用抗PD-L1抗体治疗癌症的方法与流程
用抗pd-l1抗体治疗癌症的方法
1.相关申请的交叉引用
2.本技术要求于2019年5月3日提交的美国临时专利申请号62/843,233的优先权,该美国临时专利申请的内容全文以引用方式并入本文。
3.以ascii文本文件提交序列表
4.以下提交的ascii文本文件的内容全文以引用方式并入本文:序列表的计算机可读格式(crf)(文件名:146392045040seqlist.txt,记录日期:2020年4月17日,大小:24kb)。
技术领域
5.本公开涉及与通过施用抗pd-l1抗体(例如,阿特珠单抗)来治疗癌症相关的方法、用途和试剂盒。
背景技术:
6.pd-l1在许多癌症中过度表达并且通常与不良预后相关(okazaki t等人,intern.immun.2007 19(7):813)(thompson rh等人,cancer res2006,66(7):3381)。有趣的是,与正常组织中的t淋巴细胞和外周血t淋巴细胞相反,大多数肿瘤浸润性t淋巴细胞主要表达pd-1,这表明肿瘤应答性t细胞上pd-1的上调可导致抗肿瘤免疫应答受损(blood 2009114(8):1537)。这可能是由于利用表达pd-l1的肿瘤细胞与表达pd-1的t细胞相互作用所介导的pd-l1信号传导,导致t细胞活化的减弱和逃避免疫监视(sharpe等人,nat rev 2002)(keir me等人,2008annu.rev.immunol.26:677)。因此,抑制pd-l1/pd-1相互作用可能会增强cd8+t细胞介导的肿瘤杀伤。
7.(阿特珠单抗)是一种人源化免疫球蛋白g1单克隆抗体,由两条重链和两条轻链组成。阿特珠单抗靶向肿瘤-浸润免疫细胞(ic)和肿瘤细胞上的人程序性死亡-配体1(pd-l1),并抑制其与其受体程序性死亡-1(pd-1)和b7.1的相互作用,这两种受体都可以向t细胞提供抑制信号。阿特珠单抗已在超过71个国家/地区被批准作为单一疗法用于治疗2l nsclc、2l转移性uc和/或1l不符合进行顺铂治疗条件的转移性uc。例如,阿特珠单抗已在美国或欧洲被批准用于以下适应症:治疗既往含铂化疗后的患有局部晚期或转移性尿路上皮癌(uc)的成人患者,或治疗被认为不符合进行顺铂治疗的条件且其肿瘤具有pd-l1表达≥5%的成人患者,对治疗既往化疗后的局部晚期或转移性非小细胞肺癌(nsclc)成人患者;治疗患有局部晚期或转移性uc的患者,该患者不符合进行含顺铂化疗的条件且其肿瘤表达pd-l1(pd-l1染色的ic覆盖≥5%的肿瘤区域),或者无论肿瘤pd-l1表达水平如何均不符合进行任何含铂化疗的条件,或者在进行任何含铂化疗期间或之后或在进行新辅助化疗或辅助化疗的12个月内具有疾病进展;以及治疗在进行含铂化疗期间或之后具有疾病进展的患有转移性nsclc的患者。阿特珠单抗还作为单一疗法进行开发,并与其他靶向药剂和细胞毒性剂联合用于治疗患有多种实体瘤和血液肿瘤的患者,包括肺癌、肾癌、结直肠癌和乳腺癌。
8.所有目前批准的阿特珠单抗适应症均获得批准以1200mg的剂量每3周(q3w)进行
一次静脉(iv)输注,直至出现疾病进展或不可接受的毒性。
9.本文所引用的所有参考文献,包括专利申请、专利公开和uniprotkb/swiss-prot登录号通过引用整体并入本文,如同个别参考文献各自特定地和个别地指示为通过引用并入一样。
技术实现要素:
10.除了1200mg q3w以外的给药方案将为包括阿特珠单抗在内的单一疗法和联合疗法提供更大的灵活性。例如,每4周施用一次的阿特珠单抗给药方案提供与批准的q3w方案相似的疗效和安全性水平,将会提供更大的患者便利,特别是作为维持阶段疗法的一部分。
11.在一些方面,本文提供了用于治疗人类患者中的癌症或延缓其进展的方法、试剂盒和用途,包括在两个或更多个4周或28天的周期内以1680mg的剂量向人类患者施用抗pd-l1抗体,其中在两个或更多个4周或28天周期的每个周期中,以每个周期1680mg的剂量施用抗pd-l1抗体(例如,每4周或每28天一次向人类患者施用抗pd-l1抗体)。
12.在一些方面,本文提供了用于治疗人类患者中的癌症或延缓其进展的方法、试剂盒和用途,包括在两个或更多个2周或14天的周期内以840mg的剂量向人类患者施用抗pd-l1抗体,其中在两个或更多个2周或14天周期的每个周期中,以每个周期840mg的剂量施用抗pd-l1抗体(例如,每2周或每14天一次向人类患者施用抗pd-l1抗体)。
13.在一些方面,本公开提供用于治疗患有癌症的人类患者的方法,该方法包括以每2周840mg或每4周1680mg的剂量向患者施用抗pd-l1抗体,其中抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。
14.在一些实施例中,抗pd-l1抗体在2周或4周的周期中每个周期的第1天施用。
15.在一些实施例中,抗pd-l1抗体在治疗的维持阶段内向患者施用。在一些实施例中,抗pd-l1抗体在治疗的诱导阶段内向患者施用。
16.在一些实施例中,本文所述的方法进一步包括向患者施用附加治疗剂。在一些实施例中,附加治疗剂包含化疗剂。在一些实施例中,化疗剂为针对癌症的标准护理。在一些实施例中,附加治疗剂包含抗体。
17.在一些实施例中,通过静脉输注向患者施用抗pd-l1抗体。在一些实施例中,通过历时60分钟的静脉输注向患者施用抗pd-l1抗体。在一些实施例中,在初次输注中,通过历时60分钟的静脉输注向患者施用抗pd-l1抗体,并且如果对第一次输注耐受,则在后续输注中通过历时30分钟的静脉输注向患者施用抗pd-l1抗体。在一些实施例中,通过历时30分钟的静脉输注向患者施用抗pd-l1抗体。
18.在一些实施例中,癌症选自由以下项组成的组:乳腺癌、结直肠癌、肺癌、肾细胞癌(rcc)、卵巢癌、黑素瘤和膀胱癌。在一些实施例中,乳腺癌为三阴性乳腺癌。在一个实施例中,肺癌为非小细胞肺癌或小细胞肺癌。在一些实施例中,膀胱癌为尿路上皮癌。在一些实施例中,癌症为局部晚期或转移性的。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌。
19.在一些实施例中,人类患者在施用抗pd-l1抗体之前已经使用含铂化疗进行治疗。在一些实施例中,人类患者不符合进行含铂化疗的条件。在一些实施例中,人类患者在施用抗pd-l1抗体之前已经使用辅助化疗或新辅助化疗进行治疗。
20.在一些实施例中,癌症为局部晚期或转移性非小细胞肺癌,并且其中患者在施用抗pd-l1抗体之前已经使用化疗进行治疗。
21.在一些实施例中,来自患者的癌症的样品包含肿瘤浸润免疫细胞,所述肿瘤浸润免疫细胞表达pd-l1并覆盖1%或更多的肿瘤区域,如通过免疫组化(ihc)测定的。
22.在本文所述方法的一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者。在本文所述方法的一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者,其中在既往含铂化疗之后向人类患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者,其中人类患者被认为不符合进行顺铂治疗的条件,并且其肿瘤具有≥5%的pd-l1表达。
23.在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性尿路上皮癌,其中人类患者不符合进行含顺铂的化疗的条件并且其肿瘤表达pd-l1(pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥5%的肿瘤区域),如通过由美国fda批准的测试确定的。在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性尿路上皮癌,其中人类患者无论pd-l1状态如何均不符合进行任何含铂化疗的条件。在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性尿路上皮癌,其中人类患者在进行任何含铂化疗期间或之后、或在新辅助化疗或辅助化疗的12个月内,具有疾病进展。
[0024]
在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性尿路上皮癌,其中人类患者接受了既往含铂化疗。在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性尿路上皮癌的,其中人类患者被认为不符合进行顺铂治疗的条件,并且其肿瘤具有≥5%的pd-l1表达。在一些实施例中,人类患者为成年人。
[0025]
在本文所述方法的一些实施例中,人类患者为患有转移性非鳞状非小细胞肺癌(nsclc)的成人患者,其中该方法包括施用抗pd-l1抗体、贝伐单抗、紫杉醇和卡铂,并且其中该方法为一线治疗。
[0026]
在本文所述方法的一些实施例中,人类患者为患有转移性非鳞状非小细胞肺癌(nsclc)的成人患者,其中转移性非鳞状nsclc为egfr突变的或alk阳性的,其中该包括施用抗pd-l1抗体、贝伐单抗、紫杉醇和卡铂的方法仅在合适的靶向疗法,诸如含铂疗法例如卡铂、贝伐单抗、长春氟宁、多西他赛或紫杉醇失败后才适用。在一些实施例中,转移性非鳞状nsclc为egfr突变的。在一些实施例中,转移性非鳞状nsclc为alk阳性的。
[0027]
在本文所述方法的一些实施例中,人类患者为进行既往化疗后的患有局部晚期或转移性nsclc的成人患者,其中包括施用抗pd-l1抗体的方法适用于单一疗法。
[0028]
在本文所述方法的一些实施例中,人类患者为进行既往化疗后的患有局部晚期或转移性nsclc的成人患者,其中转移性非鳞状nsclc为egfr突变的或alk阳性的,其中人类患者在进行本文所述的方法之前接受了靶向疗法,诸如含铂疗法,例如,卡铂、贝伐单抗、长春氟宁、多西他赛或紫杉醇。
[0029]
在本文所述方法的一些实施例中,人类患者患有没有egfr或alk基因组肿瘤畸变的转移性非鳞状非小细胞肺癌(nsclc)。在本文所述方法的一些实施例中,人类患者患有没
有egfr或alk基因组的转移性非鳞状非小细胞肺癌(nsclc),其中该方法包括其中所述方法包括施用抗pd-l1抗体、贝伐单抗、紫杉醇和卡铂,并且其中该方法为一线治疗。
[0030]
在本文所述方法的一些实施例中,人类患者患有转移性nsclc,其中该人类患者在进行含铂化疗期间或之后进展,其中该指示为作为单一药剂的抗pd-l1抗体。
[0031]
在本文所述方法的一些实施例中,人类患者患有具有egfr或alk基因组肿瘤畸变的转移性nsclc,其中对该人类患者的针对非小细胞肺癌的靶向治疗失败,其中该方法包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂。
[0032]
在本文所述方法的一些实施例中,人类患者患有转移性非小细胞肺癌,并且其中人类患者在进行含铂化疗期间或之后进展。在一些实施例中,该方法包括将抗pd-l1抗体作为单一药剂向人类患者施用。在一些实施例中,其中人类患者具有egfr或alk基因组肿瘤畸变,患者在靶向治疗上取得进展。在一些实施例中,其中人类患者具有egfr或alk基因组肿瘤畸变,患者在fda批准的治疗上取得进展。
[0033]
在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性非小细胞肺癌,其中人类患者已接受过既往化疗。
[0034]
在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性三阴性乳腺癌。在本文所述方法的一些实施例中,人类患者患有局部晚期或转移性三阴性乳腺癌,其是不可切除的局部晚期或转移性三阴性乳腺癌。在本文所述方法的一些实施例中,人类患者具有表达pd-l1的肿瘤(任何强度的pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥1%的肿瘤区域),如通过fda批准的测试确定的。
[0035]
在另一方面,本公开提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,所述方法包括以每2周840mg或每4周1680mg的剂量向患者施用抗pd-l1抗体,其中该抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。在一些实施例中,患者(i)不符合进行含顺铂化疗的条件,并且其肿瘤表达pd-l1(pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥5%的肿瘤区域),(ii)无论pd-l1状态如何均不符合进行任何含铂化疗的条件,或者(iii)在进行任何含铂化疗期间或之后、或在进行新辅助化疗或辅助化疗的12个月内,具有疾病进展。
[0036]
在另一方面,本公开提供了用于治疗患有非小细胞肺癌(nsclc)的人类患者的方法,所述方法包括以每2周840mg或每4周1680mg的剂量将抗pd-l1抗体作为单一药剂向患者施用,其中该抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。在一些实施例中,患者(i)患有转移性nsclc并且在进行含铂化疗期间或之后具有疾病进展,或者(ii)具有egfr或alk基因组肿瘤畸变。
[0037]
在另一方面,本公开提供了用于治疗患有非小细胞肺癌(nsclc)的人类患者的方法,所述方法包括(a)以每3周1200mg的剂量向患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇
和卡铂,施用4-6个周期的紫杉醇和卡铂;以及(b)如果停止使用贝伐单抗,则以每2周840mg或每4周1680mg的剂量向患者施用抗pd-l1抗体;其中该抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。在一些实施例中,患者患有没有egfr或alk基因组肿瘤畸变的转移性非鳞状nsclc。在一些实施例中,该方法适用于针对没有egfr或alk基因组肿瘤畸变的转移性非鳞状nsclc的一线治疗。在一些实施例中,贝伐单抗以15mg/kg施用,紫杉醇以175mg/m2或200mg/m2施用,并且卡铂以auc 6mg/ml/min施用,其中该
[0038]
在另一方面,本公开提供了用于治疗患有小细胞肺癌(sclc)的人类患者的方法,所述方法包括(a)以每3周1200mg的剂量向患者施用抗pd-l1抗体联合卡铂和依托泊苷,施用4个周期的卡铂和依托泊苷;以及(b)在完成(a)后,以每2周840mg或每4周1680mg的剂量向患者施用抗pd-l1抗体;其中该抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。在一些实施例中,患者患有广泛期小细胞肺癌(es-sclc)。在一些实施例中,在每个21天周期中,卡铂在第1天以auc 5mg/ml/min施用,并且依托泊苷在第1、2和3天以100mg/m2静脉内施用。在一些实施例中,治疗适用于一线治疗。
[0039]
在另一方面,本公开提供了用于治疗患有不可切除的局部晚期或转移性tnbc的人类患者的方法,所述方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体,其中该方法进一步包括每周几天以100mg/m2的剂量向人类患者施用紫杉醇,其中该抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列,所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。在一些实施例中,该方法包括在28天周期的第1天和第15天以840mg的剂量向人类患者施用抗pd-l1抗体,并在28天周期的第1、8和15天向人类患者施用蛋白结合型紫杉醇。在一些实施例中,人类患者具有表达pd-l1的肿瘤(pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥1%的肿瘤区域)。
[0040]
在本文所述方法的一些实施例中,癌症为乳腺癌(例如,不可切除的局部晚期或转移性tnbc),并且该方法进一步包括施用紫杉烷(例如,紫杉醇或蛋白结合型紫杉醇)联合抗pd-l1抗体(例如,阿特珠单抗)。
[0041]
在本文所述方法的一些实施例中,通过静脉输注向患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,通过历时60分钟的静脉输注向患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,在初次输注中,通过历时60分钟的静脉输注向患者施用抗pd-l1抗体,并且如果对第一次输注耐受,则在后续输注中通过历时30分钟的静脉输注向患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,通过历时30分钟的静脉输注向患者施用抗pd-l1抗体。
[0042]
在本文所述方法的一些实施例中,患者为成年患者。
[0043]
在本文所述方法的一些实施例中,抗pd-l1抗体包含重链和轻链,所述重链包含gftfsdswih(seq id no:1)的hvr-h1序列、awispyggstyyadsvkg(seq id no:2)的hvr-h2序列和rhwpggfdy(seq id no:3)的hvr-h3序列;所述轻链包含rasqdvstava(seq id no:4)的hvr-l1序列、sasflys(seq id no:5)的hvr-l2序列和qqylyhpat(seq id no:6)的hvr-l3序列。
[0044]
在本文所述方法的一些实施例中,抗pd-l1抗体的重链包含重链可变(vh)结构域,所述vh结构域包含evqlvesggglvqpggslrlscaasgftfsdswihwvrqapgkglewvawispyggstyyadsvkgrftisadtskntaylqmnslraedtavyycarrhwpggfdywgqgtlvtvss(seq id no:7)的序列,并且其中抗pd-l1抗体的轻链包含轻链可变(vl)结构域,所述vl结构域包含diqmtqspsslsasvgdrvtitcrasqdvstavawyqqkpgkapklliysasflysgvpsrfsgsgsgtdftltisslqpedfatyycqqylyhpatfgqgtkveikr(seq id no:8)的序列。
[0045]
在本文所述方法的一些实施例中,抗pd-l1抗体为阿特珠单抗。
[0046]
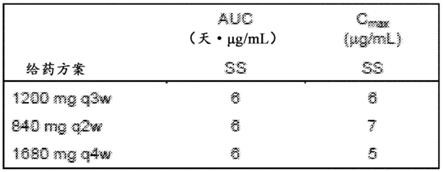
在另一方面,本公开提供了试剂盒,该试剂盒包含处于药用载体中的单位剂量的抗pd-l1抗体,用于本文所述的任何一种方法。在一些实施例中,单位剂量的抗pd-l1抗体为840mg。在一些实施例中,单位剂量的抗pd-l1抗体在14ml的包含药用载体的溶液中提供。
[0047]
应了解,本文所描述的各种实施例的一种、一些或所有特性可组合形成本发明的其它实施例。本发明的这些和其它方面对于本领域技术人员将变得显而易见。通过下面的详细描述进一步描述本发明的这些和其它实施例。
附图说明
[0048]
专利或申请文件包含至少一幅彩色附图。在提出请求并支付必要的费用后,专利局将提供带有一幅或多幅彩图的本专利或专利申请公布的拷贝。
[0049]
图1示出了针对阿特珠单抗的poppk模型鉴定的具有统计学意义的参数-协变量关系。bwt=体重(kg);i表示特定患者;albu=白蛋白(g/l);肿瘤负荷(mm);atag=抗治疗性抗体的基线后状态。
[0050]
图2提供了比较协变量(bw、白蛋白、肿瘤负荷、性别、atag)对阿特珠单抗稳态暴露参数auc
ss
(左)、c
max,ss
(中)和c
min,ss
(右)的影响的敏感性图。除bw外,没有协变量效应导致典型患者的暴露量发生超过30%的变化。atag=抗治疗性抗体的基线后状态;auc
ss
=在稳态时的血清浓度时间曲线下的面积;c
max,ss
=在稳态时观察到的最大血清浓度;c
min,ss
=在稳态时观察到的最小血清浓度。最终模型估计值(由黑色垂直线和值表示)是指在协变量等于中位数的典型患者(男性)中的阿特珠单抗1200mg q3w的预测稳态暴露。在灰色区域,深色和浅色分别代表相对于基线变化20%和30%。顶部的条示出接受1200mg q3w的群体的第10个和第90个百分位数([p10-p90])暴露范围。每个水平条代表单个协变量对暴露度量的影响。条形左端的标签表示使用协变量分布的第10个和第90个百分位数([p10-p90])的值评估的协变量。每个条的长度描述了该特定协变量对阿特珠单抗暴露的潜在影响,以及相对于基线(蓝色值)的暴露百分比变化。
