康评估问卷-残疾指数;crp=c反应蛋白;jia acr30/70/90=幼年特发性关节炎美国风湿病学会30/70/90%响应;qw=每周;q2w=每2周;se=标准误差;vas=视觉模拟量表。
[0846]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0847]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0848]
||体重组a,30-60kg,体重组b,10-《30kg。
[0849]
表27.在第12周时实现临床非活动性疾病或低疾病活动性的患者(观察的病例)*
[0850]
[0851][0852]
*crp=c反应蛋白;jadas-27-crp=使用27-关节计数和crp的幼年关节炎疾病活动性得分;lda=低疾病活动性;n=治疗期间至少一次满足标准的患者总数;qw=每周;q2w=每2周;vas=视觉模拟量表。
[0853]
剂量1,在30-60kg患者中为2.0mg/kg q2w,并且在10-《30kg患者中为2.5mg/kg q2w。
[0854]
剂量2,在30-60kg患者中为3.0mg/kg q2w,并且在10-《30kg患者中为4.0mg/kg q2w。
[0855]
§
剂量3,在30-60kg患者中为2.0mg/kg qw,并且在10-《30kg患者中为2.5mg/kg qw。
[0856]
||体重组a,30-60kg,体重组b,10-《30kg。
[0857]
**一名患者在第12周时未完成jadas-27-crp评估的所有组分,并被去掉。
[0858]
华莱士标准定义为医师全面vas《1/10,无活动性关节炎,无活动性葡萄膜炎,以及crp《10mg/l。
[0859]
一个或多个次要终点
[0860]
12周核心治疗阶段
[0861]
将在12周开放标签核心处理阶段中分析以下参数。
[0862]
■
安全性
[0863]
ο不良事件、生命体征、身体检查、实验室值
[0864]
ο可接受性评估(局部耐受性)
[0865]
■
功效
[0866]
ο第12周时的jia acr30/50/70/90/100(在没有发烧的情况下)响应率
[0867]
ο第12周时单独jia acr组分中自基线的变化
[0868]
ο第12周时幼年关节炎疾病活动性得分-27(jadas)自基线的变化
[0869]
ο糖皮质激素使用的变化将按剂量群组和体重组,对每个单独患者从基线到12周核心治疗期结束时使用糖皮质激素等效物泼尼松剂量的线形图进行描述性评估
[0870]
■
药效学
[0871]
οil-6相关生物标记物(例如高敏c反应蛋白[hs-crp]、il-6、sil-6r的血清水平)的变化
[0872]
延长阶段
[0873]
■
安全性
[0874]
ο不良事件、生命体征、身体检查、实验室值
[0875]
ο可接受性评估(局部耐受性)
[0876]
■
功效
[0877]
ο第24周、第48周和每24周直到研究结束时的jia acr30/50/70/90/100(在没有发烧的情况下)响应率,第24周、第48周和每24周直到访视结束时幼年关节炎疾病活动性得分-27自基线的变化
[0878]
ο-第24周和48周以及每24周直到研究结束时单独jiaacr组分中自基线的变化
[0879]
ο-与基线相比,第24周、第48周和每24周直到研究结束时,按剂量类别(糖皮质激素等效物泼尼松剂量20.5mg/kg、20.2mg/kg和《0.5mg/kg、《0.2mg/kg)接受糖皮质激素的患者比例
[0880]
ο-第24周、第48周和随后每24周直到研究结束时,不接受糖皮质激素且无jia加剧的患者比例
[0881]
ο-糖皮质激素使用的变化将按剂量群组和体重组,对每个单独患者从基线到治疗期结束时使用糖皮质激素等效物泼尼松剂量的线形图进行描述性评估
[0882]
探索性终点
[0883]
12周核心治疗期
[0884]
■
由于全身型jia基线患有发烧的患者在第12周时无发烧的比例
[0885]
■
由于全身型jia基线患有皮疹的患者在第12周时无皮疹的比例延长阶段
[0886]
·
第1、2和3年的生长评估,包括身高速度和身高标准偏差(sd)得分,针对的是从未接受生长激素并且在治疗第一年结束时未达到第5唐纳阶段的患者。
[0887]
·
在基线时患有发烧的患者在第24周、第48周和每24周直到研究结束时无发烧的比例。
[0888]
·
在基线时由于全身型jia患有皮疹的患者在第24周、第48周和每24周直到研究结束时无皮疹的比例。
[0889]
功效终点
[0890]
jia acr响应
[0891]
在这项研究中,将使用用以评估体征和症状的jia acr评级量表。将在第12、24、48、72、96、120、144和156周(eot)时评估jia acr 30/50/70/90/100。jia acr 30/50/70/90/100(在没有发烧的情况下)响应被定义为6个核心集变量中的3个变量自基线改善至少30%/50%/70%/90%/100%同时剩余变量中不多于1个变量恶化多于30%的患者。
[0892]
jia acr核心集包括6个变量加上sjia的发烧:
[0893]
■
疾病活动性的医师全面评估
[0894]
■
总体健康状况的患者/父母评估
[0895]
■
由儿童健康评估问卷(chaq)确定的功能能力
[0896]
■
患有活动性关节炎的关节数量(0-71个关节)
[0897]
■
活动受限的关节数量(0-67个关节)
[0898]
■
高敏c反应蛋白
[0899]
■
发烧(在前7天内)
[0900]
■
除了第3次访视(第3天)、第4次访视(第5天)、第5次访视(第8天)、第6次访视(第12天)以及eos访视之外,将在每次现场访视时评价jia acr核心集。应在acr评估前7天测量体温。
[0901]
将通过访视使用平均值、标准误差和对应的95%ci对每个剂量群组和体重组汇总jia acr30核心集变量得分和自基线的变化。取代hs-crp,使用esr的jia acr30/50/70/90/100响应也将使用相同的方法被汇总。
[0902]
疾病活动性的医师全面评估
和“100非常严重疼痛”)。
[0918]
·
计算chaq不适指数(项目67)
[0919]-测量从项目67中vas左端到响应者标记的距离,并乘以0.2,范围为0-3。不适指数得分可以被重新调整到0-100规模。
[0920]
chaq健康状况得分的第3部分测量病患的患者或父母全面评估。
[0921]
·
计算chaq健康状况得分(项目69)
[0922]-测量从项目69中vas左端到响应者标记的距离,并乘以0.2,范围为0-3。健康状况得分可以被重新调整到0-100规模。
[0923]
患有活动性关节炎的关节数量和活动受限的关节数量
[0924]
活动关节被定义为具有以下特征的关节:
[0925]
■
不是由于畸形导致的关节内肿胀,或者
[0926]
■
伴随疼痛或压痛的活动受限
[0927]
七十一(71)个关节将通过计数患有不是由于畸形导致的肿胀或者伴随疼痛或压痛或两者的活动受限的关节数量来评估活动性疾病(bazso,a.等人,2009j rheumatol.36(1):183-90;national cholesterol education program(ncep):highlights of the report of the expert panel on blood cholesterol levels in children and adolescents.pediatrics.1992;89(3);495-501)。
[0928]