[0051]
图3a-3b提供了使用来自imvigor210(图3a)和imvigor211(图3b)临床试验的阿特珠单抗数据的i期群体药代动力学(poppk)模型进行的预测校正视觉预测检查(pcvpc)。pcvpc表明,i期poppk模型足以预测来自imvigor210和imvigor211的所有患者的阿特珠单
抗pk数据。ci=置信区间。
[0052]
图4a-4b提供了使用来自birch、fir和poplar(图4a)以及oak(图4b)临床试验的合并的阿特珠单抗数据的i期poppk模型进行的预测校正视觉预测检查(pcvpc)。通过研究,pcvpc表明,i期poppk模型足以预测birch(所有群组)以及fir(所有群组)和oak中的阿特珠单抗pk数据。观察到poplar出现负的群体-水平预测和残差的趋势,但这种趋势在个体预测和残差中得到解决,这表明i期poppk模型允许在所有研究中对个体参数进行可靠和稳健的贝叶斯估计。ci=置信区间。
[0053]
图5a-5c提供了imvigor210中接受阿特珠单抗1200mg q3w的1l不符合顺铂治疗条件的尿路上皮癌患者的客观应答率对阿特珠单抗暴露度量周期1auc(图5a)、周期1c
min
(图5b)和auc
ss
(图5c)的逻辑回归。在考虑到任何暴露度量的情况下,应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系。1l=一线;auc=曲线下面积;c
min
=周期中的最低浓度;auc
ss
=稳态时的曲线下面积;ci=置信区间;cr=完全应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;pr=部分应答;q3w=每三周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0054]
图6a-6c提供了imvigor210中接受阿特珠单抗1200mg q3w的2l尿路上皮癌患者的客观应答率对阿特珠单抗暴露度量周期1auc(图6a)、周期1c
min
(图6b)和auc
ss
(图6c)的逻辑回归。在考虑到任何暴露度量的情况下,应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系。2l=二线;auc=曲线下面积;c
min
=周期中的最低浓度;auc
ss
=稳态时的曲线下面积;ci=置信区间;cr=完全应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;pr=部分应答;q3w=每三周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0055]
图7提供了imvigor211中接受1200mg阿特珠单抗的2l尿路上皮癌患者的客观应答率对阿特珠单抗暴露度量周期1auc的逻辑回归。在阿特珠单抗1200mg q3w后,没有发现与orr具有统计学意义的er关系(周期1auc)。2l=二线;auc=曲线下面积;ci=置信区间;cr=完全应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;pr=部分应答;q3w=每三周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0056]
图8a-8d提供了针对birch中接受1200mg阿特珠单抗q3w的nsclc患者,客观应答率对阿特珠单抗暴露度量周期1c
min
(图8a)、周期1auc(图8b)、auc
ss
(图8c)和患者体重(图8d)的逻辑回归。对于birch,在与应答阿特珠单抗暴露的概率增加趋势相关的暴露度量中,与auc
ss
相关的p值最低(p=0.0005343)。auc=曲线下面积;c
min
=周期中的最低浓度;auc
ss
=
稳态时的曲线下面积;ci=置信区间;c
min
=周期中的最低浓度;cr=完全应答;ic=免疫细胞;pr=部分应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;q3w=每3周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0057]
图9a-9d提供了针对oak中接受1200mg阿特珠单抗q3w的nsclc患者,客观应答率对阿特珠单抗暴露度量周期1c
min
(图9a)、周期1auc(图9b)、auc
ss
(图9c)和患者体重(图9d)的逻辑回归。对于oak,在与应答阿特珠单抗暴露的概率增加趋势相关的暴露度量中,与auc
ss
相关的p值最低。auc=曲线下面积;c
min
=周期中的最低浓度;auc
ss
=稳态时的曲线下面积;ci=置信区间;c
min
=周期中的最低浓度;cr=完全应答;ic=免疫细胞;pr=部分应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;q3w=每3周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0058]
图10a-10c提供了针对poplar中接受阿特珠单抗1200mg q3w的nsclc患者,客观应答率对阿特珠单抗暴露度量周期1c
min
(图10a)、周期1auc(图10b)和auc
ss
(图10c)的逻辑回归。在考虑到任何暴露度量的情况下,应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系。auc=曲线下面积;c
min
=周期中的最低浓度;auc
ss
=稳态时的曲线下面积;ci=置信区间;cr=完全应答;n=患者人数;p=在有应答者的比例对暴露的逻辑回归中wald检验的p值;pr=部分应答;q3w=每三周。灰色实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci。垂直线为暴露四分位数的限值。十字为患者应答事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0059]
图11a-11b提供了在校正预后因素的不平衡之后的总存活(os)模型的模拟。在跨auc
ss
三分位数和多西他赛组校正预后因素(转移部位的数量和白蛋白水平)的不平衡之后,poplar中nsclc患者的os模型的模拟(图11a)表明所有患者都将受益于阿特珠单抗治疗。在跨auc
ss
三分位数和多西他赛组校正预后因素(基线bsld、白蛋白、ecog体能状态和ldh水平)的不平衡之后,oak中nsclc患者的os模型的模拟(图11b)表明所有患者都将受益于阿特珠单抗治疗。auc
ss
=稳态时曲线下面积的中值和范围,以μg.天/ml为单位;hr=风险比;ci=置信区间;nsclc=非小细胞肺癌;q3w=每3周。
[0060]
图12提供了oak中接受1200mg阿特珠单抗q3w的nsclc患者按bw的四分位数区分的os的kaplan-meier图。kaplan-meier图表明体重较重的患者与体重较轻的患者具有相似的os。n=患者人数;nsclc=非小细胞肺癌;os=总存活;q1=第一个四分位数;q2=第二个四分位数;q3=第三个四分位数;q4=第四个四分位数;q3w=每3周;对于区间符号,包括a但不包括b。普通线和虚线为kaplan-meier估计。十字为截尾观察值。
[0061]
图13a-13b提供了针对合并的患有局部晚期或转移性nsclc或uc的患者,应答的比例(cr+pr)对阿特珠单抗暴露度量周期1auc(图13a)和周期1c
min
(图13b)的逻辑回归。对于
图13a,为便于阅读,图上未显示1个极端auc值(》15,000μg.天/ml)。显示了来自有应答者的比例对暴露的逻辑回归的wald p值。灰色实线和阴影区域代表逻辑回归斜率模型和95%pi。实心圆圈和误差条代表暴露四分位数中有应答者的比例和95%ci;垂直线为暴露四分位数的限值。十字标记(x)代表应答事件(0:否,1:是)。三角形和2头箭头分别代表接受阿特珠单抗1200mg的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。周期1auc对应于治疗开始后前3周的auc,pk参数仅基于周期1数据估计。auc=浓度-时间曲线下面积;cmin=最低(谷值)血清阿特珠单抗浓度;cr=完全应答;n=患者人数;nsclc=非小细胞肺癌;pi=预测区间;pk=药代动力学;pr=部分应答;uc=尿路上皮癌。
[0062]
图14a-14b提供了通过auc(周期1,μg.天/ml)四分位数在模拟os分布中进行的tgi-os模型的验证。用来自oak(nsclc)(图14a)和imvigor211(uc)(图14b)的截尾数据(+符号)绘制了观察到的kaplan-meier os分布。阴影区域代表os分布的95%pi。对于区间符号格式[a,b),包括a但不包括b,所以为a≤x《b。auc:浓度-时间曲线下面积(0至21天),nsclc=非小细胞肺癌;os=总存活;pi=预测区间;tgi=肿瘤生长抑制;uc=尿路上皮癌。
[0063]
图15a-15b提供了针对具有原始协变量的患者通过周期1auc四分位数在模拟hr(阿特珠单抗与比较剂对比)中进行的tgi-os模型的验证。示出了来自oak(nsclc)(图15a)和imvigor211(uc)(图15b)的os hr的森林图。观察到的hr显示为正方形,模型预测的hr显示为菱形,条形表示95%pi(1000次重复)。atezo=阿特珠单抗;auc=浓度-时间曲线下面积;chemo=化疗;c
min
=最低(谷值)血清阿特珠单抗浓度;doce=多西他赛;hr=风险比;nsclc=非小细胞肺癌;os=总存活;pi=预测区间;tgi=肿瘤生长抑制;uc=尿路上皮癌。
[0064]
图16a-16b提供了针对具有中值协变量的患者通过周期1auc四分位数预测的os hr(阿特珠单抗与比较剂对比)。示出了来自oak(nsclc)(图16a)和imvigor211(uc)(图16b)的os hr的森林图。模型预测的hr显示为菱形,条形表示95%的pi(1000次重复)。atezo=阿特珠单抗;auc=浓度-时间曲线下面积;chemo=化疗;doce=多西他赛;hr=风险比;nsclc=非小细胞肺癌;os=总存活;pi=预测区间;uc=尿路上皮癌。
[0065]
图17a-17c提供了针对在pcd4989g研究(尿路上皮癌群组)和imvigor210研究(群组1和2)中阿特珠单抗剂量为15mg/kg和1200mg q3w的患者,经历级别≥3的ae的患者比例对阿特珠单抗暴露度量周期1auc(图17a)、周期1c
max
(图17b)和auc
ss
(图17c)的逻辑回归。对aeg35(级别≥3的ae)发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的er关系。auc=浓度-时间曲线下的面积;c
max
=血清中的最大浓度;auc
ss
=稳态时的auc;ae=不良事件;ci=置信区间;n=患者人数;p=发生率对暴露的逻辑回归中wald检验的p值;q3w=每三周。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0066]
图18a-18b提供了针对imvigor211研究中接受阿特珠单抗1200mg q3w患者,经历级别≥3的ae的患者比例对阿特珠单抗暴露度量周期1auc(图18a)和周期1c
max
(图18b)的逻辑回归。对aeg35发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的er关系。auc=浓度-时间曲线下的面积;c
max
=血清中的最大浓度;ae=不良事件;ci=置信区间;n=患者人数;p=发生率对暴露的逻辑回归中wald检验的p值;q3w=每三周。粗实线和
阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0067]
图19a-19c提供了针对在pcd4989g研究(尿路上皮癌群组)和imvigor210研究(群组1和2)中阿特珠单抗剂量为15mg/kg和1200mg q3w的患者,经历aesi的患者比例对阿特珠单抗暴露度量周期1auc(图19a)、周期1c
max
(图19b)和auc
ss
(图19c)的逻辑回归。aesi的发生率未显示与所调查的任何暴露度量有任何统计学上显著的er关系。auc=浓度-时间曲线下的面积;c
max
=血清中的最大浓度;auc
ss
=稳态时的auc;aesi=特别关注的不良事件;ci置信区间;n=患者人数;p=发生率对暴露的逻辑回归中wald检验的p值;q3w=每三周。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0068]
图20a-20b提供了针对imvigor211研究中接受阿特珠单抗1200mg q3w患者,经历aesi的患者比例对阿特珠单抗暴露度量周期1auc(图20a)和周期1c
max
(图20b)的逻辑回归。对aesi发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的er关系。auc=浓度-时间曲线下的面积;c
max
=血清中的最大浓度;aesi=特别关注的不良事件;n=患者人数;p=发生率对暴露的逻辑回归中wald检验的p值;q3w=每三周。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0069]
图21a-21c提供了针对在pcd4989(nsclc群组)、birch、poplar和fir研究中阿特珠单抗剂量为1mg/kg至20mg/kg(包括1200mg固定剂量)的nsclc患者,经历级别≥3的ae的患者比例对阿特珠单抗暴露度量周期1auc(图21a)、周期1c
max
(图21b)和auc
ss
(图21c)的逻辑回归。对aeg35发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的正er关系。auc=浓度-时间曲线下面积;c
max
=血清中最大浓度;auc
ss
=稳态时的auc;ae=不良事件;aeg35=级别3至5的不良事件;ci=置信区间;n=患者人数;nsclc=非小细胞肺癌;p=发生率对暴露的逻辑回归中wald检验的p值。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0070]
图22a-22c提供了针对oak研究中接受阿特珠单抗1200mg q3w的患者,经历级别≥3的ae的患者比例对阿特珠单抗暴露度量周期1auc(图22a)、周期1c
max
(图22b)或auc
ss
(图22c)的逻辑回归。对aeg35发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的正er关系。auc=浓度-时间曲线下面积;c
max
=血清中最大浓度;auc
ss
=稳态时的auc;ae=不良事件;aeg35=级别3至5的不良事件;ci=置信区间;n=患者人数;nsclc=非小细胞肺癌;p=发生率对暴露的逻辑回归中wald检验的p值。粗实线和阴影区域代表逻辑
回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0071]
图23a-23c提供了针对在pcd4989(nsclc群组)、birch、poplar和fir研究中阿特珠单抗剂量为1mg/kg至20mg/kg(包括1200mg固定剂量)的nsclc患者,经历aesi的患者比例对阿特珠单抗暴露度量周期1auc(图23a)、周期1c
max
(图23b)和auc
ss
(图23c)的逻辑回归。对pcd4989g、birch、poplar和fir中nsclc患者的合并分析的aesi发生率的分析未显示与周期1auc(图23a)或c
max
(图23b)有任何统计学上显著的er关系,但是确实与auc
ss
具有统计学上显著的关系(图23c)。auc=浓度-时间曲线下面积;auc
ss
=稳态时的浓度-时间曲线下面积;c
max
=血清中最大浓度;aesi=任何级别的特别关注的不良事件;ci=置信区间;n=患者人数;nsclc=非小细胞肺癌;p=发生率对暴露的逻辑回归中wald检验的p值。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0072]
图24a-24c提供了针对oak研究中接受阿特珠单抗1200mg q3w的患者,经历aesi的患者比例对阿特珠单抗暴露度量周期1auc(图24a)、周期1c
max
(图24b)和auc
ss
(图24c)的逻辑回归。对aesi发生率的分析未显示与所调查的任何暴露度量有任何统计学上显著的er关系。auc=浓度-时间曲线下面积;c
max
=血清中最大浓度;auc
ss
=稳态时的浓度-时间曲线下面积;aesi=任何级别的特别关注的不良事件;ci=置信区间;n=患者人数;nsclc=非小细胞肺癌;p=发生率对暴露的逻辑回归中wald检验的p值。粗实线和阴影区域代表逻辑回归斜率模型和95%预测区间。实心圆圈和误差条代表暴露四分位数中的发生率和95%ci。垂直线为暴露四分位数的限值。十字为ae事件(0:否;1:是)。三角形和双头箭头分别代表接受1200mg阿特珠单抗的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。
[0073]
图25a-25b提供了患有局部晚期或转移性nsclc或uc的患者安全性的合并暴露-应答分析。将指示的ae频率([a,c]级别≥3ae(图25a);[b,d]aesi(图25b))对auc周期1作图。为便于阅读,图中未显示2个极端auc值(》15,000μg.天/ml)。显示了来自ae发生率对暴露的逻辑回归的wald p值。灰色实线和阴影区域代表逻辑回归斜率模型和95%pi。实心圆圈和误差条代表暴露四分位数中ae的比例和95%ci;垂直线为暴露四分位数的限值。十字标记(x)代表ae事件(0:否,1:是)。三角形和2头箭头分别代表接受阿特珠单抗1200mg的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。周期1auc对应于治疗开始后前3周的auc,pk参数仅基于周期1数据估计。ae=不良事件;aesi=特别关注的不良事件;auc=浓度-时间曲线下面积;c
max
=最大血清阿特珠单抗浓度;n=患者人数;nsclc=非小细胞肺癌;pi=预测区间;pk=药代动力学;uc=尿路上皮癌。
[0074]
图26a-26b提供了患有局部晚期或转移性nsclc或uc的患者安全性的合并暴露-应答分析。将指示的ae频率([a,c]级别≥3ae(图26a);[b,d]aesi(图26b))对周期1的c
max
作图。为便于阅读,图中未显示2个极端c
max
值(》1500μg/ml)。显示了来自ae发生率对暴露的逻辑回归的wald p值。灰色实线和阴影区域代表逻辑回归斜率模型和95%pi。实心圆圈和误差条代表暴露四分位数中ae的比例和95%ci;垂直线为暴露四分位数的限值。十字标记(x)
代表ae事件(0:否,1:是)。三角形和2头箭头分别代表接受阿特珠单抗1200mg的患者在第10个和第90个百分位数之间的平均暴露和暴露区间。周期1auc对应于治疗开始后前3周的auc,pk参数仅基于周期1数据估计。ae=不良事件;aesi=特别关注的不良事件;auc=浓度-时间曲线下面积;c
max
=最大血清阿特珠单抗浓度;n=患者人数;nsclc=非小细胞肺癌;pi=预测区间;pk=药代动力学;uc=尿路上皮癌。
[0075]
图27说明了指定给药方案(840mg q2w、1200mg q3w、1680mg q4w和20mg/kg q3w)的模拟阿特珠单抗暴露曲线。绘制几何平均值。阴影区域代表90%pi。线:几何平均数;面积:90%预测区间(500名患者)。显示了28天期间的pk曲线,显示2个剂量的1200mg q3w、20mg/kg q3w和840mg q2w;以及1个剂量的1680mg q4w。周期1和稳态时相应的预测c
max
和c
min
值在表7中列出。pi=预测区间;q2w=每2周;q3w=每3周;q4w=每4周。
[0076]
图28示出了在研究pcd4989g中接受20mg/kg阿特珠单抗q3w的个体患者的最大观察到的c
max
浓度的直方图。
[0077]
图29提供了使用1期poppk模型在tnbc(impassion130)中预测校正的阿特珠单抗数据的vpc。数据以半对数标度绘制。该图中未显示两个《1μg/ml的群体预测浓度。n=样品数;obs=观察的;pi=预测区间;poppk=群体药代动力学;pred=预测;sim=模拟;tnbc=三阴性乳腺癌;vpc=视觉性能检查。
[0078]
图30提供了接受阿特珠单抗1200mg q3w iv或20mg/kg iv q3w(阿特珠单抗治疗的安全性可评估患者)的不良事件的总体总结。阿特珠单抗以20mg/kg q3w剂量给药的总体安全性与以1200mg q3w固定剂量给药时观察到的相似。
[0079]
图31提供了基于食蟹猴重复给药毒性研究的安全范围。auc=浓度-时间曲线下面积;c
max
=观察到的最大浓度;q2w=每2周;q3w=每3周;q4w=每4周;ss=稳态。
具体实施方式
[0080]
i.定义
[0081]
在详细描述本发明之前,应当理解,本发明不限于特定的组合物或生物学系统,这些组合物或生物学系统当然可以变化。另外应当了解,本文使用的术语只是为了描述特定实施例的目的,并非旨在进行限制。
[0082]
如在本说明书和所附权利要求中所用,单数形式“一个”、“一种”、“该”和“所述”包括复数指代物,除非上下文另外明确规定。因此,例如,对“分子”的提及任选地包括两个或更多此类分子的组合等。
[0083]
如本文所用的术语“约”是指为此技术领域中的技术人员容易知晓的相应值的常见误差范围。在本文中提及“约”值或参数包括(且描述)涉及该值或参数本身的实施例。
[0084]
应当理解,本文所述的发明的方面和实施例包括“包含”、“由以下组成”及“基本上由以下组成”所指的方面和实施例。
[0085]
如本文所用,术语“治疗”是指旨在于临床病理学的进程期间改变所治疗的个体或细胞的自然进程的临床干预。理想的治疗效果包括降低疾病进展速度、减缓或减轻疾病状态以及缓解或改善预后。例如,如果减轻或消除了与癌症有关的一种或多种症状,包括但不限于减少癌细胞的增殖(或破坏)、减轻疾病所致的症状、提高患有该疾病的人的生活质量、减少治疗该疾病所需的其他药物的剂量和/或延长个体的存活,则成功地“治疗”了个体。
[0086]
如本文所用,“延缓疾病的进展”意指延缓、阻碍、减缓、迟滞、稳定和/或推迟疾病(诸如癌症)的发展。这种延迟可以具有不同的时间长度,这取决于病史和/或待治疗的个体。对于本领域技术人员显而易见的是,充分或显著延迟实际上可以涵盖预防,因为个体不会患该病。例如,晚期癌症,诸如转移的发展,可能被延迟。
[0087]“持续应答”是指在停止治疗后对减少肿瘤生长的持续效果。例如,与给药阶段开始时的大小相比,肿瘤大小可以保持相同或更小。在一些实施例中,持续应答的持续时间至少与治疗持续时间相同、至少为治疗持续时间的1.5倍、2.0倍、2.5倍或3.0倍长度。
[0088]
术语“药物制剂”是指处于允许活性成分的生物学活性有效的形式,并且不含对于将被施用制剂的受试者具有不可接受的毒性的另外组分的制备物。此类制剂为无菌制剂。“药用”赋形剂(载体、添加剂)是指可合理地施用于受试哺乳动物以提供有效剂量的所用活性成分的赋形剂。
[0089]
如本文所用,“与...结合”(in conjunction with)是指除一种治疗方式之外还施用另一种治疗方式。这样,“与
……
结合”是指在向个体施用一种治疗方式之前,之中或之后施用另一种治疗方式。
[0090]
如本文所用,术语“肿瘤”是指所有赘生性细胞生长和增殖,无论是恶性还是良性,以及所有前癌性和癌性细胞和组织。术语“癌症”、“癌性”、“细胞增生性疾病”、“增生性疾病”和“肿瘤”在本文中并不互相排斥。
[0091]
如本文所用,“癌症”和“癌”是指或描述哺乳动物中通常以细胞生长不受控制为特征的生理状况。该定义包括良性和恶性癌症以及休眠肿瘤或微转移。癌症的实例包括但不限于癌、淋巴瘤、母细胞瘤、肉瘤和白血病。此类癌症的更具体实例包括但不限于鳞状细胞癌、肺癌(包括小细胞肺癌、非小细胞肺癌、肺腺癌和肺鳞癌)、黑素瘤、肾细胞癌、腹膜癌、肝细胞癌、胃癌或胃部癌症(包括胃肠道癌)、胰腺癌、成胶质细胞瘤、宫颈癌、卵巢癌、肝癌、膀胱癌、肝细胞瘤、乳腺癌、结肠癌、结直肠癌、子宫内膜癌或子宫癌、唾液腺癌、肾脏癌症或肾癌、肝癌、前列腺癌、外阴癌、甲状腺癌、肝细胞癌和各种类型的头颈癌,以及b细胞淋巴瘤(包括低级别/滤泡性非霍奇金淋巴瘤(nhl)、小淋巴细胞(sl)nhl、中级别/滤泡性nhl、中级别弥散性nhl、高级别免疫原性nhl、高级别淋巴母细胞性nhl、高级别小非裂解细胞性nhl、巨大肿块nhl、套细胞淋巴瘤、aids相关淋巴瘤和华氏巨球蛋白血症)、慢性淋巴细胞性白血病(cll)、急性淋巴细胞白血病(all)、毛细胞白血病、慢性粒细胞性白血病和移植后的淋巴增生性疾病(ptld),以及与斑痣性错构瘤、水肿(诸如与脑肿瘤有关的)、meigs综合征相关的异常血管增生。癌症的实例可包括任何上述癌症类型的原发性肿瘤或源自任何上述癌症类型的第二位点的转移性肿瘤。
[0092]
如本文所用,“转移”是指癌症从其原发部位扩散到身体的其他部位。癌细胞可以脱离原发肿瘤,渗入淋巴管和血管,在血流中循环,并在身体其他部位的正常组织的远处病灶中生长(转移)。转移可以是局部的或远处的。转移是一个连续的过程,取决于肿瘤细胞从原发肿瘤脱落,穿过血流,并在远处停止。在新部位,细胞建立血液供应,并可以生长形成危及生命的肿块。肿瘤细胞内的刺激性和抑制性分子通路调节这种行为,并且肿瘤细胞与远处宿主细胞之间的相互作用也很重要。
[0093]
如本文所用,术语“细胞毒性剂”是指对细胞有害(例如,造成细胞死亡、抑制增殖或以其他方式阻碍细胞功能)的任何试剂。细胞毒性剂包括但不限于放射性同位素(例如,
at
211
、i
131
、i
125
、y
90
、re
186
、re
188
、sm
153
、bi
212
、p
32
、pb
212
和lu的放射性同位素);化疗剂;生长抑制剂;酶及其片段,诸如溶核酶;以及毒素,诸如细菌、真菌、植物或动物来源的小分子毒素或酶活性毒素,包括其片段和/或变体。示例性的细胞毒性剂可以选自抗微管剂、铂配位络合物、烷基化剂、抗生素剂、拓扑异构酶ii抑制剂、抗代谢剂、拓扑异构酶i抑制剂、激素和激素类似物、信号转导途径抑制剂、非受体酪氨酸激酶血管生成抑制剂、免疫治疗剂、促凋亡剂、ldh-a抑制剂、脂肪酸生物合成抑制剂、细胞周期信号传导抑制剂、hdac抑制剂、蛋白酶体抑制剂和癌症代谢抑制剂。在一个实施例中,细胞毒性剂是紫杉烷。在一个实施例中,紫杉烷为紫杉醇或多西他赛。在一个实施例中,细胞毒性剂为铂剂。在一个实施例中,细胞毒性剂是egfr的拮抗剂。在一个实施例中,egfr的拮抗剂是n-(3-乙炔基苯基)-6,7-双(2-甲氧基乙氧基)喹唑啉-4-胺(例如厄洛替尼)。在一个实施例中,细胞毒性剂是raf抑制剂。在一个实施例中,raf抑制剂是braf和/或craf抑制剂。在一个实施例中,raf抑制剂是维罗非尼。在一个实施例中,细胞毒性剂是pi3k抑制剂。
[0094]“化疗剂”包括可用于治疗癌症的化合物。化疗剂的示例包括厄洛替尼(genentech/osi pharm.)、硼替佐米(millennium pharm.)、双硫仑、表没食子儿茶素没食子酸酯、盐孢子酰胺a、卡非佐米、17-aag(格尔德霉素)、根赤壳菌素、乳酸脱氢酶a(ldh-a)、氟维司群(阿斯利康(astrazeneca))、舒尼替布(辉瑞(pfizer)/sugen)、来曲唑(诺华(novartis))、甲磺酸伊马替尼(诺华)、非那沙酯(诺华)、奥沙利铂(赛诺菲(sanofi))、5-fu(5-氟尿嘧啶)、亚叶酸、雷帕霉素(西罗莫司,惠氏(wyeth))、拉帕替尼(gsk572016,葛兰素史克(glaxo smith kline))、罗纳法米(lonafamib)(sch 66336)、索拉非尼(拜耳实验室(bayer labs))、吉非替尼(阿斯利康)、ag1478;烷基化剂诸如噻替派和环磷酰胺;烷基磺酸盐,诸如白消安、英丙舒凡和哌泊舒凡;氮杂环丙烷类,诸如苯佐替派(benzodopa)、卡波醌、美妥替哌(meturedopa)和乌瑞替哌(uredopa);乙亚胺类和甲基蜜胺类,包括六甲蜜胺、三亚乙基蜜胺、三亚乙基磷酰胺、三亚乙基硫代磷酰胺和三羟甲基蜜胺;番荔枝内酯类(尤其是布拉他辛(bullatacin)和布拉他辛酮(bullatacinone));喜树碱(包括拓扑替康和伊立替康);苔藓抑素;卡利他汀(callystatin);cc-1065(包括其阿多来新(adozelesin)、卡折来新(carzelesin)和比折来新(bizelesin)合成类似物);念珠藻素(cryptophycin)(特别是念珠藻素1和念珠藻素8);肾上腺皮质类固醇(包括泼尼松和泼尼松龙);醋酸环丙孕酮;5α-还原酶(包括非那雄胺和度他雄胺);伏立诺他、罗米地辛、泛比司他、丙戊酸、莫西司他(mocetinostat)、多拉他汀(dolastatin);阿地白介素、滑石、杜卡霉素(包括合成类似kw-2189和cb1-tm1);五加苷素(eleutherobin);水鬼蕉碱;匍枝珊瑚醇(sarcodictyin);海绵抑素;氮芥类,诸如苯丁酸氮芥、氯苯哌嗪、氯磷酰胺、雌莫司汀、异环磷酰胺、甲氮芥、盐酸甲氧氮芥、美法仑、新氮芥(novembichin)、苯芥胆甾醇、泼尼氮芥、曲洛磷胺(trofosfamide)、乌拉莫司汀(uracil mustard);亚硝基脲,诸如卡莫司汀、氯脲霉素、福莫司汀、洛莫斯汀、尼莫斯汀和拉尼莫斯汀;抗生素,诸如烯二炔抗生素(例如加利车霉素(calicheamicin),尤其是加利车霉素γ1i和加利车霉素ω1i(angew chem.intl.ed.engl.1994 33:183-186);达内霉素
(dynemicin),包括达内霉素a;双膦酸盐,诸如氯膦酸盐;艾司米星;以及新抑癌菌素(neocarzinostatin)发色团和相关的发色蛋白烯二炔类抗生素发色团;阿克拉霉素(aclacinomysin)、放线菌素(actinomycin)、氨茴霉素(authramycin)、偶氮丝氨酸(azaserine)、博来霉素、放线菌素(cactinomycin)、卡柔比星(carabicin)、洋红霉素(caminomycin)、嗜癌素(carzinophilin)、色霉素(chromomycinis)、更生霉素、道诺霉素、地托比星(detorubicin)、6-叠氮-5-氧代-l-正亮氨酸、(阿霉素)、吗啉代-阿霉素、氰基吗啉代-阿霉素、2-吡咯啉合-阿霉素和脱氧阿霉素、表柔比星、伊索比星、伊达比星、马塞罗霉素(marcellomycin);丝裂霉素,诸如丝裂霉素c、霉酚酸、诺加霉素、橄榄霉素、培洛霉素、甲基丝裂霉素、嘌呤霉素、三铁阿霉素(quelamycin)、罗多比星(rodorubicin)、链黑菌素、链脲佐菌素、杀结核菌素、乌苯美司、净司他汀、佐柔比星;抗代谢物,诸如甲氨蝶呤和5-氟尿嘧啶(5-fu);叶酸类似物,诸如二甲叶酸(denopterin)、甲氨蝶呤、喋罗呤(pteropterin)、三甲蝶呤;嘌呤类似物,诸如氟达拉滨、6-巯基嘌呤、硫咪嘌呤(thiamiprine)、硫鸟嘌呤;嘧啶类似物,诸如安西他滨、阿扎胞苷、6
‑‑
氮杂尿苷、卡莫氟、阿糖胞苷、双脱氧尿苷、多西氟啶、依诺他宾、氟尿苷;雄激素,诸如卡普睾酮、丙酸屈他雄酮、环硫雄醇、美雄烷、睾内酯;抗肾上腺素类药物,诸如氨鲁米特、米托坦、曲洛斯坦;叶酸补充剂,诸如亚叶酸;醋葡醛内酯;醛磷酰胺糖苷;氨基乙酰丙酸;恩尿嘧啶;安吖啶;倍曲布西(bestrabucil);比生群;依达曲沙(edatraxate);地磷酰胺(defofamine);秋水仙胺;亚胺醌;依洛尼塞(elfomithine);依利醋铵;埃博霉素;乙环氧啶;硝酸镓;羟基脲;香菇多糖;氯尼达明(lonidainine);美登木素生物碱,诸如美登素和安丝菌素;米托胍腙;米托蒽醌;莫哌达醇(mopidamnol);二胺硝吖啶(nitraerine);喷司他丁;蛋氨氮芥(phenamet);吡柔比星;洛索蒽醌(losoxantrone);鬼臼酸;2-乙基肼;甲基苄肼;多糖复合物(美国俄勒冈州尤金市的jhs天然产物公司(jhs natural products,eugene,oreg.));雷佐生;根霉素(rhizoxin);裂裥菌素(sizofuran);锗螺胺;细交链孢菌酮酸;三亚胺醌;2,2',2
″‑
三氯三乙胺;单端孢霉烯族毒素(尤其是t-2毒素、维拉库林a(verracurin a)、漆斑菌素a和蛇形菌素(anguidine));尿烷;长春地辛;达卡巴嗪;甘露醇氮芥;二溴甘露醇;二溴卫矛醇;哌泊溴烷;加西托星(gacytosine);阿拉伯糖苷(“ara-c”);环磷酰胺;噻替派;紫杉烷类,例如taxol(紫杉醇;新泽西州普林斯顿的百时美施贵宝癌症专科(bristol-myers squibb oncology,princeton,n.j.)、(不含氢化蓖麻油(cremophor))、紫杉醇的白蛋白工程化纳米颗粒制剂(伊利诺伊州邵伯克的美国制药伙伴公司(american pharmaceutical partners,schaumberg,ill.))和(多西紫杉醇、多西他赛;赛诺菲-安万特(sanofi-aventis));苯丁酸氮芥;(吉西他滨);6-硫鸟嘌呤;巯基嘌呤;甲氨蝶呤;铂类似物,诸如顺铂和卡铂;长春碱;依托泊苷(vp-16);异环磷酰胺;米托蒽醌;长春新碱;(长春瑞滨);诺万隆(novantrone);替尼泊苷;依达曲塞;道诺霉素;氨基蝶呤;卡培他滨();伊班膦酸盐;cpt-11;拓扑异构酶抑制剂rfs 2000;二氟甲基鸟氨酸(dmfo);维甲酸类,诸如视黄酸;以及以上任一项的药用盐、酸和衍生物。
[0095]
化疗剂还包括(i)起到调节或抑制激素作用于肿瘤作用的抗激素剂,诸如抗雌激素剂和选择性雌激素受体调节剂(serm),包括例如他莫昔芬(包括枸橼酸
他莫昔芬)、雷洛昔芬、屈洛昔芬、碘氧芬(iodoxyfene)、4-羟基他莫昔芬、曲沃昔芬、雷洛昔芬(keoxifene)、ly117018、奥那司酮和(枸橼酸托瑞米芬);(ii)抑制酶芳香化酶的芳香化酶抑制剂,其可调节肾上腺的雌激素产生,例如4(5)-咪唑类、氨基戊二酰亚胺、(醋酸甲地孕酮)、(依西美坦;辉瑞)、福美他尼(formestanie)、法曲唑、(伏罗唑)、(来曲唑;诺华)和(阿那曲唑;阿斯利康);(iii)抗雄激素类,诸如氟他米特、尼鲁米特、比卡鲁胺、亮丙瑞林和戈舍瑞林;布舍瑞林、曲普瑞林、醋酸甲羟孕酮、己烯雌酚、倍美力、氟甲睾酮、所有反式维甲酸、芬维a胺以及曲沙他滨(1,3-二氧戊环核苷胞嘧啶类似物);(iv)蛋白激酶抑制剂;(v)脂质激酶抑制剂;(vi)反义寡核苷酸,特别是那些抑制受异常细胞增殖牵连的信号传导途径中的基因表达的寡核苷酸,例如pkc-α、ralf和h-ras;(vii)核酶,诸如vegf表达抑制剂(例如)和her2表达抑制剂;(viii)疫苗,诸如基因疗法疫苗,例如和和ril-2;拓扑异构酶1抑制剂,诸如诸如rmrh;和(ix)以上任一项的药用盐、酸和衍生物。
[0096]
化疗剂还包括抗体,例如阿仑单抗(campath)、贝伐单抗(genentech);西妥昔单抗(imclone);帕尼单抗(amgen)、利妥昔单抗(rituximab)(genentech/biogen idec)、帕妥珠单抗(2c4,genentech)、曲妥珠单抗(trastuzumab)(genentech)、托西莫单抗(tositumomab)(bexxar,corixia)和抗体药物缀合物、吉妥珠单抗奥佐米星(wyeth)。与本发明的化合物联合的具有治疗潜力的其他人源化单克隆抗体包括:阿泊珠单抗(apolizumab)、阿塞珠单抗、阿利珠单抗、巴比妥珠单抗、莫比伐珠单抗(bivatuzumab mertansine)、莫坎妥珠单抗(cantuzumab mertansine)、西利珠单抗(cedelizumab)、塞妥珠单抗(certolizumab pegol)、西孚昔珠单抗(cidfusituzumab)、西土珠单抗(cidtuzumab)、达利珠单抗、依库珠单抗(eculizumab)、依法珠单抗(efalizumab)、依帕珠单抗(epratuzumab)、厄利珠单抗(erlizumab)、泛维珠单抗(felvizumab)、芳妥珠单抗(fontolizumab)、吉妥珠单抗奥唑米星、奥英妥珠单抗(inotuzumab ozogamicin)、伊匹单抗、拉贝珠单抗(labetuzumab)、林妥珠单抗、马妥珠单抗、美泊利单抗、莫维珠单抗、莫妥维珠单抗(motovizumab)、那他珠单抗、尼妥珠单抗、诺罗维珠单抗(nolovizumab)、奴马维珠单抗(numavizumab)、奥瑞珠单抗(ocrelizumab)、奥玛珠单抗、帕丽珠单抗、帕考珠单抗(pascolizumab)、培孚昔单抗(pecfusituzumab)、培土珠单抗(pectuzumab)、培克珠单抗(pexelizumab)、拉利维珠单抗(ralivizumab)、兰尼单抗、瑞丽维珠单抗(reslivizumab)、瑞利珠单抗(reslizumab)、瑞希维珠单抗(resyvizumab)、罗维珠单抗(rovelizumab)、卢丽珠单抗(ruplizumab)、西罗珠单抗、西利珠单抗、松妥珠单抗(sontuzumab)、替珠单抗(tacatuzumab tetraxetan)、他度珠单抗(tadocizumab)、他利珠单抗、替非珠单抗(tefibazumab)、托珠单抗、托利珠单抗(toralizumab)、西莫白介素单抗(tucotuzumab celmoleukin)、土库昔珠单抗(tucusituzumab)、乌马维珠单抗(umavizumab)、乌珠单抗、尤特克单抗(ustekinumab)、维西珠单抗和抗白介素-12(abt-874/j695,惠氏研究和雅培实验室)(抗白介素-12是一种重组的人特有序列全长igg1λ抗
体,经基因修饰以识别白介素-12p40蛋白)。
[0097]
化疗剂另选地包括“egfr抑制剂”,是指与egfr结合或直接相互作用并且阻止或降低其信号传导活性的化合物,并且替代地称为“egfr拮抗剂”。此类试剂的实例包括与egfr结合的抗体和小分子。与egfr结合的抗体的示例包括mab 579(atcc crl hb 8506)、mab 455(atcc crl hb8507)、mab 225(atcc crl 8508)、mab 528(atcc crl 8509)(参见,美国专利号4,943,533,mendelsohn等人)及其变体,例如嵌合的225(c225或西妥昔单抗;)和重塑的人225(h225)(参见,wo 96/40210,imclone systems inc.);imc-11f8,一种靶向egfr的完全人抗体(imclone);结合ii型突变egfr的抗体(美国专利号5,212,290);如美国专利号5,891,996中所述结合egfr的人源化和嵌合抗体;以及结合egfr的人抗体,卡诸如abx-egf或帕尼单抗(参见wo98/50433,安尼克斯(abgenix)/amgen);emd 55900(stragliotto等人eur.j.cancer 32a:636-640(1996));emd7200(马妥珠单抗),一种针对egfr的人源化egfr抗体,与egf和tgf-α竞争而与egfr结合(emd/默克公司(merck));人egfr抗体,humax-egfr(genmab);完全人抗体,称为e1.1、e2.4、e2.5、e6.2、e6.4、e2.11、e6.3和e7.6.3,并在us 6,235,883中进行了描述;mdx-447(梅达雷克斯公司(medarex inc));以及mab 806或人源化mab 806(johns等人,j.biol.chem.279(29):30375-30384(2004))。可以将抗egfr抗体与细胞毒性剂缀合,从而产生免疫缀合物(参见,例如,ep659439a2,默克专利公司(merck patent gmbh))。egfr拮抗剂包括小分子,例如美国专利号5,616,582、5,457,105、5,475,001、5,654,307、5,679,683、6,084,095、6,265,410、6,455,534、6,521,620、6,596,726、6,713,484、5,770,599、6,140,332、5,866,572、6,399,602、6,344,459、6,602,863、6,391,874、6,344,455、5,760,041、6,002,008和5,747,498,以及以下pct出版物:wo98/14451、wo98/50038、wo99/09016和wo99/24037中所述描述的化合物。特定的小分子egfr拮抗剂包括osi-774(cp-358774,厄洛替尼,genentech/osi pharmaceuticals);pd 183805(ci 1033,2-丙烯酰胺,n-[4-[(3-氯-4-氟苯基)氨基]-7-[3-(4-吗啉基)丙氧基]-6-喹唑啉基]-,二盐酸盐,辉瑞公司);zd1839,吉非替尼4-(3'-氯-4'-氟苯胺基)-7-甲氧基-6-(3-吗啉代丙氧基)喹唑啉,阿斯利康);zm105180(6-氨基-4-(3-甲基苯基-氨基)-喹唑啉,捷利康公司(zeneca));bibx-1382(n8-(3-氯-4-氟-苯基)-n2-(1-甲基-哌啶-4-基)-嘧啶并[5,4-d]嘧啶-2,8-二胺,勃林格殷格翰(boehringer ingelheim));pki-166((r)-4-[4-[(1-苯乙基)氨基]-1h-吡咯并[2,3-d]嘧啶-6-基]-苯酚);(r)-6-(4-羟苯基)-4-[(1-苯乙基)氨基]-7h-吡咯并[2,3-d]嘧啶);cl-387785(n-[4-[(3-溴苯基)氨基]-6-喹唑啉基]-2-丁炔酰胺);ekb-569(n-[4-[(3-氯-4-氟苯基)氨基]-3-氰基-7-乙氧基-6-喹啉基]-4-(二甲基氨基)-2-丁烯酰胺)(惠氏);ag1478(辉瑞);ag1571(su 5271;辉瑞);双重egfr/her2酪氨酸激酶抑制剂,诸如拉帕替尼(gsk572016或n-[3-氯-4-[(3氟苯基)甲氧基]苯基]-6[5[[[(2甲基磺酰基)乙基]氨基]甲基]-2-呋喃基]-4-喹唑啉胺)。