颈椎(计为1个关节)、颞下颌(2个关节,右侧和左侧)、胸锁(2个关节)、肩锁(2个关节)、肩(2个关节)、肘(2个关节)、腕(2个关节)、掌指(总共10个关节,每侧5个)、近端指间(总共10个关节,每侧5个)、远端指间(总共8个关节,每侧4个)、髋(2个关节)、膝(2个关节)、踝(2个关节)、距下(2个关节)、跗跖(2个关节)、跖趾(10个关节,每侧5个)和脚趾间(10个关节,每侧5个)总共71个关节。
[0929]
除了胸锁关节(n=2)和肩锁关节(n=2)之外,将针对活动受限检查的六十七(67)个关节与针对活动性疾病检查的关节相同。将由培训过的评估员进行关节的正式计数。关节压痛被定义为由评估者的拇指和食指施加的关节压力引起的疼痛。
[0930]
高敏c反应蛋白
[0931]
对于保持在所选择剂量的患者,将在以下时间评价高敏c反应蛋白:第1次访视(第-28天至第-1天,最多31天)、基线第2次访视(第0周,第1天)、第7次访视(第2周)、第8次访视(第4周)、第9次访视(第6周)、第10次访视(第8周)、第11次访视(第10周)、第12次访视(第12周)以及延长阶段的每次访视、第13次访视(第16周)至第27次访视eot(第156周);第101次访视(第0周)至第108次访视(第12周)、第109次访视(第24周)、第112次访视(第48周)、第114次访视(第72周)、第116次访视(第96周)、第118次访视(第120周)和第27次访视eot。对于pk时间表1组中《30kg的患者(b组),将在第3天和第8天测量hs-crp。对于pk时间表2组中《30kg的患者(b组),将在第5天和第12天测量hs-crp。高敏c反应蛋白水平与il-6r活性是直接相关的。预期主动剂量方案将对crp水平具有显著的降低作用。
[0932]
幼年关节炎疾病活动性评分
[0933]
jadas包括4项测量:
[0934]
1.疾病活动性的医师全面评估,在10cm vas上测量,其中0=无活动性且10=最大活动性
[0935]
2.健康状况的父母/患者全面评估,在10cm vas上测量,其中0=非常好且10=非常差
[0936]
3.患有活动性疾病的关节计数
[0937]
4.根据以下公式将红细胞沉降率归一化为0-10级:
[0938]
[esr(mm/小时)-20]/10
[0939]
在进行计算之前,将《20mm/小时的esr值转换为0,并且将》120mm/小时的esr值转换为120。
[0940]
jadas将被计算为其4个组分得分的简单线性和(consolaro,a.等人,2009arthritis rheum.61(5):658-623)。将通过访视(包括数量、平均值、标准误差、sd、中值、最小值和最大值)对每个剂量群组和体重组(如果可能)汇总jadas-27的总得分和自基线的变化。
[0941]
jadas-27包括以下关节:颈椎、肘、腕、掌指关节(从第一至第三)、近端指间关节、髋、膝和脚踝。jadas被认为是评估jia中疾病活动性的有效工具,并有可能应用于标准临床护理、观察性研究和临床试验(23)。对于保持在所选择剂量的患者,将在以下时间计算jadas-27:第12次访视(第12周)、第15次访视(第24周)、第18次访视(第48周)、
[0942]
第20次访视(第72周)、第22次访视(第96周)、第24次访视(第120周)、第26次访视(第144周)和第27次访视eot(第156周);对于变为所选择剂量的患者,将在以下时间计算:第101次访视(第0周)至第106次访视(第12周)、第109次访视(第24周)、第112次访视(第48周)、第114次访视(第72周)、第116次访视(第96周)、第118次访视(第120周)和第27次访视eot。
[0943]
sjia的全身性特征
[0944]
sjia的全身性特征(包括发烧、短暂鲑鱼色红斑疹、全身淋巴结肿大、肝肿大、脾肿大、浆膜炎和其他并发症)将在第1次筛查访视(第-28天至第-1天,最多31天)(以确认诊断)、第2次访视(第0周)、第12次访视(第12周)和第27次访视(第156周)时收集;将在第15次访视(第24周)、第18次访视(第48周)和每24周直到研究结束时仅收集发烧、短暂鲑鱼色红斑疹。
[0945]
当评估jia acr核心组分时,将在所有访视时收集常见的全身性特征(包括与sjia相关的发烧和皮疹)。
[0946]
与sjia相关的皮疹是指短暂鲑鱼色红斑疹。
[0947]
无皮疹被定义为在评估日之前的7天内,患者日记中没有记录与sjia相关的皮疹。
[0948]
没有发烧被定义为当评估jia acr核心组分时,在访视前的7天内,无体温测量值≥37.5℃。
[0949]
治疗对发烧和皮疹的影响将相比于基线在第12周时进行评估。
[0950]
将通过访视(即,第12、24、48周和每24周直到研究结束)对每个剂量群组、总体上以及按体重组(如果有足够的数据)汇总在基线时由于全身型jia患有发烧的患者无发烧的比例。
[0951]
将通过访视(即,第12、24、48周和每24周直到研究结束)对每个剂量群组、总体上以及按体重组(如果有足够的数据)汇总在基线时由于全身型jia患有皮疹的患者无皮疹的比例。
[0952]
糖皮质激素使用评估
[0953]
将对于每个剂量群组和体重组汇总按剂量类别(20.5mg/kg、20.2mg/kg和《0.5mg/kg、《0.2mg/kg)接受糖皮质激素的患者比例,以及在第24、48、72和104周时不接受糖皮质激素且无jia加剧的患者比例,以与基线时的比例进行比较。特定时间点(即第0、24、48、72或104周)的糖皮质激素等效物泼尼松剂量(mg/kg/天)将基于所述时间点之前2周内接受的糖皮质激素进行计算。特定时间点(即第24、48、72或104周)的jia加剧被定义为在前次访视与此时间点之间jia acr50响应未被维持,或发烧(任何体温测量值≥37.5℃),或esr 220mm/h。
[0954]
将通过访视(平均值、sd、中值等)按剂量群组和体重组汇总糖皮质激素等效物泼尼松剂量(原始值和自基线的变化)。此外,在治疗期期间(核心和延长两者)每次访视时的糖皮质激素等效物泼尼松剂量将按剂量群组和体重组为每个单独患者绘制。
[0955]
生长评估
[0956]
对于从未接受生长激素并且在治疗第一年结束时未达到第5唐纳阶段的患者,将使用身高速度和身高sd得分评估对生长的影响。
[0957]
身高速度(每年厘米数)将基于身高变化和身高测量之间的时间间隔来计算。假设测量值是在基线前9个月与24个月之间获得的,则将使用基线前最接近1年测量的身高来估计治疗前身高速度。随后,将计算治疗第1年和第2年的身高速度。将使用最接近1年和2年截止点的身高测量值。
[0958]
将使用壁挂式测距仪在站立位置测量身高。在获得身高测量值时,儿童将在以下情况下被测量:不穿鞋子,不戴帽子或发饰,同时头部、肩部、臀部和脚跟与墙壁成固定的直角并且双脚并拢。由于每次测量时儿童的姿势会有轻微的变化,因此将对每个儿童进行三次连续测量。三次测量的平均值将被认为是儿童的真实身高,前提是测量值在0.3cm以内。如果测量相距》0.3cm,将重新测量,并当它们在可接受范围内时取平均值。用于生长速度计算的测量将如评估时间表中所指示地进行。
[0959]
将经由现场验证的方法测量身高。给定患者在间隔期间的预期正常身高速度将被计算为针对患者的年龄和性别世界卫生组织(who;世界卫生组织.儿童生长标准:who anthro(2011年版本)and macros.url:http://www.who.int/childgrowth/software/en/)平均身高的差异除以年龄变化。将身高速度与治疗前时以及治疗的第1、2和3年期间的预期正常身高速度进行比较。
[0960]
身高sd得分将使用who标准(同上)进行计算(参见下文)。参考群体的平均sd得分为0,并且-2与+2之间的值通常被视为正常范围:sd得分=(观察值-参考群体的中值)/参考群体的sd值。