[0098]
化疗剂还包括“酪氨酸激酶抑制剂”,包括上段所述的egfr靶向药物;小分子her2酪氨酸激酶抑制剂,诸如可从武田制药公司(takeda)获得的tak165;cp-724714,一种erbb2受体酪氨酸激酶的口服选择性抑制剂(辉瑞和osi);双重her抑制剂,诸如ekb-569(可从惠氏获得),其可优先结合egfr但同时抑制过表达her2和egfr的细胞;拉帕替尼(gsk572016;可从葛兰素史克公司获得),一种口服her2和egfr酪氨酸激酶抑制剂;pki-166(可从诺华公
司获得);泛her抑制剂,诸如卡那替尼(ci-1033;法玛西亚公司(pharmacia));raf-1抑制剂,诸如可从isis制药公司获得的抑制raf-1信号传导的反义剂isis-5132;非her靶向的tk抑制剂,诸如甲磺酸伊马替尼(可从葛兰素史克公司获得);多靶向酪氨酸激酶抑制剂,诸如舒尼替尼(可从辉瑞获得);vegf受体酪氨酸激酶抑制剂,诸如瓦他拉尼(ptk787/zk222584,可从诺华/先灵公司(schering ag)获得);mapk细胞外调节的激酶i抑制剂ci-1040(可从法玛西亚公司获得);喹唑啉类,诸如pd 153035,4-(3-氯苯胺基)喹唑啉;吡啶并嘧啶类;嘧啶并嘧啶类;吡咯并嘧啶类,诸如cgp 59326、cgp 60261和cgp 62706;吡唑并嘧啶类,4-(苯氨基)-7h-吡咯并[2,3-d]嘧啶;姜黄素(二氟甲酰甲烷,4,5-双(4-氟苯胺基)邻苯二甲酰亚胺);含有硝基噻吩部分的酪氨酸酪氨酸;pd-0183805(华纳-兰伯特公司(warner-lamber));反义分子(例如与her编码核酸结合的分子);喹啉类(美国专利号5,804,396);酪氨酸磷酸化抑制剂(美国专利号5,804,396);zd6474(阿斯利康);ptk-787(诺华/先灵公司);泛her抑制剂,诸如ci-1033(辉瑞);affinitac(isis 3521;isis/礼来制药公司(lilly));甲磺酸伊马替尼pki 166(诺华公司);gw2016(葛兰素史克公司);ci-1033(辉瑞);ekb-569(惠氏);塞马替尼(辉瑞);zd6474(阿斯利康);ptk-787(诺华/先灵公司);inc-1c11(imclone),、雷帕霉素(西罗莫司,);或以下任何专利出版物中所述:美国专利号5,804,396、wo 1999/09016(american cyanamid)、wo 1998/43960(american cyanamid)、wo 1997/38983(warner lambert)、wo 1999/06378(warner lambert)、wo 1999/06396(warner lambert)、wo 1996/30347(pfizer,inc)、wo 1996/33978(zeneca)、wo 1996/3397(zeneca)和wo1996/33980(zeneca)。
[0099]
化疗剂还包括地塞米松、干扰素、秋水仙碱、美托品、环孢素、两性霉素、甲硝唑、阿仑单抗、阿利维甲酸、别嘌呤醇、氨磷汀、三氧化二砷、天冬酰胺酶、卡介苗活菌、贝伐单抗、蓓萨罗丁、克拉屈滨、氯法拉滨、阿法达贝泊汀、地尼白介素、右雷佐生、阿法依泊汀、厄洛替尼、非格司亭、醋酸组氨瑞林、替伊莫单抗、干扰素α-2a、干扰素α-2b、来那度胺、左旋咪唑、美司钠、甲氧沙林、诺龙、奈拉滨、诺非妥莫单抗(nofetumomab)、奥普瑞白介素、帕利夫明、帕米磷酸二钠、聚乙二醇化腺苷脱氨酶、培门冬酶、聚乙二醇非格司亭、培美曲塞二钠、普卡霉素、卟吩姆钠、奎纳克林、拉布立酶、沙格司亭、替莫唑胺、vm-26、6-tg、托瑞米芬、维甲酸、atra、戊柔比星、唑来膦酸盐和唑来膦酸,及其药用盐。
[0100]
化疗剂还包括氢化可的松、醋酸氢化可的松、醋酸可的松、特戊酸硫氢可的松、曲安奈德、曲安奈德醇、莫米松、安西奈德、布地奈德、地奈德、醋酸氟氢松、肤轻松、倍他米松、磷酸倍他米松钠、地塞米松、磷酸地塞米松钠、氟可龙、氢化可的松-17-丁酸酯、氢化可的松-17-戊酸酯、二丙酸阿氯米松(aclometasone dipropionate)、戊酸倍他米松、二丙酸倍他米松、泼尼卡酯、氯倍他松-17-丁酸酯、氯倍他索-17-丙酸酯、氟可龙己酸酯、氟可龙戊酸酯和醋酸氟泼尼定;免疫选择性抗炎肽(imsaid),诸如苯丙氨酸-谷氨酰胺-甘氨酸(feg)及其d-异构体形式(feg)(伊姆兰生物治疗剂有限公司(imulan biotherapeutics,llc));抗风湿药物,诸如硫唑嘌呤、环孢素(环孢霉素a)、d-青霉胺、金盐、羟氯喹、来氟米特、米诺环素、柳氮磺吡啶;肿瘤坏死因子α(tnfα)阻断剂,诸如依那西普(enbrel)、英夫利昔单抗(remicade)、阿达木单抗(humira)、赛妥珠单抗(cimzia)、戈利木单抗(simponi);白介素1(il-1)阻断剂,诸如阿那白滞素(kineret);t细胞共刺激阻断剂,诸如阿巴西普(orencia);白介素6(il-6)阻断剂,诸如托珠单抗白介素13(il-13)阻断剂,诸如来
瑞珠单抗(lebrikizumab);干扰素α(ifn)阻断剂,诸如罗那珠单抗;β7整联蛋白阻断剂,诸如rhumabβ7;ige途径阻断剂,诸如抗m1引物;分泌的同源三聚体lta3和膜结合异源三聚体lta1/β2阻断剂,诸如抗淋巴毒素α(lta);放射性同位素(例如at
211
、i
131
、i
125
、y
90
、re
186
、re
188
、sm
153
、bi
212
、p
32
、pb
212
和lu的放射性同位素);各种各样的试验药物,诸如硫铂、ps-341、苯基丁酸酯、et-18-och3或法呢基转移酶抑制剂(l-739749,l-744832);多酚类物质,诸如槲皮素、白藜芦醇、苦味酚、表没食子儿茶素没食子酸酯、茶黄素、黄烷醇、原花青素、桦木酸及其衍生物;自噬抑制剂,诸如氯喹;δ-9-四氢大麻酚(屈大麻酚,);β-拉帕酮;拉帕酚;秋水仙碱;桦木酸;乙酰喜树碱、莨菪亭(scopolectin)和9-氨基喜树碱);鬼臼毒素;替加氟贝沙罗汀双膦酸盐,诸如氯膦酸盐(例如,或)、依替膦酸盐ne-58095、唑来膦酸/唑来膦酸盐阿仑膦酸盐帕米膦酸盐替罗膦酸盐或利塞膦酸盐和表皮生长因子受体(egf-r);疫苗,诸如疫苗;哌立福辛;cox-2抑制剂(例如塞来昔布或依托昔布);蛋白体抑制剂(例如ps341);cci-779;替吡法尼(r11577);奥拉非尼,abt510;bcl-2抑制剂,诸如奥利美森钠(oblimersen sodium)匹杉琼(pixantrone);法呢基转移酶抑制剂,诸如洛那法尼(lonafarnib)(sch 6636,sarasar
tm
);以及以上任一项的药用盐、酸或衍生物;以及上述两种或多种的组合,例如chop(环磷酰胺、阿霉素、长春新碱和泼尼松龙的联合疗法的缩写);以及folfox(奥沙利铂(eloxatin
tm
)与5-fu和亚叶酸钙联合治疗方案的缩写)。
[0101]
化疗剂还包括具有镇痛、解热和抗炎作用的非甾体抗炎药。nsaid包括环氧化酶的非选择性抑制剂。nsaid的具体实例包括阿司匹林、丙酸衍生物(例如布洛芬、非诺洛芬、酮洛芬、氟比洛芬、恶丙嗪(oxaprozin)和萘普生)、乙酸衍生物(例如吲哚美辛、舒林酸、依托度酸、双氯芬酸)、烯醇酸衍生物(例如吡罗昔康、美洛昔康、替诺昔康、屈昔康(droxicam)、氯诺昔康和伊索昔康)、芬那酸(fenamic acid)衍生物(例如甲芬那酸、甲氯芬那酸、氟芬那酸、托芬那酸)和cox-2抑制剂(例如塞来昔布、依托考昔(etoricoxib)、鲁美昔布(lumiracoxib)、帕瑞昔布、罗非考昔(rofecoxib)、罗非考昔和戊地昔布(valdecoxib))。nsaid可适用于减轻病症的症状,诸如类风湿性关节炎、骨关节炎、炎性关节炎、强直性脊柱炎、银屑病关节炎、莱特综合征、急性痛风、痛经、转移性骨痛、头痛和偏头痛、术后疼痛、由于炎症和组织损伤、发热、肠梗阻和肾绞痛引起的轻度至中度疼痛。
[0102]
当在本文中使用时,“生长抑制剂”是指在体外或体内抑制细胞生长的化合物或组合物。在一个实施例中,生长抑制剂是生长抑制抗体,其防止或减少表达该抗体结合的抗原的细胞的增殖。在另一个实施例中,生长抑制剂可以是显著降低s期细胞百分比的抑制剂。生长抑制剂的实例包括阻断细胞周期进程(在s期以外的地方)的试剂,例如诱导g1停滞和m期停滞的试剂。经典的m期阻滞剂包括长春花(长春新碱和长春碱)、紫杉烷类和拓扑异构酶ii抑制剂(例如阿霉素、表柔比星、柔红霉素、依托泊苷和博来霉素)。那些阻滞g1的试剂也溢出到s期阻滞中,例如dna烷基化剂,诸如他莫昔芬、泼尼松、达卡巴嗪、氮芥、顺铂、甲氨蝶呤、5-氟尿嘧啶和ara-c。进一步的信息可见于mendelsohn和israel编辑的《癌症的分子基础》(the molecular basis of cancer)中murakami等人所著的第1章,标题为“细胞周期调
节,癌基因和抗肿瘤药”(w.b.saunders,philadelphia,1995),例如,第13页。紫杉烷类(紫杉醇和多西他赛)都是抗癌药,均来源于紫杉。多西他赛(rhone-poulenc rorer)源自欧洲紫杉,是紫杉醇的半合成类似物(bristol-myers squibb)。紫杉醇和多西他赛促进微管蛋白二聚体的微管装配,并通过防止解聚作用稳定微管,从而抑制细胞的有丝分裂。
[0103]“放疗”是指使用定向γ射线或β射线以诱导对细胞的足够损害,从而限制细胞正常发挥功能或完全破坏细胞的能力。将理解的是,在本领域中将有许多已知方法可以确定治疗的剂量和持续时间。典型治疗为一次施用,典型剂量范围为每天10到200单位(gray)。
[0104]
用于治疗目的的“受试者”或“个体”是指被分类为哺乳动物的任何动物,包括人、家畜和农场动物以及动物园动物、运动动物或宠物,诸如狗、马、猫、牛等。优选地,哺乳动物是人。
[0105]
本文的术语“抗体”以最广泛的含义使用,并且具体地覆盖单克隆抗体(包括全长单克隆抗体)、多克隆抗体、多特异性抗体(例如,双特异性抗体)和抗体片段,只要它们表现出所需的抗原结合活性即可。
[0106]“经分离的”抗体是已经鉴定并且自其自然环境的组分中分离和/或回收的抗体。其自然环境的污染物组分是会干扰抗体研究、诊断或治疗用途的材料,并且可以包括酶、激素和其它蛋白质或非蛋白质溶质。在一些实施例中,将抗体纯化至(1)大于抗体重量的95%(例如通过lowry方法测定),在一些实施例中,大于99%重量;(2)足以获得n末端或内部氨基酸序列的至少15个残基的程度(例如通过使用旋转杯测序仪),或(3)均质(在还原或非还原条件下进行sds-page,使用例如考马斯蓝或银染)。经分离的抗体包括重组细胞内的原位抗体,因为不会存在抗体天然环境的至少一种成分。然而,通常,分离的抗体将通过至少一个纯化步骤来制备。
[0107]“天然抗体”通常是约150,000道尔顿的异源四聚体糖蛋白,由两条相同的轻(l)链和两条相同的重(h)链组成。每条轻链通过一个共价二硫键与重链相连,而二硫键的数目在不同免疫球蛋白同种型的重链之间变化。每条重链和轻链还具有规则间隔的链内二硫键。每条重链在一末端具有可变结构域(vh),其后是多个恒定结构域。每条轻链在一末端(vl)具有可变结构域,在另一末端具有恒定结构域;轻链的恒定结构域与重链的第一恒定结构域对齐,并且轻链可变结构域与重链的可变结构域对齐。据信特定的氨基酸残基在轻链和重链可变结构域之间形成界面。
[0108]
术语“恒定结构域”是指免疫球蛋白分子的一部分,该部分相对于免疫球蛋白的另一部分(即可变结构域,其包含抗原结合位点)具有更保守的氨基酸序列。恒定结构域包含重链的ch1、ch2和ch3结构域(统称为ch)和轻链的chl(或cl)结构域。
[0109]
抗体的“可变区”或“可变结构域”是指抗体的重链或轻链的氨基末端结构域。重链的可变结构域可称为“vh”。轻链的可变结构域可称为“vl”。这些结构域通常是抗体中变化最大的部分,并且包含抗原结合位点。
[0110]
术语“可变的”是指以下事实:可变结构域的某些部分在抗体之间的序列差异很大,并用于每种特定抗体对其特定抗原的结合和特异性。然而,可变性并非在抗体的可变结构域中均匀分布。它集中在轻链和重链可变结构域中的三个称为高变区(hvr)的区段中。可变结构域中保守性更高的部分称为构架区(fr)。天然重链和轻链的可变结构域各自包含四
个fr区,其主要采用β折叠结构,由三个hvr连接,这三个hvr形成连接β折叠结构的环并且在一些情况下形成β折叠结构的一部分。每条链中的hvr通过fr区紧密保持在一起,并且与另一条链中的hvr一起,有助于抗体的抗原结合位点的形成(参见kabat等人,《具有免疫学意义的蛋白质序列》(sequences of proteins of immunological interest),第五版,美国卫生与公众服务部,国立卫生研究院,马里兰州贝塞斯达(1991))。恒定结构域不直接参与抗体与抗原的结合,但具有各自效应物功能,诸如抗体参与抗体依赖性细胞毒性作用。
[0111]
来自任何哺乳动物物种抗体(免疫球蛋白)的“轻链”基于其恒定结构域的氨基酸序列,可以配属为两种明显不同的类型中的一种,这两种类型分别称为卡帕(“κ”)和兰姆达(“λ”)。
[0112]
如本文所用,术语igg“同种型”或“亚类”是指由免疫球蛋白恒定区的化学和抗原特征定义的免疫球蛋白的任何亚类。
[0113]
根据其重链恒定结构域的氨基酸序列,可以将抗体(免疫球蛋白)分为不同的类别。免疫球蛋白主要分为五类:iga、igd、ige、igg和igm,并且它们中的一些可以进一步分为亚类(同型),例如,igg1、igg2、igg3、igg4、iga1和iga2。对应于不同类别的免疫球蛋白的重链恒定结构域分别称为α、γ、ε、γ和μ。不同种类的免疫球蛋白的亚基结构和三维构型是众所周知的,并在例如以下文献中有一般描述:abbas等人,《细胞和分子免疫学》(cellular and mol.immunology),第4版(w.b.saunders,co.,2000)。抗体可以是较大融合分子的一部分,该融合分子是通过抗体与一个或多个其他蛋白质或肽的共价或非共价缔合形成的。
[0114]
术语“全长抗体”、“完整抗体”和“全抗体”在本文中可互换使用,是指其基本上完整形式的抗体而不是如下文定义的抗体片段。该术语特别是指具有包含fc区的重链的抗体。
[0115]
出于本文目的的“裸抗体”是未与药物部分或放射性标记缀合的抗体。
[0116]“抗体片段”包含完整抗体的一部分,优选包含其抗原结合区。在一些实施例中,本文所述的抗体片段是抗原结合片段。抗体片段的示例包括fab、fab'、f(ab')2和fv片段;双体抗体;线性抗体;单链抗体分子;和由抗体片段形成的多特异性抗体。
[0117]
木瓜蛋白酶消化抗体产生两个相同抗原结合片段,称为“fab”片段,每个片段都有单个抗原结合位点和残留的“fc”片段,其名称反映其容易结晶的能力。胃蛋白酶处理产生的f(ab')2片段具有两个抗原结合位点并且仍能与抗原交联。
[0118]“fv”是包含完全的抗原结合位点的最小抗体片段。在一个实施例中,双链fv物类由紧密和非共价缔合的一个重链和一个轻链可变结构域的二聚体组成。在单链fv(scfv)物类中,一个重链可变结构域和一个轻链可变结构域可通过柔性肽连接基共价连接,使得轻链和重链可缔合成类似于在双链fv物类中的“二聚体”结构。以此构型,每个可变结构域的三个hvr相互作用以在vh-vl二聚体的表面上限定抗原结合位点。六个hvr共同对抗体赋予抗原结合特异性。但是,即使单个可变结构域(或仅包含三个对抗原具有特异性的hvr的fv的一半)也具有识别和结合抗原的能力,尽管其亲和力低于完整结合位点。
[0119]
fab片段含有重链可变结构域和轻链可变结构域且亦含有轻链的恒定结构域和重链的第一恒定结构域(ch1)。fab'片段与fab片段的不同之处在于fab'片段在重链ch1结构域的羧基末端添加了一些残基,这些残基包括来自抗体铰链区的一个或多个半胱氨酸。fab'-sh是本文中关于其中恒定结构域的半胱氨酸残基带有游离硫醇基的fab'的命名。f
1991/10741;jakobovits等人,proc.natl.acad.sci.usa 90:2551(1993);jakobovits等人,nature 362:255-258(1993);bruggemann等人,year in immunol.7:33(1993);美国专利号5,545,807、5,545,806、5,569,825、5,625,126、5,633,425和5,661,016;marks等人,bio/technology 10:779-783(1992);lonberg等人,nature 368:856-859(1994);morrison,nature 368:812-813(1994);fishwild等人,nature biotechnol.14:845-851(1996);neuberger,nature biotechnol.14:826(1996);以及lonberg和huszar,intern.rev.immunol.13:65-93(1995))。
[0124]
本文中的单克隆抗体具体地包括“嵌合”抗体,其中重链和/或轻链的一部分与来自特定物种或属于特定抗体类别或亚类的抗体中的相应序列相同或同源,而一条或多条链的其余部分与来自另一物种或属于另一抗体类别或亚类的抗体中的相应序列以及这些抗体的片段相同或同源,只要它们表现出所需的生物学活性即可(参见例如美国专利号4,816,567和morrison等人,proc.natl.acad.sci.usa 81:6851-6855(1984))。嵌合抗体包括抗体,其中抗体的抗原结合区源自通过例如用目标抗原免疫猕猴产生的抗体。
[0125]“人源化”形式的非人(例如,鼠)抗体为包含来源于非人免疫球蛋白的最小序列的嵌合抗体。在一个实施例中,人源化抗体是人类免疫球蛋白(受体抗体),其中来自受体hvr的残基被来自非人类物种(供体抗体)例如小鼠、大鼠、兔或具有所需特异性、亲和力和/或能力的非人类灵长类动物的hvr的残基取代。在一些情况下,人类免疫球蛋白的fr残基被相应的非人类残基取代。此外,人源化抗体可包含受体抗体或供体抗体中不存在的残基。可以进行这些修饰以进一步改善抗体性能。总体上,人源化抗体将基本上包含所有中的至少一个可变结构域,通常是两个可变结构域,其中所有或基本上所有高变环对应于非人类免疫球蛋白的高变环,并且所有或基本上所有的fr为人类免疫球蛋白序列的fr。人源化抗体还将任选地包含免疫球蛋白恒定区(fc)的至少一部分,该免疫球蛋白通常为人类免疫球蛋白。更多详情参见例如jones等人,nature 321:522-525(1986);riechmann等人,nature 332:323-329(1988);和presta,curr.op.struct.biol.2:593-596(1992)。另见例如vaswani和hamilton,ann.allergy,asthma&immunol.1:105-115(1998);harris,biochem.soc.transactions 23:1035-1038(1995);hurle and gross,curr.op.biotech.5:428-433(1994);和美国专利号6,982,321和7,087,409。