[0961]
为了评估对生长的影响,将使用平均值、sd和95%ci描述性地汇总治疗第1、2和3年的身高sd得分自基线的变化。在治疗前时以及治疗的第1、2和3年期间,将汇总身高速度超过who预期的患者比例。将平均身高速度与治疗前时以及第1、2和3年期间的预期who正常身高速度进行比较。
[0962]
身体检查
[0963]
对于保持在所选择剂量的患者,将在以下时间进行完整的身体检查:第1次筛查访视(第-28天至第-1天,最多31天)、第2次访视(第1天,第0周)、第8次访视(第4周)、第9次访
视(第6周)、第12次访视(第12周)、第15次访视(第24周)、第18次访视(第48周)、第20次访视(第72周)、第22次访视(第96周)、第24次访视(第120周)、第26次访视(第144周)和第27次访视eot(第156周);对于变为所选择剂量的患者,将在以下时间进行:第101次访视(第0周)、第104次访视(第6周)、第106次访视(第12周)、第109次访视(第24周)、第112次访视(第48周)、第114次访视(第72周)、第116次访视(第96周)、第118次访视(第120周)、第27次访视eot和第28次访视eos。任何临床显著异常如果在第1次访视(第-28天至第-1天,最多31天)时观察到并已知,则应在患者ecrf中报告为病史,并且如果在第2次访视(第1天,第0周)时和随后的访视期间观察到,则应报告为ae。在筛查访视时已知的任何临床显著的身体检查异常应报告为病史,而不是ae。筛查访视后报告的临床显著异常或自基线的恶化应报告为ae。
[0964]
体重
[0965]
体重应在患者穿着内衣或很轻的衣服(没有外套或配饰)并且不穿鞋子且膀胱为空的情况下测量。建议在整个研究中使用同一量表。体重应精确到0.1kg。
[0966]
建议在整个研究中使用同一量表。对于保持在所选择剂量的患者,将在以下时间收集体重:第1次筛查访视(第-28天至第-1天,最多31天)、基线第2次访视(第1天,第0周)、第12次访视(第12周)和延长阶段期间每次访视;对于变为所选择剂量的患者,将在以下时间进行:第101次访视(第0周)至第119次访视(132周)、第27次访视eot和第28次访视eos时(除了第102次访视(第2周)和第104次访视(第6周)之外)。
[0967]
药效学参数
[0968]
将通过测量以下生物标记物来评估萨瑞鲁单抗的药效学作用:hs-crp、il-6、sil-6r。
[0969]
评估时间表
[0970]
血液采集的取样时间表可见于研究流程表(参见表17-表19)。将在基线第2次访视(第0周)和第12次访视(第12周)时测量il-6和总sil-6r。对于在核心治疗阶段期间过早中断研究治疗的患者,将在eot评估时测量il-6、和总sil-6r。
[0971]
药物基因组学
[0972]
父母一方或双方或者一个或多个法定监护人将被要求签署一份单独的用于唾液样品收集的icf。用于基因组研究的脱氧核糖核酸样品将按照国际协调理事会(ich)指南15的定义进行双重编码。脱氧核糖核酸样品可以在临床研究报告(csr)的最终日期后保存长达15年,并可以用于研究目的。
[0973]
基因组分析的目的是鉴定基因组与临床或生物标记物对目标调节、疾病预后和进展或其他临床结果测量的响应之间的关系。这些数据可以与从其他研究中收集的数据一起使用或相组合,以鉴定可以预测响应和阐明疾病机制的基因组标记物。分析可以包括候选基因和周围基因组区域的序列测定或单核苷酸多态性研究。还可以进行全基因组研究,包括(但不限于)单核苷酸多态性分析、基因组测序和转录组测序。如有需要,还可以进行基因组分析,以鉴定与毒性相关的标记物。如果患者及其父母或法定监护人不希望参与药物基因组样品收集,则患者仍有资格纳入本研究。
[0974]
为药物浓度或ada测定所收集的任何未使用或遗留的血清样品可以被保存并用于与sjia相关的探索性生物标记物研究、用抗体抑制il-6rα途径、治疗响应(pd和或预测性)、