[0126]“人类抗体”是具有对应于由人类产生的抗体的氨基酸序列的抗体和/或使用本文所公开的用于制备人类抗体的任何技术制得的抗体。人抗体的该定义特别地排除了包含非人抗原结合残基的人源化抗体。可以使用本领域已知的各种技术产生人类抗体,包括噬菌体展示文库。hoogenboom和winter,《分子生物学杂志》(j.mol.biol.),227:381(1991);marks等人,《分子生物学杂志》(j.mol.biol.),222:581(1991)。还可用于制备人单克隆抗体的方法如以下文献所述:cole等人,《单克隆抗体与癌症治疗》(monoclonal antibodies and cancer therapy),alan r.liss,第77页(1985);boerner等人,《免疫学杂志》(j.immunol.),147(1):86-95(1991)。另参见van dijk和van de winkel,《药理学新见》(curr.opin.pharmacol.),5:368-74(2001)。可以通过向转基因动物施用抗原来制备人抗体,该转基因动物已经修饰以对抗原攻击产生应答而产生此类抗体,但其内源基因座已失效,例如,免疫异种小鼠(参见例如,有关xenomousetm技术的美国专利no.6,075,181和6,
150,584)。另参见例如li等人,《美国国家科学院院刊》(proc.natl.acad.sci.usa),103:3557-3562(2006)关于通过人b细胞杂交瘤技术产生的人抗体。
[0127]“物种依赖性抗体”是对来自第一哺乳动物物种的抗原具有比对来自第二哺乳动物物种的该抗原同系物更强的结合亲和力的抗体。通常,物种依赖性抗体与人抗原“特异性结合”(例如,其结合亲和力(kd)值不超过约1
×
10-7
m,优选不超过约1
×
10-8
m,优选不超过约1
×
10-9
m),但对来自第二非人哺乳动物物种的该抗原同系物的结合亲和力比其对该人抗原的结合亲和力弱至少约50倍或至少约500倍或至少约1000倍。物种依赖性抗体可以是如上定义的各种抗体中的任何一种,但是优选地是人源化或人抗体。
[0128]
如本文所用的术语“高变区”、“hvr”或“hv”是指在序列上高变和/或形成结构上限定的环的抗体可变结构域的区域。通常,抗体包含六个hvr;三个在vh中(h1、h2、h3),并且三个在vl中(l1、l2、l3)。在天然抗体中,h3和l3在六个hvr中表现出最多的多样性,尤其是h3被认为在赋予抗体精细特异性方面起着独特的作用。参见例如:xu等人,immunity 13:37-45(2000);johnson和wu,methods in molecular biology 248:1-25(lo,ed.,human press,totowa,n.j.,2003)。实际上,仅由重链组成的天然存在的骆驼科动物抗体在不存在轻链的情况下是有功能并稳定的。参见例如:hamers-casterman等人,nature 363:446-448(1993);sheriff等人,nature struct.biol.3:733-736(1996)。
[0129]
许多hvr描述得到应用,并且包含于本文中。kabat互补决定区(cdr)基于序列变异性并且是最常用的(kabat等人,《具有免疫学意义的蛋白质序列》(sequences of proteins of immunological interest),第5版,美国卫生与公众服务部,国立卫生研究院,马里兰州贝塞斯达(1991))。相反,chothia指的是结构环的位置(chothia和lesk j.mol.biol.196:901-917(1987))。abm hvr表示kabat hvr和chothia结构环之间的折衷,并且被牛津分子公司(oxford molecular)的abm抗体建模软件采用。“接触”hvr基于可用的复杂晶体结构的分析结果。这些hvr中的每个的残基如下文所述。
[0130][0131]
hvr可以包括以下“扩展hvr”:vl中的24-36或24-34(l1)、46-56或50-56(l2)和89-97或89-96(l3),以及vh中的26-35(h1)、50-65或49-65(h2)和93-102、94-102或95-102(h3)。对于这些定义中的每一个,可变结构域残基均根据上述kabat等人的方法进行编号。
[0132]
hvr可以包括以下“扩展hvr”:vl中的24-36或24-34(l1)、46-56或50-56(l2)和89-97或89-96(l3),以及vh中的26-35(h1)、50-65或49-65(h2)和93-102、94-102或95-102(h3)。对于这些定义中的每一个,可变结构域残基均根据上述kabat等人的方法进行编号。
[0133]“框架”或“fr”残基是除本文定义的hvr残基以外的那些可变结构域残基。
[0134]
术语“kabat所述的可变结构域残基编号”或“kabat所述的氨基酸位置编号”及其变型是指在上述kabat等人的文献中提出的用于重链可变结构域或轻链可变结构域的编号系统。使用该编号系统,实际线性氨基酸序列可能包含较少或附加的氨基酸,其对应于对可变结构域的fr或hvr的缩短或插入。例如,重链可变结构域可在h2的残基52之后包括单个氨基酸插入片段(根据kabat编号的残基52a)以及重链fr残基82之后的插入残基(例如,根据kabat编号的残基82a、82b和82c等)。可通过将抗体序列与“标准”kabat编号序列的同源性区域进行比对来确定给定抗体的残基的kabat编号。
[0135]
当提及可变结构域中的残基(大约是轻链的残基1-107和重链的残基1-113)时,通常使用kabat编号系统(例如,kabat等人,《具有免疫学意义的蛋白质序列》(sequences of proteins of immunological interest)。第5版,美国卫生与公众服务部,国立卫生研究院,马里兰州贝塞斯达(1991))。当提及免疫球蛋白重链恒定区中的残基时,通常使用“eu编号系统”或“eu索引”(例如,上述kabat等人所报道的eu索引)。“kabat所述的eu索引”是指人类igg1 eu抗体的残基编号。
[0136]
如本文所用,术语“结合”、“特异性结合”或“具有特异性”是指可测量和可再现的相互作用,诸如靶与抗体之间的结合,在分子(包括生物分子)的异质群体的存在下,其确定靶的存在。例如,与靶标(其可以是表位)结合或特异性结合的抗体是与其结合其他靶标相比具有更大亲和力、亲合力、更容易和/或持续时间更长的结合该靶标的抗体。在一个实施例中,抗体与无关靶标的结合程度为该抗体与抗原结合的小于约10%,例如,通过放射免疫分析(ria)所测量。在某些实施例中,与靶标特异性地结合的抗体的解离常数(kd)为≤1μm、≤100nm、≤10nm、≤1nm或≤0.1nm。在某些实施例中,抗体与蛋白上的表位特异性地结合,该表位在不同物类的蛋白之间具有保守性。在另一个实施例中,特异性结合可以包括但不要求排他结合。
[0137]
患者对药物和治疗的“有效响应”或患者的“响应性”和类似措词是指赋予处于患疾病或病症诸如癌症的风险下或患有该疾病或病症的患者的临床或治疗有益效果。在一个实施例中,这种益处包括以下一个或多个:延长存活(包括总存活和无进展存活);导致客观应答(包括完全应答或部分应答);或改善癌症的体征或症状。
[0138]
对治疗“没有有效响应”的患者是指没有以下任一项的患者:延长存活(包括总存活和无进展存活);导致客观响应(包括完全响应或部分响应);或改善癌症的征兆或症状。
[0139]“功能性fc区”具有天然序列fc区的“效应子功能”。示例性的“效应子功能”包括c1q结合;cdc;fc受体结合;adcc;吞噬作用;细胞表面受体(例如,b细胞受体;bcr)的下调等。此类效应子功能通常需要fc区与结合域(例如,抗体可变结构域)组合,并且可使用例如本文定义中所公开的各种测定方法进行评估。
[0140]
如本文所用,术语“样品”是指获自或衍生自目的受试者和/或个体的组合物,其包含例如待基于物理、生化、化学和/或生理特征进行表征和/或鉴定的细胞和/或其他分子实体。例如,短语“疾病样品”及其变型是指从目标受试者获得的任何样品,其预期或已知含有待表征的细胞和/或分子实体。样品包括但不限于原代或培养的细胞或细胞系、细胞上清液、细胞裂解液、血小板、血清、血浆、玻璃体液、淋巴液、滑液、卵泡液、精液、羊水、乳汁、全血、血液来源的细胞、尿液、脑脊液、唾液、痰、眼泪、汗液、粘液、肿瘤溶解产物和组织培养基、组织提取物诸如均质化的组织、肿瘤组织、细胞提取物以及它们的组合。在一些实施例
中,样品是从个体的癌症获得的样品(例如,肿瘤样品),其包含肿瘤细胞并且任选地包含肿瘤浸润免疫细胞。例如,样品可为包埋在石蜡块中的肿瘤标本,或者包括新鲜切割的、连续未染色的切片。在一些实施例中,样品来自活检,并且包括50个或更多活肿瘤细胞(例如,来自芯针活检并且任选地包埋于石蜡块中;切除、切口、穿孔或活检钳活检;或肿瘤组织切除)。
[0141]“组织样品”或“细胞样品”是指从受试者或个体的组织获得的相似细胞的集合。组织或细胞样品的来源可以是来自新鲜的、冷冻的和/或保存的器官、组织样品、活组织检查和/或吸出物的实体组织;血液或任何血液成分,例如血浆;体液,例如脑脊髓液、羊水、腹膜液或间质液;受试者妊娠或发育中任何时候的细胞。组织样品也可以是原代或培养的细胞或细胞系。可选地,组织或细胞样品获自疾病组织/器官。组织样品可以包含在自然环境天然不与组织混合的化合物,例如防腐剂、抗凝剂、缓冲剂、固定剂、营养物、抗生素等。
[0142]“具有人效应细胞”的癌症或生物样本是,在诊断测试中,样本中存在人效应细胞(例如,浸润的人效应细胞)的癌症或生物样本。
[0143]“具有表达fcr的细胞”的癌症或生物学样本是,在诊断测试中,样本中存在表达fcr的细胞(例如,浸润的表达fcr的细胞)的癌症或生物样本。在一些实施例中,fcr是fcγr。在一些实施例中,fcr是活化fcγr。
[0144]
ii.治疗方法
[0145]
本文提供用于治疗个体中的癌症或延缓其进展的方法,包括以两个或更多个4周或28天的周期向个体施用本公开的抗pd-l1抗体。在一些实施例中,抗pd-l1抗体以每个周期1680mg的剂量施用(例如,抗pd-l1抗体以每4周或每28天1680mg的剂量施用)。在一些实施例中,抗pd-l1抗体为阿特珠单抗。
[0146]
本文提供用于治疗个体中的癌症或延缓其进展的方法,包括以两个或更多个2周或14天的周期向个体施用本公开的抗pd-l1抗体。在一些实施例中,抗pd-l1抗体以每个周期840mg的剂量施用(例如,抗pd-l1抗体以每2周或每14天840mg的剂量施用)。在一些实施例中,抗pd-l1抗体为阿特珠单抗。
[0147]
在一些实施例中,抗pd-l1抗体在两个或更多个周期中每个周期的约第1天施用。在一些实施例中,抗pd-l1抗体在两个或更多个周期中每个周期的第1天施用。
[0148]
在一些实施例中,抗pd-l1抗体在两个或更多个周期的每一个中以1680mg或840mg的剂量施用。
[0149]
在一些实施例中,本公开的治疗包括诱导阶段和维持阶段(或“维持疗法”)。如本领域已知的,维持阶段或维持疗法可以指在诱导阶段或初始疗法之后提供的一种或多种治疗,例如以防止癌症复发。在一些实施例中,维持阶段或维持疗法可在比诱导阶段或初始疗法更长的时间段内给予。在一些实施例中,维持阶段或维持疗法的特征可以在于比诱导阶段或初始疗法更少的副作用或毒性(例如,与短期和/或长期使用相关),从而允许使用更长的持续时间。在一些实施例中,本公开的抗pd-l1抗体可以作为诱导阶段或初始疗法、维持阶段或维持疗法或两者的一部分向个体施用。在一些实施例中,向个体施用维持阶段或维持疗法,直至出现疾病进展或不可接受的毒性。
[0150]
在一些实施例中,用于治疗患有癌症的人类患者的方法包括向人类患者施用诱导阶段,随后向人类患者施用维持阶段。在一些实施例中,用于治疗患有癌症的人类患者的方
法包括向人类患者施用诱导阶段,然后施用一种或多种附加治疗剂,诸如贝伐单抗、紫杉醇和卡铂中的一种或多种。
[0151]
在一些实施例中,本公开的抗pd-l1抗体在治疗的维持阶段向个体施用。例如,在一些实施例中,本公开的方法包括,在治疗的诱导阶段向个体施用4-6个周期(例如,4、5或6个周期)的本公开的一种或多种化学疗法(例如,紫杉醇和卡铂,或者卡铂和依托泊苷),然后在治疗的维持阶段向个体施用抗pd-l1抗体,例如,如本文所述。在一些实施例中,在治疗的维持阶段之前,在治疗的诱导阶段向个体施用本公开的抗pd-l1抗体。
[0152]
在一些实施例中,在治疗的诱导阶段期间,在一个或多个2周或14天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,以840mg的剂量在一个或多个2周或14天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在一个或多个4周或28天周期的第1天和第15天,以840mg的剂量向个体施用本公开的抗pd-l1抗体。
[0153]
在一些实施例中,在治疗的诱导阶段期间,在一个或多个3周或21天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,在一个或多个3周或21天周期中的大约第1天向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,在一个或多个3周或21天周期中的第1天向个体施用本公开的抗pd-l1抗体。
[0154]
在一些实施例中,在治疗的诱导阶段期间,以1200mg的剂量在一个或多个3周或21天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,以1200mg的剂量在一个或多个3周或21天周期中的第1天向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段,以1200mg的剂量在一个或多个3周或21天周期的每个周期内向个体施用本公开的抗pd-l1抗体。
[0155]
在根据本文所述的任何实施例的一些实施例中,该方法进一步包括在用一种或多种化疗或其他抗肿瘤药物(例如,卡铂和依托泊苷,或者卡铂、紫杉醇和贝伐单抗)治疗之前,在一个或多个3周或21天周期中,以1200mg的剂量向个体施用本公开的抗pd-l1抗体(例如,阿特珠单抗)。
[0156]
在一些实施例中,在治疗的诱导阶段期间,在一个或多个4周或28天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,在一个或多个4周或28天周期中的大约第1天向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,在一个或多个4周或28天周期中的第1天向个体施用本公开的抗pd-l1抗体。
[0157]
在一些实施例中,在治疗的诱导阶段期间,以1680mg的剂量在一个或多个4周或28天周期中向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段期间,以1680mg的剂量在一个或多个4周或28天周期中的第1天向个体施用本公开的抗pd-l1抗体。在一些实施例中,在治疗的诱导阶段,以1680mg的剂量在一个或多个4周或28天周期的每个周期内向个体施用本公开的抗pd-l1抗体。
[0158]
在一些实施例中,在一个或多个4周或28天周期中,以1680mg的剂量向个体静脉内施用抗pd-l1抗体(例如,阿特珠单抗),历时30(
±
15分钟)。在一些实施例中,在一个或多个4周或28天周期中的第1天,以1680mg的剂量向个体静脉内施用抗pd-l1抗体(例如,阿特珠单抗),历时30(
±
15分钟)。在一些实施例中,在一个或多个4周或28天周期中,以1680mg的剂量向个体静脉内施用抗pd-l1抗体(例如,阿特珠单抗),历时60(
±
15分钟)。在一些实施例中,在一个或多个4周或28天周期中的第1天,以1680mg的剂量向个体静脉内施用抗pd-l1
抗体(例如,阿特珠单抗),历时60(
±
15分钟)。在一些实施例中,在治疗的诱导阶段期间,在一个或多个4周或28天周期中的第1天,以1680mg的剂量向个体静脉内施用抗pd-l1抗体(例如,阿特珠单抗),历时60(
±
15分钟)。在一些实施例中,在治疗的维持阶段期间,在一个或多个4周或28天周期中的第1天,以1680mg的剂量向个体静脉内施用抗pd-l1抗体(例如,阿特珠单抗),历时60(
±
15分钟)。
[0159]
在一些实施例中,方法可以进一步包括附加疗法。在一些实施例中,该方法可以进一步包括向个体施用附加治疗剂。附加疗法可以是放射疗法、手术(例如,乳房肿瘤切除术和乳房切除术)、化学疗法、基因疗法、dna疗法、病毒疗法、rna疗法、免疫疗法、骨髓移植、纳米疗法、单克隆抗体疗法、或上述疗法的组合。附加疗法可以是辅助疗法或新辅助疗法的形式。在一些实施例中,附加药剂包含化疗剂。在一些实施例中,化疗剂为针对待治疗的癌症的标准护理。在一些实施例中,附加疗法是施用小分子酶抑制剂或抗转移剂。在一些实施例中,附加疗法为施用副作用限制剂(例如,旨在减少治疗副作用的发生和/或严重性的药剂,如抗恶心剂等)。在一些实施例中,附加疗法为放射疗法。在一些实施例中,附加疗法为手术。在一些实施例中,附加疗法为放射疗法和手术的组合。在一些实施例中,附加疗法为γ射线辐照。
[0160]
在一些实施例中,附加疗法包含紫杉烷。在一些实施例中,在治疗的诱导阶段期间施用附加疗法。紫杉烷类(例如,紫杉醇和多西他赛)是普遍使用的抗癌药,最初来源于紫杉树。紫杉烷类促进微管蛋白二聚体的微管装配,并通过防止解聚作用稳定微管,从而抑制有丝分裂和细胞死亡。多西他赛是紫杉醇的半合成类似物。
[0161]
紫杉醇是在本文所述方法中使用的示例性紫杉烷。原料药的化学名称为5β,20-环氧-1,2α,4,7β,10β,13α-六羟基紫衫-11-烯-9-酮4,10-二醋酸酯2-苯甲酸酯13-(2r,3s)-n-苄基-3-苯基异丝氨酸酯,分子式为c
47h51
no
14
,并且分子量为853.9。本文提及紫杉烷类诸如紫杉醇还包括其缀合物,诸如白蛋白结合型紫杉醇,一种以销售的白蛋白结合形式的紫杉醇。
[0162]
紫杉醇具有以下化学结构:
[0163][0164]
紫杉醇可以作为为等商购获得。多西他赛可作为等商购获得。
[0165]
在一些实施例中,附加疗法包含拓扑异构酶ii抑制剂。在一些实施例中,在治疗的
诱导阶段期间施用附加疗法。拓扑异构酶ii的抑制剂(例如依托泊苷(vp-16)、替尼泊苷、阿霉素、道诺霉素、米托蒽醌、安吖啶、玫瑰树碱、金精三羧酸和hu-331)也是广泛使用的抗肿瘤药物,其在形成酶介导的dna断裂之后稳定化拓扑异构酶ii:dna共价复合物(即,“裂解复合物”)。这种裂解复合物的聚集诱导细胞死亡途径。
[0166]
依托泊苷是用于本文所述方法的示例性拓扑异构酶ii抑制剂。依托泊苷通常作为前药依托泊苷磷酸酯施用,其化学名称为:4'-去甲基表鬼臼毒素9-[4,6-o-(r)-亚乙基-β-吡喃葡萄糖苷],4'(磷酸二氢酯)。
[0167]
依托泊苷磷酸酯具有以下结构:
[0168][0169]
依托泊苷磷酸酯,即依托泊苷的磷酸酯,是鬼臼毒素的半合成衍生物,通过去磷酸化转化为依托泊苷。依托泊苷通过与dna拓扑异构酶ii相互作用或形成自由基来诱导dna链断裂,从而导致细胞周期阻滞(主要在细胞周期的g2阶段)和细胞死亡。依托泊苷可作为toposar
tm
、vp-16、actitop、aside、bioposide、ctop、cytop、eposed、eside、ethopul、etolon、etonis、etoplast、etosid、etovel、fytop、fytosid、lastet、nzytop、oncoside、placid、posid、retopson、tevaside、topok、toposide等获得。
[0170]
在一些实施例中,附加疗法包含抗代谢物。在一些实施例中,在治疗的诱导阶段期间施用附加疗法。抗代谢物(例如培美曲塞、5-氟尿嘧啶、6-巯基嘌呤、卡培他滨、阿糖胞苷、氟尿苷、氟达拉滨、羟基脲、甲氨蝶呤等)是广泛使用的抗肿瘤药物,会干扰一种或多种dna合成所需的酶。抗代谢物通常通过多种机制起作用,包括例如掺入核酸,从而触发细胞凋亡,或例如竞争参与核苷酸合成的酶的结合位点,从而耗尽dna和/或rna复制和细胞增殖所需的供给。
[0171]
培美曲塞是在本文所述方法中使用的示例性抗代谢物。培美曲塞是叶酸类似物。药物培美曲塞七水合二钠具有化学名称l-谷氨酸,n-[4-[2-(2-氨基-4,7-二氢-4-氧代-1h-吡咯并[2,3-d]嘧啶-5基)乙基]苯甲酰基]-二钠盐七水合物,分子式为c
20h19
n5na2o6·
7h2o,分子量为597.49。
[0172]
培美曲塞七水合二钠具有以下结构:
[0173][0174]
培美曲塞抑制胸腺嘧啶和嘌呤合成中使用的多种叶酸依赖性酶,即胸苷酸合酶(ts),二氢叶酸还原酶(dhfr)和甘氨酰胺核糖核苷酸甲酰基转移酶(garft)(参见shih等人,(1997)cancer res.57:1116-23)。培美曲塞通过抑制嘌呤和嘧啶前体核苷酸的形成,阻止了正常细胞和癌细胞两者的生长和存活所需的dna和rna的形成。培美曲塞可以以商品名giopem、pexate、pemanat、pemex、pemmet、pexate、relitrexed、temeran、ciambra等来商购。
[0175]
在一些实施例中,附加疗法包含vegf拮抗剂,例如抗vegf抗体。在一些实施例中,在治疗的诱导阶段和/或治疗的维持阶段期间施用附加疗法。在一些实施例中,抗vegf抗体可以为人抗体或人源化抗体。在一些实施例中,抗vegf抗体可以为单克隆抗体。vegf拮抗剂的其他实例包括但不限于与vegf特异性结合的可溶性vegf受体或可溶性vegf受体片段、vegf受体分子或其vegf结合片段(例如,vegf受体的可溶性形式)和嵌合vegf受体蛋白。
[0176]
待用于产生vegf抗体的vegf抗原可以为例如vegf
165
分子以及vegf的其他同工型或其含有所需表位的片段。在一个实施例中,所需表位为贝伐单抗识别的表位,其结合至与由杂交瘤atcc hb 10709产生的单克隆抗vegf抗体a4.6.1相同的表位(本文中称为“表位a.4.6.1”)。可用于产生本发明的抗vegf抗体的其他形式的vegf对本领域技术人员来说是显而易见的。
[0177]
可用于本发明的方法中的抗vegf抗体包括以足够的亲和力和特异性结合vegf并且可以降低或抑制vegf的生物活性的任何抗体或其抗原结合片段。抗vegf抗体通常不会与其他vegf同源物(诸如vegf-b或vegf-c)结合,也不会与其他生长因子(诸如plgf、pdgf或bfgf)结合。
[0178]
在某些实施例中,抗vegf抗体包括但不限于与由杂交瘤atcc hb10709产生的单克隆抗vegf抗体a4.6.1结合相同表位的单克隆抗体;根据presta等人(1997)cancer res.57:4593-4599产生的重组人源化抗vegf单克隆抗体。在一个实施例中,抗vegf抗体为“贝伐单抗(bv)”,也称为“rhumab vegf”或它包含突变的人igg1框架区和来自鼠抗hvegf单克隆抗体a.4.6.1的阻断人vegf与其受体结合的抗原结合互补决定区。贝伐珠单抗的约93%的氨基酸序列,包括大部分框架区,来源于人igg1,并且约7%的序列来源于鼠抗体a4.6.1。
[0179]
在一些实施例中,抗vegf抗体为贝伐单抗。贝伐单抗是fda批准的第一种抗血管生成疗法,并且被批准用于治疗转移性结直肠癌(一线和二线治疗,联合基于5-fu的静脉化疗)、晚期非鳞状非小细胞肺癌(nsclc)(不可切除的、局部晚期、复发性或转移性nsclc的一线治疗,联合卡铂和紫杉醇)和转移性her2阴性乳腺癌(先前未治疗的转移性her2阴性乳腺癌,联合紫杉醇)。
[0180]
贝伐单抗和其他人源化抗vegf抗体进一步描述于2005年2月26日授权的美国专利
号6,884,879。另外的抗体包括g6或b20系列抗体(例如,g6-31、b20-4.1),如pct公开号wo2005/012359、pct公开号wo2005/044853和美国专利申请60/991,302中所述,这些专利申请的内容通过引用明确并入本文。关于另外的抗体,参见美国专利号7,060,269、6,582,959、6,703,020、6,054,297;wo98/45332;wo 96/30046;wo94/10202;ep 0666868b1;美国专利申请公布号2006009360、20050186208、20030206899、20030190317、20030203409和20050112126;以及popkov等人,journal of immunological methods288:149-164(2004)。其他抗体包括与人vegf上的功能表位结合的抗体,该表位包含残基f17、m18、d19、y21、y25、q89、i191、k101、e103和c104,或者替代地,包含残基f17、y21、q22、y25、d63、i83和q89。
[0181]
在本发明的一个实施例中,抗vegf抗体具有包含以下氨基酸序列的轻链可变区:
[0182]
diqmtqspss lsasvgdrvt itcsasqdis nylnwyqqkp gkapkvliyf tsslhsgvps rfsgsgsgtd ftltisslqp edfatyycqq ystvpwtfgq gtkveikr.(seq id no:11);和/或包含以下氨基酸序列的重链可变区:evqlvesggg lvqpggslrl scaasgytft nygmnwvrqa pgkglewvgw intytgepty aadfkrrftf sldtskstay lqmnslraed tavyycakyp hyygsshwyf dvwgqgtlvt vss(seq id no:12)。
[0183]
在一些实施例中,抗vegf抗体包含贝伐单抗的一个、两个、三个、四个、五个或六个高变区(hvr)序列。在一些实施例中,抗vegf抗体包含选自以下项的一个、两个、三个、四个、五个或六个高变区(hvr)序列:(a)包含gytftnygmn(seq id no:13)的氨基酸序列的hvr-h1、(b)包含wintytgeptyaadfkr(seq id no:14)的氨基酸序列的hvr-h2、(c)包含yphyygsshwyfdv(seq id no:19)的氨基酸序列的hvr-h3、(d)包含sasqdisnyln(seq id no:20)的氨基酸序列的hvr-l1、(e)包含ftsslhs(seq id no:21)的氨基酸序列的hvr-l2和(f)包含qqystvpwt(seq id no:22)的氨基酸序列的hvr-l3。在一些实施例中,抗vegf抗体包含美国专利号6,884,879中描述的抗体的一个、两个、三个、四个、五个或六个高变区(hvr)序列。在一些实施例中,抗vegf抗体包含轻链可变区的一个、两个或三个高变区(hvr)序列,所述轻链可变区包含以下氨基酸序列:diqmtqspss lsasvgdrvt itcsasqdis nylnwyqqkp gkapkvliyf tsslhsgvps rfsgsgsgtd ftltisslqp edfatyycqq ystvpwtfgq gtkveikr.(seq id no:11);和/或重链可变区的一个、两个或三个高变区(hvr)序列,所述重链可变区包含以下氨基酸序列:evqlvesggg lvqpggslrl scaasgytft nygmnwvrqa pgkglewvgw intytgepty aadfkrrftf sldtskstay lqmnslraed tavyycakyp hyygsshwyf dvwgqgtlvt vss(seq id no:12)。
[0184]“g6系列抗体”为衍生自根据pct公开号wo2005/012359的图7、24-26和34-35中任一项的g6抗体或g6衍生抗体的序列的抗vegf抗体,其全部公开内容通过引用明确并入本文。还参见pct公开号wo2005/044853,其全部公开内容通过引用明确并入本文。在一个实施例中,g6系列抗体结合人vegf上包含残基f17、y21、q22、y25、d63、i83和q89的功能表位。
[0185]“b20系列抗体”为衍生自根据pct公开号wo2005/012359的图27-29中任一项的b20抗体或b20衍生抗体的序列的抗vegf抗体,其全部公开内容通过引用明确并入本文。还参见pct公开号wo2005/044853和美国专利申请60/991,302,这些专利申请的内容通过引用明确并入本文。在一个实施例中,b20系列抗体结合人vegf上的功能表位,其包含残基f17、m18、d19、y21、y25、q89、i91、k101、e103和c104。
[0186]“功能表位”(当用于vegf表位时)是指抗原的对抗体结合有积极贡献的氨基酸残
基。抗原的任何一个有积极贡献的残基的突变(例如,野生型vegf由丙氨酸引起的突变或同源突变)将破坏抗体的结合,使得抗体的相对亲和力比(ic50突变vegf/ic50野生型vegf)将大于5(参见wo2005/012359的实例2)。在一个实施例中,相对亲和力比通过溶液结合噬菌体展示elisa来确定。简而言之,将96孔maxisorp免疫板(nunc)在4℃用fab形式的待测抗体在pbs中以2μg/ml的浓度包被过夜,并用pbs、0.5%bsa和0.05%tween20(pbt)在室温封闭2小时。将展示hvegf丙氨酸点突变体(残基8-109形式)或野生型hvegf(8-109)的噬菌体在pbt中的系列稀释液首先在包被fab的板上室温孵育15分钟,然后用pbs、0.05%tween20(pbst)洗涤板。结合的噬菌体用抗m13单克隆抗体辣根过氧化物酶(amersham pharmacia)缀合物在pbt中以1:5000稀释,用3,3',5,5'-四甲基联苯胺(tmb,kirkegaard&perry labs,gaithersburg,md.)底物显影约5分钟,用1.0m h3po4淬灭,并在450nm处进行分光光度计读数。ic50值的比率(ic50,ala/ic50,wt)代表结合亲和力(相对结合亲和力)降低的倍数。
[0187]
在一些实施例中,附加疗法包含铂剂或含铂化疗。在一些实施例中,在治疗的诱导阶段期间施用附加疗法。铂剂/含铂化疗(诸如例如顺铂、卡铂、奥沙利铂和沙铂(staraplatin))是广泛使用的抗肿瘤药物,其引起dna交联为单加合物、链间交联物、链内交联物或dna蛋白交联物。铂剂通常作用于鸟嘌呤的相邻n-7位置,形成1,2链内交联(poklar等人(1996).proc.natl.acad.sci.u.s.a.93(15):7606 11;rudd等人(1995).cancer chemother.pharmacol.35(4):323 6)。所得的交联抑制癌细胞中的dna修复和/或dna合成。
[0188]
卡铂是用于本文所述方法中的示例性铂配位化合物。卡铂的化学名称为铂,二胺[1,1-环丁烷二羧基(2-)-o,o
′
]-,(sp-4-2),卡铂具有以下结构式:
[0189][0190]
卡铂是分子式为c6h
12
n2o4pt的晶体粉末,分子量为371.25。它以约14mg/ml的率溶于水,1%溶液的ph为5至7。它实际上不溶于乙醇、丙酮和二甲基乙酰胺。卡铂主要产生链间dna交联,这种作用是细胞周期非特异性的。卡铂可以以商品名biocarn、blastocarb、blastoplatin、carbokem、carbomax、carbopa、carboplan、carboteen、carbotinal、cytocarb、ducarb、karplat、kemocarb、naproplat、neoplatin、niscarbo、oncocarbin、tevacarb、womastin和其它来商购。
[0191]
顺铂是本文所述方法中使用的另一种示例性铂配位化合物。顺铂的化学名称为二氨二氯合铂(dichloroplatinum diammoniate),顺铂具有以下结构式:
[0192][0193]
顺铂是一种无机水溶性铂络合物,分子式为pt(nh3)2cl2,分子量为300.046。水解后,它与dna反应产生链内和链间交联。这些交联似乎削弱了dna的复制和转录。顺铂的细胞毒性与细胞周期g2期的细胞停滞有关。顺铂可以以商品名-aq、cddp、cisplan、cisplat、platikem、plationco、practicis、platicis、blastolem、cismax、cisplan、cisplatinum、cisteen、duplat、kemoplat、oncoplatin-aq、platinex、platin、tevaplatin和其它来商购。
[0194]
在一些实施例中,在治疗的诱导阶段期间向个体施用附加疗法或药剂。在一些实施例中,在治疗的维持阶段期间向个体施用附加疗法或药剂。例如,在一些实施例中,在治疗的维持阶段期间向个体施用抗体。
[0195]
在一些实施例中,在使用本文所述方法治疗之前,个体已经用含铂化疗进行治疗,例如,如上文所述。在一些实施例中,个体不符合进行含铂化疗的条件,例如,如上文所述。
[0196]
在一些实施例中,在使用本文所述方法治疗之前,个体已经用辅助化疗或新辅助化疗进行治疗。在一些实施例中,癌症为局部晚期或转移性非小细胞肺癌,并且个体在使用本文所述方法治疗之前已经用化疗进行治疗。
[0197]
在一些实施例中,来自个体的癌症的样品包含表达pd-l1的肿瘤浸润免疫细胞。在一些实施例中,来自个体的癌症的样品包含肿瘤浸润免疫细胞,所述肿瘤浸润免疫细胞表达pd-l1并覆盖1%或更多的肿瘤区域。在一些实施例中,表达pd-l1的肿瘤浸润免疫细胞经由免疫组织化学测定法,例如ventana sp142测定法进行测定。
[0198]
在一些实施例中,个体为“pd-l1高”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤细胞总计≥样品中肿瘤细胞总数的50%,则患者为“pd-l1高”。在一些实施例中,将治疗前样品中≥50%的肿瘤细胞上的pd-l1表达定义/评分为“tc3”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤浸润免疫细胞总计≥样品中肿瘤浸润免疫细胞总数的10%,则患者为“pd-l1高”。在一些实施例中,将治疗前样品中≥10%的肿瘤浸润免疫细胞上的pd-l1表达定义/评分为“ic3”。在一些实施例中,治疗前样品是新鲜的肿瘤样品。在一些实施例中,治疗前样品是福尔马林固定石蜡包埋的(ffpe)肿瘤样品。在一些实施例中,通过免疫组织化学测定法确定在治疗前样品中肿瘤细胞和/或肿瘤浸润免疫细胞上的pd-l1表达水平。在一些实施例中,免疫组织化学测定是ventana sp142测定。
[0199]
在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤细胞总计为样品中肿瘤细胞的总数的1%至《5%,则患者为“pd-l1低”。在一些实施例中,将治疗前样品中1%至《5%的肿瘤细胞上的pd-l1表达定义/评分为“tc1”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤细胞总计为样品中肿瘤细胞的总数的5%至《50%,则患者为“pd-l1低”。在一些实施例中,将治疗前样品中5%至《50%的肿瘤细胞上的pd-l1表达定义/评分为“tc2”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤浸润免疫细胞总计为样品中肿瘤浸润免疫细胞总数的1%至《5%,则患者为“pd-l1低”。在一些实施例中,将在治疗前样品中1%至《5%的肿瘤浸润免疫细胞上pd-l1表达定义/评分为“ic1”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤浸润免疫细胞总计为样品中肿瘤浸润免疫细胞总数的5%至《10%,则患者为“pd-l1低”。在一些实施例中,将在治疗前样品中5%至《10%的肿瘤浸润免疫细胞上pd-l1表达定义/评分为“ic2”。在一些实施例中,治疗前样品是新鲜的肿瘤样品。在一些实施例中,治疗前样品是福尔马林固定石蜡包埋的(ffpe)肿瘤样品。在一些实施例中,通过免疫组织化学测定法确定在治疗前样品中肿瘤细胞和/或肿瘤浸润免疫细胞上的pd-l1表达水平。在一些实施例中,免疫组织化
学测定是ventana sp142测定。
[0200]
在一些实施例中,个体为“pd-l1阴性”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤细胞总计为样品中肿瘤细胞总数的《1%,则患者为“pd-l1阴性”。在一些实施例中,将在治疗前样品中《1%的肿瘤细胞上pd-l1表达定义分为“tc0”。在一些实施例中,如果来自患者的治疗前样品中表达pd-l1的肿瘤浸润免疫细胞总计为样品中肿瘤浸润免疫细胞总数的《1%,则患者为“pd-l1阴性”。在一些实施例中,将在治疗前样品中《1%的肿瘤浸润免疫细胞上pd-l1表达定义分为“ic0”。在一些实施例中,治疗前样品是新鲜的肿瘤样品。在一些实施例中,治疗前样品是福尔马林固定石蜡包埋的(ffpe)肿瘤样品。在一些实施例中,通过免疫组织化学测定法确定治疗前样品中肿瘤细胞和/或肿瘤浸润免疫细胞中的pd-l1表达水平。在一些实施例中,免疫组织化学测定是ventana sp142测定。
[0201]
在一些实施例中,tc0、tc1、tc2、tc3、ic0、ic1、ic2和ic3定义/评分如下表所总结:
[0202]
示例性肿瘤细胞(tc)和肿瘤浸润免疫细胞(ic)评分定义*
[0203][0204]
ic,肿瘤浸润免疫细胞;pd-l1,程序性死亡配体1;tc,肿瘤细胞。
[0205]
来自socinski m,等人the n engl j med.atezolizumab for first-line treatment of metastatic nonsquamous nsclc.2018;378:2288-301。
[0206]
在另一方面,个体患有表达(已例如在诊断测试中显示出表达)pd-l1生物标志物的癌症。在一些实施例中,患者的癌症表达低pd-l1生物标志物。在一些实施例中,患者的癌症表达高pd-l1生物标志物。在任何方法、测定法和/或试剂盒的一些实施例中,当pd-l1生物标志物占样品的0%时,其不存在于样品中。
[0207]
在一些实施例中,本文提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,其中该人类患者不符合进行含顺铂化疗的条件并且其肿瘤表达pd-l1(pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥5%的肿瘤区域),如通过fda批准的测试确定的。在本文所述方法的一些实施例中,本文提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,其中该人类患者无论pd-l1状态如何均不符合进行任何含铂化疗的条件。在本文所述方法的一些实施例中,本文提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,其中人类患者在进行任何含铂化疗期间或之后、或在新辅助化疗或辅助化疗的12个月内,具有疾病进展。
[0208]
在一些实施例中,本文提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,其中该方法包括在既往含铂化疗之后向人类患者施用抗pd-l1抗体。在一些实施例中,本文提供了用于治疗患有局部晚期或转移性尿路上皮癌的人类患者的方法,其中该方法包括向该人类患者施用抗pd-l1抗体,并且其中该人类患者被认为不符合进行顺铂治疗的条件,并且其肿瘤具有pd-l1表达≥5%。在一些实施例中,人类患者为成年人。
[0209]
在一些实施例中,本文提供了用于治疗患有没有egfr或alk基因组肿瘤畸变的转移性非小细胞肺癌的人类患者的方法。在一些实施例中,该方法包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂。
[0210]
在一些实施例中,本文提供了用于治疗患有具有egfr和/或alk基因组肿瘤畸变的转移性非小细胞肺癌的人类患者的方法,其中该方法包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂,其中对该人类患者进行的针对非小细胞肺癌的靶向疗法失败。
[0211]
在一些实施例中,本文提供了用于治疗患有转移性非小细胞肺癌的人类患者的方法,并且其中该人类患者在进行含铂化疗期间或之后进展。在一些实施例中,该方法包括将抗pd-l1抗体作为单一药剂向人类患者施用。在一些实施例中,其中人类患者具有egfr或alk基因组肿瘤畸变,患者在靶向治疗上取得进展。在一些实施例中,其中人类患者具有egfr或alk基因组肿瘤畸变,患者在fda批准的治疗上取得进展。
[0212]