调查意外ae、或鉴定与毒性相关的标记物。样品可以在csr日期后保存长达15年,或基于当地法规保存。
[0975]
表28:从非选择剂量变为所选择剂量的患者在eot访视(第27次访视)之前的最后一次治疗中访视
[0976][0977]
表29:sjia的特征
[0978][0979]
缩写:hla-b27=人白细胞抗原-b27,ilar=国际风湿病学协会联盟,jia=幼年特发性关节炎,rf=类风湿因子。
[0980]
a具体排除标准:a=银屑病或一级亲属的银屑病;b=存在hla-b27,男性,并且年龄大于6岁;
[0981]
c=强直性脊柱炎、肌腱端炎相关关节炎、伴有慢性炎性肠病的骶髂关节炎、
[0982]
一级亲属的莱特尔病;d=至少相隔3个月至少2次出现rf。
[0983]
表30:研究者报告的teae
[0984][0985]
py,患者-年;uln:正常上限
[0986]
表31:显示按照研究中所记录的最低值嗜中性粒细胞计数,3级和4级嗜中性粒细胞减少症的表。
[0987][0988]a预先指定的实验室监测,anc,绝对嗜中性粒细胞计数;lln,正常下限
[0989]
[0990]
[0991][0992]
i幼年特发性关节炎美国风湿病学会(jia acr)核心集包括:医师对疾病严重程度的全面评估、患者或父母对总体健康状况的全面评估、患有活动性关节炎(定义为不是由于
畸形导致的关节内肿胀,或者伴随疼痛、压痛或两者的活动受限)的关节数量、活动受限的关节数量、儿童健康评估问卷(chaq)和hs-crp。如果患者在第48周前过早中断治疗,则hs-crp必须由中心实验室在eot时进行。
[0993]
j幼年关节炎疾病活动性得分评分
[0994]
k血液学(在药物给予之前应抽血):血红蛋白、血细胞比容、红细胞(rbc)计数和形态学(如果rbc计数异常)、白细胞(wbc)分类(嗜中性粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞)、血小板计数、绝对嗜中性粒细胞计数(anc)。如果患者在第48周前过早中断治疗,则所述测试必须由中心实验室在eot时进行。
[0995]
l化学(在药物给予前应抽血):将测试alt、ast、alp、总胆红素、结合胆红素、未结合胆红素和白蛋白。完整的化学检查应在第27次访视eot(对于第三部分为第96周)时完成。如果患者在第48周前过早中断治疗,则所述测试必须由中心实验室在eot时进行。
[0996]
m血脂(在药物给予前应抽血):甘油三酯(tg)、总胆固醇、高密度脂蛋白(hdl)胆固醇、低密度脂蛋白(ldl)胆固醇。需要患者在测试前禁食至少8小时。在eot时,所述测试将仅针对在第48周前过早中断治疗的患者进行,并且必须由中心实验室进行。
[0997]
n在eot时,ana/抗dsdna抗体测试将仅针对在第48周前过早中断治疗的患者进行,并且必须由中心实验室进行。
[0998]
o对于已经开始月经来潮的女性,应在每次安排的访视时暴露于imp之前和eot时进行尿液妊娠测试。可在当地进行尿液妊娠测试。
[0999]
p在治疗期期间的给药日,将在imp给予之前采集血液样品。如果患者出现sae,则在可能的情况下,应在所述事件开始和结束时或者接近开始和结束时采集血液样品以测定萨瑞鲁单抗浓度和抗药物抗体(ada)评估。
research.arch dis child 2018;103:695.
[1026]
25.gill kl,machavaram kk,rose rh,chetty m.potential sources of inter-subject variability in monoclonal antibody pharmacokinetics.clin pharmacokinet 2016;55:789
–
805.