在一些实施例中,本文提供了用于治疗患有局部晚期或转移性非小细胞肺癌的人类患者的方法,其中该方法包括在既往化疗之后向人类患者施用抗pd-l1抗体。
[0213]
在一些实施例中,本文提供了用于治疗患有局部晚期或转移性三阴性乳腺癌的人类患者的方法。在一些实施例中,癌症为不可切除的局部晚期或转移性三阴性乳腺癌。在一些实施例中,肿瘤表达pd-l1(任何强度的pd-l1染色的肿瘤浸润免疫细胞[ic],覆盖≥1%的肿瘤区域),如通过fda批准的测试确定的。在一些实施例中,该方法包括向人类患者施用抗pd-l1抗体联合蛋白结合型紫衫醇。
[0214]
在任何方法、测定法和/或试剂盒的一些实施例中,当pd-l1生物标志物占样品的0%以上时,其存在于样品中。在一些实施例中,pd-l1生物标志物存在于至少1%的样品中。在一些实施例中,pd-l1生物标志物存在于至少5%的样品中。在一些实施例中,pd-l1生物标志物存在于至少10%的样品中。
[0215]
在任何方法、测定和/或试剂盒的一些实施例中,使用选自由facs、western印迹、elisa、免疫沉淀、免疫组织化学、免疫荧光、放射免疫测定、斑点杂交、免疫检测方法、hplc、表面等离子体共振、光谱、质谱、hplc、qpcr、rt-qpcr、多重qpcr或rt-qpcr、rna-seq、微阵列分析、sage、massarray技术和fish及其组合组成的组中的方法,在样品中检测pd-l1生物标志物。
[0216]
在任何方法、测定和/或试剂盒的一些实施例中,通过蛋白质表达在样品中检测pd-l1生物标志物。在一些实施例中,蛋白表达通过免疫组织化学(ihc)确定。在一些实施例中,使用抗pd-l1抗体检测pd-l1生物标志物。在一些实施例中,通过ihc检测pd-l1生物标志物为弱染色强度。在一些实施例中,通过ihc检测pd-l1生物标志物为中等染色强度。在一些实施例中,通过ihc检测pd-l1生物标志物为强染色强度。在一些实施例中,在肿瘤细胞、肿瘤浸润免疫细胞、基质细胞及其任何组合上检测pd-l1生物标志物。在一些实施例中,染色是膜染色、细胞质染色或其组合。在一些实施例中,免疫组织化学测定是ventana sp142测
定。
[0217]
在任何方法、测定和/或试剂盒的一些实施例中,将pd-l1生物标志物的不存在检测为样品中不存在染色或无染色。在任何方法、测定和/或试剂盒的一些实施例中,将pd-l1生物标志物的存在检测为样品中的任何染色。
[0218]
在根据本文所述任何实施例的一些实施例中,个体为人。
[0219]
在一些实施例中,抗pd-l1抗体通过静脉内、肌肉内、皮下、局部、口服、透皮、腹膜内、眶内、通过植入、通过吸入、鞘内、心室内或鼻内施用。在一些实施例中,抗pd-l1抗体通过静脉输注施用。在一些实施例中,抗pd-l1抗体通过历时30分钟或历时60分钟的静脉输注施用。在一些实施例中,第一剂量的抗pd-l1抗体通过历时60分钟的脉输注施用,并且后续剂量的抗pd-l1抗体通过历时30分钟的静脉输注施用(例如,如果对第一剂量耐受)。
[0220]
在根据本文所述任何实施例的一些实施例中,待通过本公开的方法治疗的癌症包括但不限于结直肠癌、肾细胞癌症(例如,肾细胞癌)、黑素瘤、膀胱癌、卵巢癌、乳腺癌(例如,三阴性乳腺癌、her2阳性乳腺癌或激素受体阳性癌症)和非小细胞肺癌(例如,鳞状非小细胞肺癌或非鳞状非小细胞肺癌)。在一些实施例中,待通过本公开的方法治疗的癌症包括但不限于癌、淋巴瘤、母细胞瘤、肉瘤和白血病。在一些实施例中,待通过本公开的方法治疗的癌症包括但不限于鳞状细胞癌、肺癌(包括小细胞肺癌、非小细胞肺癌、肺腺癌和肺鳞癌)、黑素瘤、肾细胞癌、腹膜癌、肝细胞癌、胃癌或胃部癌症(包括胃肠道癌)、胰腺癌、成胶质细胞瘤、宫颈癌、卵巢癌、肝癌、膀胱癌、肝细胞瘤、乳腺癌、结肠癌、结直肠癌、子宫内膜癌或子宫癌、唾液腺癌、肾脏癌症或肾癌、肝癌、前列腺癌、外阴癌、甲状腺癌、肝细胞癌和各种类型的头颈癌,以及b细胞淋巴瘤(包括低级别/滤泡性非霍奇金淋巴瘤(nhl)、小淋巴细胞(sl)nhl、中级别/滤泡性nhl、中级别弥散性nhl、高级别免疫原性nhl、高级别淋巴母细胞性nhl、高级别小非裂解细胞性nhl、巨大肿块nhl、套细胞淋巴瘤、aids相关淋巴瘤和华氏巨球蛋白血症)、慢性淋巴细胞性白血病(cll)、急性淋巴细胞白血病(all)、毛细胞白血病、慢性粒细胞性白血病和移植后的淋巴增生性疾病(ptld),以及与斑痣性错构瘤、水肿(诸如与脑肿瘤有关的)、meigs综合征相关的异常血管增生。在一些实施例中,癌症可以为早期癌症或晚期癌症。在一些实施例中,癌症可以为原发性肿瘤。在一些实施例中,癌症可以为源自任何上述类型癌症的第二位点处的转移性肿瘤。
[0221]
在一些实施例中,待通过本公开的方法治疗的癌症选自由以下项组成的组:乳腺癌、结直肠癌、肺癌、肾细胞癌(rcc)、卵巢癌、黑素瘤和膀胱癌。在一些实施例中,乳腺癌为三阴性乳腺癌,例如,癌症为雌激素受体阴性(er阴性)、孕激素受体阴性(pr阴性)和her2阴性。在一个实施例中,肺癌是非小细胞肺癌(nsclc)。在一些实施例中,肺癌为小细胞肺癌(sclc)。在一些实施例中,膀胱癌为尿路上皮癌。
[0222]
在一些实施例中,癌症为局部晚期或转移性的。
[0223]
在一些实施例中,癌症为局部晚期或转移性尿路上皮癌。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌,并且在使用本文所述方法治疗之前,个体已经用含铂化疗进行治疗。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌,并且个体不符合进行含铂化疗的条件。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌,个体不符合进行含铂化疗(例如,含有顺铂)的条件,并且癌症表达pd-l1(例如,从癌症获得的样品显示表达pd-l1的肿瘤浸润免疫细胞覆盖5%或更多的肿瘤区域,这可以例如使用免疫组织化学测定
确定)。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌,并且在使用本文所述方法治疗之前,个体在进行含铂化疗治疗期间或之后已出现疾病进展。在一些实施例中,癌症为局部晚期或转移性尿路上皮癌,并且在使用本文所述方法治疗之前,个体在用新辅助化疗或辅助化疗治疗的12个月内已出现疾病进展。
[0224]
在一些实施例中,癌症为nsclc。在一些实施例中,癌症为转移性非鳞状nsclc。在一些实施例中,癌症为没有egfr或alk基因组肿瘤畸变或突变的nsclc。在一些实施例中,癌症是没有egfr或alk基因组肿瘤畸变或突变的nsclc(例如,转移性非鳞状nsclc),并且该方法进一步包括施用抗vegf抗体(例如,贝伐单抗)、紫杉烷(例如,紫杉醇或蛋白结合型紫衫醇)和含铂化疗(例如,卡铂)联合抗pd-l1抗体(例如,阿特珠单抗)。
[0225]
在一些实施例中,癌症为局部晚期或转移性nsclc。在一些实施例中,癌症为局部晚期或转移性nsclc,并且在使用本文所述方法治疗之前,个体已经用化疗进行治疗。在一些实施例中,癌症为局部晚期或转移性nsclc,癌症具有egfr活化或alk阳性突变,并且在使用本文所述方法治疗之前,个体已经用靶向疗法进行治疗。在一些实施例中,癌症为局部晚期或转移性nsclc,癌症具有egfr活化或alk阳性突变,并且在使用本文所述方法治疗之前,个体在用靶向疗法进行治疗时已出现疾病进展。在一些实施例中,癌症为局部晚期或转移性nsclc,并且在使用本文所述方法治疗之前,个体在进行含铂化疗治疗期间或之后已出现疾病进展。
[0226]
各种活化egfr突变是本领域已知的。egfr基因编码表皮生长因子受体,也称为v-erb-b、erbb、erbb1、her1和sa7。在一些实施例中,egfr突变导致egfr的过表达(例如,基因扩增或egfr基因拷贝数增加)。在一些实施例中,egfr突变包括egfr基因外显子18、19、20或21中的点突变或缺失。已知的egfr突变包括但不限于外显子19缺失、外显子20插入、l858r、t790m、s768i、g719a、g719c、g719s、l861q、c797s、外显子19插入、a763_y764insfqea和激酶结构域的重复。其他egfr突变在例如肿瘤学和血液学中的遗传学和细胞遗传学图谱(参见atlasgeneticsoncology.org/genes/gc_egfr.html)和omim基因id:131550中有描述。用于检测egfr突变的示例性测定包括,例如,直接测序、变性高效液相色谱(dhplc)、高分辨率熔解分析(hrma)、焦磷酸测序、聚合酶链反应(pcr)以检测关注的特定突变或以靶向特定目标区域、片段长度分析、基于阳离子缀合聚合物(ccp)的荧光共振能量转移(fret)、smartamp、肽核酸(pna)介导的pcr钳位、ihc、arms、实时pcr和下一代测序。参见例如ellison,g.等人(2013)j.clin.pathol.66:79-89。
[0227]
各种alk突变是本领域已知的。alk基因编码间变性淋巴瘤激酶(alk)受体酪氨酸激酶,也称为cd246和nblst3。在一些实施例中,alk突变包含alk基因中的重排或易位,例如,产生融合基因,诸如eml4-alk、kif5b-alk、klc1-alk或tfg-alk。alk突变包括但不限于e13;a20(v10)、e20;a20(v2)、e6a/b;a20(v3a/b)、e14;a20(v4)、e2a/b;a20(v6)、e14;a20(v7)、e15;a20(v4)、e18;a20(v5)、kif5b-alk、klc1-alk和tfg-alk。其他alk突变在shackelford,r.e.等人(2014)genes cancer 5:1-14中有描述。用于检测alk突变的示例性测定包括例如pcr、逆转录酶pcr(rt-pcr)、微阵列或外显子阵列分析、荧光原位杂交(fish)(例如,使用alk断裂或分裂信号探针;参见kwak,e.l.等人(2010)n.engl.j.med.363:1693-1703)、ihc、cdna末端的5'快速扩增(race)分析和下一代测序。参见例如shackelford,r.e.等人(2014)genes cancer 5:1-14。
[0228]
在一个实施例中,癌症为乳腺癌。在一些实施例中,癌症为三阴性乳腺癌(tnbc)。在一些实施例中,癌症为tnbc(例如,不可切除的局部晚期或转移性tnbc),并且该方法进一步包括施用紫杉烷(例如,紫杉醇或蛋白结合型紫杉醇)联合抗pd-l1抗体(例如,阿特珠单抗)。在一些实施例中,癌症为tnbc,并且癌症表达pd-l1(例如,从癌症获得的样品显示表达pd-l1的肿瘤浸润免疫细胞覆盖1%或更多的肿瘤区域,这可以例如使用免疫组织化学测定确定)。在一些实施例中,癌症为tnbc,癌症表达pd-l1(例如,从癌症获得的样品显示表达pd-l1的肿瘤浸润免疫细胞覆盖1%或更多的肿瘤区域,这可以例如使用免疫组织化学测定确定),并且该方法进一步包括施用抗pd-l1抗体(例如,阿特珠单抗)联合紫杉烷(例如,紫杉醇或蛋白质结合的紫杉醇)。
[0229]
在一些实施例中,癌症为小细胞肺癌(sclc)。在一些实施例中,癌症为广泛期sclc(es-sclc)。在一些实施例中,癌症为广泛期sclc(es-sclc),并且该方法进一步包括施用含铂化疗(例如,卡铂)和拓扑异构酶ii抑制剂(例如依托泊苷)联合抗pd-l1抗体(例如,阿特珠单抗)。
[0230]
在一些实施例中,包括但不限于治疗nsclc,该方法包括向个体施用4-6个周期的紫杉烷(例如,紫杉醇或蛋白结合型紫衫醇)、含铂化疗(例如,卡铂)和任选的抗vegf抗体(例如,贝伐单抗),然后在两个或更多个4周的周期内以1680mg的剂量向个体施用抗pd-l1抗体(例如,阿特珠单抗)。
[0231]
在一些实施例中,包括但不限于治疗sclc,该方法包括向个体施用4个周期的含铂化疗(例如,卡铂)和拓扑异构酶ii抑制剂(例如,依托泊苷),然后在两个或更多个4周的周期内以1680mg的剂量向个体施用抗pd-l1抗体(例如,阿特珠单抗)。
[0232]
在一些实施例中,本文提供了用于治疗患有癌症的人类患者的方法,其中癌症为广泛期小细胞肺癌。在一些实施例中,该方法包括施用抗pd-l1抗体联合卡铂和依托泊苷。在一些实施例中,该方法为一线治疗。
[0233]
在一些实施例中,人类患者先前未进行治疗,例如,先前未使用化疗剂进行治疗。在一些实施例中,人类患者患有尿路上皮癌并且先前未进行针对尿路上皮癌的治疗,例如,先前未使用化疗剂进行治疗。在一些实施例中,癌症为先前未治疗的癌症,例如,先前未使用化疗剂进行治疗的癌症。在一些实施例中,癌症为初治的局部晚期或转移性尿路上皮癌。在一些实施例中,人类患者不符合进行顺铂治疗的条件。在一些实施例中,人类患者不符合进行顺铂治疗的条件,并且癌症为初治的局部晚期或转移性尿路上皮癌。
[0234]
示例性治疗方法
[0235]
在一些实施例中,该方法包括在两个或更多个4周或28天的周期内以1680mg的剂量向人类患者施用抗pd-l1抗体,其中在两个或更多个4周或28天周期的每个周期中,以每个周期1680mg的剂量向该人类患者施用抗pd-l1抗体(例如,每4周或每28天一次向人类患者施用抗pd-l1抗体)。
[0236]
在一些实施例中,该方法包括在两个或更多个2周或14天的周期内以840mg的剂量向人类患者施用抗pd-l1抗体,其中在两个或更多个2周或14天周期的每个周期中,以每个周期840mg的剂量向该人类患者施用抗pd-l1抗体(例如,每2周或每14天一次向人类患者施用抗pd-l1抗体)。
[0237]
在本文所述方法的一些实施例中,人类患者患有尿路上皮癌。在本文所述方法的
一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者,其中该成人患者不符合进行含顺铂的化疗的条件并且其肿瘤表达pd-l1(pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥5%的肿瘤区域),如通过由美国fda批准的测试确定的。在本文所述方法的一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者,其中该成人患者无论pd-l1状态如何均不符合进行任何含铂化疗的条件。在本文所述方法的一些实施例中,人类患者为患有局部晚期或转移性尿路上皮癌的成人患者,其中该成人患者在进行任何含铂化疗期间或之后、或在新辅助化疗或辅助化疗的12个月内,具有疾病进展。
[0238]
在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体,并且其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体,其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性,并且其中如果对第一次输注抗pd-l1抗体耐受,全部后续输注可以历时30分钟递送。
[0239]
在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体,并且其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,人类患者患有尿路上皮癌,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体,其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性,并且其中如果对第一次输注抗pd-l1抗体耐受,全部后续输注可以历时30分钟递送。
[0240]
在本文所述方法的一些实施例中,人类患者患有非小细胞肺癌(nsclc)。在本文所述方法的一些实施例中,人类患者为成人患者,其中所述成人患者患有转移性非鳞状nsclc。在本文所述方法的一些实施例中,成人患者患有转移性非鳞状nsclc,其中该方法包括向成人患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂。在本文所述方法的一些实施例中,该方法为患有转移性非鳞状nsclc且没有egfr或alk基因组肿瘤畸变的成人患者的一线治疗。
[0241]
在本文所述方法的一些实施例中,人类患者为成人患者,其中成人患者患有转移性nsclc,其中成人患者在进行含铂化疗期间或之后具有疾病进展。在本文所述方法的一些实施例中,人类患者患有nsclc,其中人类患者具有egfr或alk基因组肿瘤畸变,并且其中在人类患者在根据本文所述方法施用抗pd-l1抗体之前,进行fda批准的针对nsclc具有这些畸变的的治疗时具有疾病进展。在本文所述方法的一些实施例中,该包括施用抗pd-l1抗体的方法为单药治疗。
[0242]
在本文所述方法的一些实施例中,人类患者为成人患者,其中该成人患者患有没有egfr或alk基因组肿瘤畸变的转移性非鳞状nsclc,并且其中该方法包括施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂。在本文所述方法的一些实施例中,该方法适用于患有没有egfr或alk基因组肿瘤畸变的转移性非鳞状nsclc成年患者的一线治疗。
[0243]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中施用抗pd-l1抗体直至出现疾病进展或不可接受的毒性。
[0244]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中当在同一天向人类患者施用时,抗pd-l1抗体在化疗或其他抗肿瘤药物之前施用。
[0245]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每2周840mg、每3周1200mg或每4周1680mg的剂量施用抗pd-l1抗体作为单一药剂。
[0246]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体,并且其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每2周840mg的剂量向人类患者施用抗pd-l1抗体,其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性,并且其中如果对第一次输注抗pd-l1抗体耐受,全部后续输注可以历时30分钟递送。在本文所述方法的一些实施例中,施用抗pd-l1抗体联合标准护理剂量的贝伐单抗、标准护理剂量的紫杉醇和标准护理剂量的卡铂,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,施用抗pd-l1抗体联合15mg/kg剂量的贝伐单抗、175mg/m2或200mg/m2剂量的紫杉醇和auc 6mg/ml/min剂量的卡铂,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,其中施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂,当在同一天施用时,抗pd-l1抗体在其他抗肿瘤药物之前施用。在本文所述方法的一些实施例中,在包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂的方法的4-6个周期完成后,如果贝伐单抗停用,则该方法包括进一步以每2周840mg的剂量施用抗pd-l1抗体,静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,在包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂的方法的4-6个周期完成后,如果贝伐单抗停用,则该方法包括进一步以每4周1680mg的剂量施用抗pd-l1抗体,静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,抗pd-l1抗体的初始输注历时60分钟。在本文所述方法的一些实施例中,如果对抗pd-l1抗体的初始输注耐受,则所有后续输注均历时30分钟递送。