[1027]
26.kojima t,yabe y,kaneko a,hirano y,ishikawa h,hayashi m,et al.monitoring c-reactive protein levels to predict favourable clinical outcomes from tocilizumab treatment in patients with rheumatoid arthritis.modern rheumatology 2013;23:977
–
85.
[1028]
27.madhok r,crilly a,watson j,capell ha.serum interleukin 6 levels in rheumatoid arthritis:correlations with clinical and laboratory indices of disease activity.ann rheum dis 1993;52:232
–
4.
[1029]
28.nishimoto n,terao k,mima t,nakahara h,takagi n,kakehi t.mechanisms and pathologic significances in increase in serum interleukin-6(il-6)and soluble il-6 receptor after administration of an anti-il-6 receptor antibody,tocilizumab,in patients with rheumatoid arthritis and castleman disease.blood 2008;112:3959
–
64.
[1030]
29.emery p,keystone e,tony hp,cantagrel a,van vollenhoven r,sanchez a,et al.il-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals:results from a 24-week multicentrerandomised placebo-controlled trial.ann rheum dis 2008;67:1516
–
23.
[1031]
30.fleischmann r,van adelsberg j,lin y,castelar-pinheiro gd,brzezicki j,hrycaj p,et al.sarilumab and nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis and inadequate response or intolerance to tumor necrosis factor inhibitors.arthritis rheumatol(hoboken)2017;69:277
–
90.
[1032]
31.genovese mc,fleischmann r,kivitz aj,rell-bakalarska m,martincova r,fiore s,et al.sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate:results of a phase iii study.arthritis rheumatol(hoboken)2015;67:1424
–
37.
[1033]
32.burmester gr,lin y,patel r,van adelsberg j,mangan ek,graham nm,et al.efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis(monarch):a randomised,double-blind,parallel-group phase iii trial.ann rheum dis 2017;76:840
–
47.
[1034]
33.emery p,rondon j,parrino j,lin y,pena-rossi c,van hoogstraten h,et al.safety and tolerability of subcutaneous sarilumab and intravenous tocilizumab in patients with rheumatoid arthritis.rheumatology(oxford)2018;58:849
–
58.
[1035]
34.pardeo m,wang j,ruperto n,alexeeva e,chasnyk v,schneider r,et al.neutropenia during tocilizumab treatment is not associated with infection risk in systemic or polyarticular-course juvenile idiopathic arthritis.j rheumatol 2019.available:https://www-ncbi-nlm-nih-gov/pubmed/30824645[accessed 29august 2019].
[1036]
35.actemra (tocilizumab)european medicines agency assessment report.available:
[1037]
https://www-ema-europa-eu/en/documents/variation-report/roactemra-h-c-955-ii-0072-epar-assessment-report-variation_en.pdf[accessed 28 may 2019].
[1038]
36.hsieh mm,everhart je,byrd-holt dd,tisdale jf,rodgers gp.prevalence of neutropenia in the u.s.population:age,sex,smoking status,and ethnic differences.ann intern med 2007;146:486
–
92.
[1039]
37.moots rj,sebba a,rigby w,ostor a,porter-brown b,donaldson f,et al.effect of tocilizumab on neutrophils in adult patients with rheumatoid arthritis:pooled analysis of data from phase 3 and 4 clinical trials.rheumatology(oxford)2017;56:541
–
49.
[1040]
38.genovese mc,van adelsberg j,fan c,graham nmh,van hoogstraten h,parrino j,et al.two years of sarilumab in patients with rheumatoid arthritis and an inadequate response to mtx:safety,efficacy and radiographic outcomes.rheumatology 2018;57:1423
–
31.
[1041]
39.european medicines agency.ethical considerations for clinical trials on medicinal products conducted with the paediatric population.recommendations of the ad hoc group for the development of implementing guidelines for directive 2001/20/ec relating to good clinical practice in the conduct of clinical trials on med.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1