[0247]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体。在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体,并且其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,人类患者患有nsclc,其中该方法包括以每4周1680mg的剂量向人类患者施用抗pd-l1抗体,其中抗pd-l1抗体历时60分钟静脉内施用,直至出现疾病进展或不可接受的毒性,并且其中如果对第一次输注抗pd-l1抗体耐受,全部后续输注可以历时30分钟递送。在本文所述方法的一些实施例中,施用抗pd-l1抗体联合标准护理剂量的贝伐单抗、标准护理剂量的紫杉醇和标准护理剂量的卡铂,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,施用抗pd-l1抗体联合15mg/kg剂量的贝伐单抗、175mg/m2或200mg/m2剂量的紫杉醇和auc 6mg/ml/min剂量的卡铂,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,其中施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂,
当在同一天施用时,抗pd-l1抗体在其他抗肿瘤药物之前施用。在本文所述方法的一些实施例中,在包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂的方法的4-6个周期完成后,如果贝伐单抗停用,则该方法包括进一步以每2周840mg的剂量施用抗pd-l1抗体,静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,在包括向人类患者施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂的方法的4-6个周期完成后,如果贝伐单抗停用,则该方法包括进一步以每4周1680mg的剂量施用抗pd-l1抗体,静脉内施用,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,抗pd-l1抗体的初始输注历时60分钟。在本文所述方法的一些实施例中,如果对抗pd-l1抗体的初始输注耐受,则所有后续输注均历时30分钟递送。
[0248]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中施用抗pd-l1抗体联合贝伐单抗、紫杉醇和卡铂,该抗pd-l1抗体以每3周1200mg的剂量在化疗或其他抗肿瘤药物之前施用。
[0249]
在本文所述方法的一些实施例中,人类患者患有nsclc,其中在完成4-6个周期的紫杉醇和卡铂后,如果贝伐单抗停用,则以每2周840mg、每3周1200mg或每4周1680mg的剂量施用抗pd-l1抗体。
[0250]
在本文所述方法的一些实施例中,人类患者为成人患者,其中该成人患者患有三阴性乳腺癌(tnbc)。在本文所述方法的一些实施例中,人类患者为成人患者,其中该成人患者患有不可切除的局部晚期或转移性tnbc,其中不可切除的局部晚期或转移性tnbc的肿瘤表达pd-l1(任何强度的pd-l1染色的肿瘤浸润免疫细胞[ic]覆盖≥1%的肿瘤区域),如根据美国fda批准的测试确定的。
[0251]
在本文所述方法的一些实施例中,成人患者患有转移性tnbc,其中该方法包括以840mg的剂量施用抗pd-l1抗体,然后以100mg/m2的剂量施用蛋白结合型紫衫醇,其中对于每个28天周期,在第1天和第15天施用抗pd-l1抗体,在第1、8和15天施用蛋白结合型紫杉醇,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,成人患者患有局部晚期或转移性tnbc,其中该方法包括以840mg的剂量施用抗pd-l1抗体并且以100mg/m2的剂量施用蛋白结合型紫衫醇,其中抗pd-l1抗体作为静脉输注施用,历时60分钟,然后施用100mg/m2蛋白结合型紫衫醇,其中对于每个28天周期,在第1天和第15天施用抗pd-l1抗体,在第1、8和15天施用蛋白结合型紫衫醇,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,抗pd-l1抗体的初始输注历时60分钟进行输注。在本文所述方法的一些实施例中,如果对抗pd-l1抗体的历时60分钟的初始输注耐受,则所有后续输注可以历时30分钟递送。
[0252]
在本文所述方法的一些实施例中,人类患者是成人患者,其中所述成人患者患有广泛期小细胞肺癌(es-sclc)。在本文所述方法的一些实施例中,成人患者患有es-sclc,并且其中成人患者适合使用本文所述的方法进行一线治疗,所述方法包含抗pd-l1抗体联合卡铂和依托泊苷。
[0253]
在本文所述方法的一些实施例中,人类患者患有sclc,其中在完成4个周期的卡铂和依托泊苷后,该方法包括向人类患者施用包含以每2周840mg、每3周1200mg或每4周1680mg的剂量施用的抗pd-l1抗体的治疗。在本文所述方法的一些实施例中,人类患者患有sclc,其中人类患者已接受4个周期的包含卡铂和依托泊苷的初始治疗,其中在完成4个周
期的初始治疗后,该方法包括向人类患者施用包含以每2周840mg的剂量静脉内施用的抗pd-l1抗体的治疗,直至出现疾病进展或不可接受的毒性。在本文所述方法的一些实施例中,人类患者患有sclc,其中人类患者已接受4个周期的包含卡铂和依托泊苷的初始治疗,其中在完成4个周期的初始治疗后,该方法包括向人类患者施用包含以每4周1680mg的剂量静脉内施用的抗pd-l1抗体的治疗,直至出现疾病进展或不可接受的毒性。在一些实施例中,初始治疗进一步包括以每3周1200mg的剂量施用抗pd-l1抗体。在本文所述方法的一些实施例中,抗pd-l1抗体的初始输注历时60分钟进行输注。在本文所述方法的一些实施例中,如果对抗pd-l1抗体的历时60分钟的初始输注耐受,则所有后续输注可以历时30分钟递送。
[0254]
在本文所述方法的一些实施例中,人类患者患有sclc,其中当施用抗pd-l1抗体与卡铂和依托泊苷时,在化疗之前以每3周1200mg的剂量施用抗pd-l1抗体。
[0255]
在本文所述方法的一些实施例中,人类患者患有sclc,其中当在同一天向人类患者施用时,抗pd-l1抗体在化疗之前施用。
[0256]
iii.抗pd-l1抗体
[0257]
多种抗pd-l1抗体被考虑用于本公开和本文所述的方法中。在本文的任何实施例中,分离的抗pd-l1抗体可以结合人pd-l1,例如uniprotkb/swiss-prot登录号q9nzq7.1中所示的人pd-l1,或其变体。“pd-l1”的替代名称包括b7-h1、b7-4、cd274和b7-h。
[0258]
在一些实施例中,抗pd-l1抗体能够抑制pd-l1和pd-1之间和/或pd-l1和b7-1之间的结合。在一些实施例中,抗pd-l1抗体是单克隆抗体。在一些实施例中,抗pd-l1抗体是选自由fab、fab'-sh、fv、scfv和(fab')2片段组成的组的抗体片段。在一些实施例中,抗pd-l1抗体为人源化抗体。在一些实施例中,抗pd-l1抗体是人抗体。可用于本发明方法的抗pd-l1抗体的实例及其制备方法在pct专利申请wo 2010/077634 a1和美国专利号8,217,149中描述,其通过引用并入本文。
[0259]
在一些实施例中,抗pd-l1抗体包含重链可变区和轻链可变区,其中:
[0260]
(a)重链可变区包含hvr-h1、hvr-h2和hvr-h3,其序列分别为gftfsdswih(seq id no:1)、awispyggstyyadsvkg(seqid no:2)和rhwpggfdy(seq id no:3),并且
[0261]
(b)轻链可变区包含hvr-l1、hvr-l2和hvr-l3,其序列分别为rasqdvstava(seq id no:4)、sasflys(seq id no:5)和qqylyhpat(seq id no:6)。
[0262]
在一些实施例中,抗pd-l1抗体是mpdl3280a,也称为阿特珠单抗和(cas登记号:1422185-06-5)。在一些实施例中,抗pd-l1抗体包含重链和轻链序列,其中:
[0263]
(a)重链可变区序列包含以下氨基酸序列:
[0264]
evqlvesggglvqpggslrlscaasgftfsdswihwvrqapgkglewvawispyggstyyadsvkgrftisadtskntaylqmnslraedtavyycarrhwpggfdywgqgtlvtvss(seq id no:7),并且
[0265]
(b)轻链可变区序列包含以下氨基酸序列:
[0266]
diqmtqspsslsasvgdrvtitcrasqdvstavawyqqkpgkapklliysasflysgvpsrfsgsgsgtdftltisslqpedfatyycqqylyhpatfgqgtkveikr(seq id no:8)。
[0267]
在一些实施例中,抗pd-l1抗体包含重链和轻链序列,其中:
[0268]
(a)重链包含以下氨基酸序列:
[0269]
evqlvesggglvqpggslrlscaasgftfsdswihwvrqapgkglewvawispyggstyyadsvkgrftisadtskntaylqmnslraedtavyycarrhwpggfdywgqgtlvtvssastkgpsvfplapsskstsggtaalgclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslssvvtvpssslgtqtyicnvnhkpsntkvdkkvepkscdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqyastyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsreemtknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspg(seq id no:9),和
[0270]
(b)轻链包含以下氨基酸序列:
[0271]
diqmtqspsslsasvgdrvtitcrasqdvstavawyqqkpgkapklliysasflysgvpsrfsgsgsgtdftltisslqpedfatyycqqylyhpatfgqgtkveikrtvaapsvfifppsdeqlksgtasvvcllnnfypreakvqwkvdnalqsgnsqesvteqdskdstyslsstltlskadyekhkvyacevthqglsspvtksfnrgec(seq id no:10)。
[0272]
在一些实施例中,抗pd-l1抗体是阿维单抗(avelumab)(cas登记号:1537032-82-8)。阿维单抗,也称为msb0010718c,是人单克隆igg1抗pd-l1抗体(merck kgaa,pfizer)。在一些实施例中,抗pd-l1抗体包含重链和轻链序列,其中:
[0273]
(a)重链包含以下氨基酸序列:
[0274]
evqllesggglvqpggslrlscaasgftfssyimmwvrqapgkglewvssiypsggitfyadtvkgrftisrdnskntlylqmnslraedtavyycariklgtvttvdywgqgtlvtvssastkgpsvfplapsskstsggtaalgclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslssvvtvpssslgtqtyicnvnhkpsntkvdkkvepkscdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspg(seq id no:15),和
[0275]
(b)轻链包含以下氨基酸序列:
[0276]
qsaltqpasvsgspgqsitisctgtssdvggynyvswyqqhpgkapklmiydvsnrpsgvsnrfsgsksgntasltisglqaedeadyycssytssstrvfgtgtkvtvlgqpkanptvtlfppsseelqankatlvclisdfypgavtvawkadgspvkagvettkpskqsnnkyaassylsltpeqwkshrsyscqvthegstvektvaptecs(seq id no:16)。
[0277]
在一些实施例中,抗pd-l1抗体包含来自seq id no:15和seq id no:16的六个hvr序列(例如,来自seq id no:15的三个重链hvr和来自seq id no:16的三个轻链hvr)。在一些实施例中,抗pd-l1抗体包含来自seq id no:15的重链可变结构域和来自seq id no:16的轻链可变结构域。
[0278]
在一些实施例中,抗pd-l1抗体是德瓦鲁单抗(durvalumab)(cas登记号:1428935-60-7)。德瓦鲁单抗,也称为medi4736,是wo2011/066389和us2013/034559中所述的fc优化的人单克隆igg1κ抗pd-l1抗体(medimmune,astrazeneca)。在一些实施例中,抗pd-l1抗体包含重链和轻链序列,其中:
[0279]
(a)重链包含以下氨基酸序列:
[0280]
evqlvesggglvqpggslrlscaasgftfsrywmswvrqapgkglewvanikqdgsekyyvdsvkgrftisrdnaknslylqmnslraedtavyycareggwfgelafdywgqgtlvtvssastkgpsvfplapsskstsggta
algclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslssvvtvpssslgtqtyicnvnhkpsntkvdkrvepkscdkthtcppcpapefeggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpasiektiskakgqprepqvytlppsreemtknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspg(seq id no:17),和
[0281]
(b)轻链包含以下氨基酸序列:
[0282]
eivltqspgtlslspgeratlscrasqrvsssylawyqqkpgqaprlliydassratgipdrfsgsgsgtdftltisrlepedfavyycqqygslpwtfgqgtkveikrtvaapsvfifppsdeqlksgtasvvcllnnfypreakvqwkvdnalqsgnsqesvteqdskdstyslsstltlskadyekhkvyacevthqglsspvtksfnrgec(seq id no:18)。
[0283]
在一些实施例中,抗pd-l1抗体包含来自seq id no:17和seq id no:18的六个hvr序列(例如,来自seq id no:17的三个重链hvr和来自seq id no:18的三个轻链hvr)。在一些实施例中,抗pd-l1抗体包含来自seq id no:17的重链可变结构域和来自seq id no:18的轻链可变结构域。
[0284]
在一些实施例中,抗pd-l1抗体是mdx-1105(bristol myers squibb)。mdx-1105,也称为bms-936559,是wo2007/005874中所述的抗pd-l1抗体。
[0285]
在一些实施例中,抗pd-l1抗体是ly3300054(eli lilly)。
[0286]
在一些实施例中,抗pd-l1抗体是sti-a1014(sorrento)。sti-a1014是人抗pd-l1抗体。
[0287]
在一些实施例中,抗pd-l1抗体是kn035(suzhou alphamab)。kn035是从骆驼噬菌体展示文库生成的单结构域抗体(dab)。
[0288]
在一些实施例中,抗pd-l1抗体包含可裂解的部分或连接基,当被裂解时(例如,通过肿瘤微环境中的蛋白酶),该部分或连接基激活抗体抗原结合结构域(例如,通过去除非结合的空间部分)以使其结合其抗原。在一些实施例中,抗pd-l1抗体是cx-072(cytomx therapeutics)。
[0289]
在一些实施例中,pd-l1抗体包含来自以下文献所述的pd-l1抗体的六个hvr序列(例如,三个重链hvr和三个轻链hvr)和/或重链可变结构域和轻链可变结构域:us20160108123(转让给novartis)、wo2016/000619(申请人:beigene)、wo2012/145493(申请人:amplimmune)、us9205148(转让给medimmune)、wo2013/181634(申请人:sorrento)和wo2016/061142(申请人:novartis)。
[0290]
在更进一步具体方面,抗体进一步包含人或鼠恒定区。在另一方面,人恒定区选自由igg1、igg2、igg2、igg3、igg4组成的组。在更进一步具体方面,人恒定区是igg1。在又一个方面,鼠恒定区选自由igg1、igg2a、igg2b、igg3组成的组。在另一方面,鼠恒定区为igg2a。
[0291]
在更进一步具体方面,抗体具有降低的或最小的效应子功能。在又一个具体方面,最小的效应子功能来自“无效应子的fc突变”或无糖基化突变。在另一实施例中,无效应子fc突变是恒定区中的n297a或d265a/n297a取代。在一些实施例中,分离的抗pd-l1抗体是去糖基化的。抗体的糖基化通常是n-连接或o-连接的。n-连接是指碳水化合物部分连接至天冬酰胺残基的侧链。三肽序列天冬酰胺-x-丝氨酸和天冬酰胺-x-苏氨酸,其中x是脯氨酸以外的任何氨基酸,是将碳水化合物部分酶促连接至天冬酰胺侧链的识别序列。因此,多肽中
这些三肽序列中任一个的存在产生潜在的糖基化位点。o-连接的糖基化是指糖n-乙酰半乳糖胺、半乳糖或木糖中的一种连接至羟基氨基酸,该羟基氨基酸最通常为丝氨酸或苏氨酸,但也可以使用5-羟脯氨酸或5-羟基赖氨酸。通过改变氨基酸序列以除去上述三肽序列之一(对于n-连接的糖基化位点),可以方便地从抗体上除去糖基化位点。可以通过将糖基化位点内的天冬酰胺、丝氨酸或苏氨酸残基取代为另一个氨基酸残基(例如,甘氨酸、丙氨酸或保守取代)来进行变异。
[0292]
在又一实施例中,本公开提供了包含任何上述抗pd-l1抗体与至少一种药用的载体联合的组合物。可以使用本文所述或本领域已知的任何药用的载体。
[0293]
iv.抗体制备
[0294]
本文所述的抗体是使用本领域可用于产生抗体的技术制备的,其示例性方法在以下部分中更详细地描述。
[0295]
该抗体针对关注的抗原(例如pd-l1,诸如人pd-l1)。优选地,抗原是生物学上重要的多肽,并且向患有病症的哺乳动物施用抗体可以在该哺乳动物中产生治疗益处。
[0296]
在某些实施例中,本文提供的抗体的解离常数(kd)为≤1μm、≤150nm、≤100nm、≤50nm、≤10nm、≤1nm、≤0.1nm、≤0.01nm或≤0.001nm(例如,10-8m或更低,例如10-8m至10-13m,例如,10-9m至10-13m)。
[0297]
在一个实施例中,通过用如以下测定所述的fab形式的目标抗体及其抗原进行放射性标记的抗原结合测定(ria)来测量kd。通过在未标记的抗原滴定系列存在下用最小浓度(125i)标记的抗原平衡fab,然后用抗fab抗体包被的板捕获结合的抗原,来测量fab对抗原的溶液结合亲和力(参见,例如,chen等人,j.mol.biol.293:865-881(1999))。为了确定用于测定的条件,用在50mm碳酸钠(ph 9.6)中5μg/ml捕获抗fab抗体(cappel labs)包被微孔板(thermo scientific)过夜,随后在室温(大约23℃)下用在pbs中2%(w/v)牛血清白蛋白阻断二至五小时。在非吸附板(nunc#269620)中,将100pm或26pm[125i]-抗原与目的fab的系列稀释液混合。然后将目的fab孵育过夜;然而,孵育可以持续更长时间(例如,约65小时)以确保达到平衡。此后,将混合物转移至捕获板以在室温下孵育(例如,一小时)。随后移除溶液并且用在pbs中的0.1%聚山梨酯20洗涤该板八次。当板已干燥时,添加150μl/孔的闪烁体(microscint-20tm;packard),并且在topcount tmγ计数器(packard)上对板计数十分钟。选择给出小于或等于20%最大结合的各fab的浓度以用于竞争性结合测定中。
[0298]
根据另一实施例,在25℃下,用经固定化的抗原cm5芯片,在大约10响应单位(ru)下,使用-2000或-3000(biacore,inc.,piscataway,nj),通过表面等离子体共振测定来测量kd。简而言之,根据供应商说明书,用n-乙基-n'-(3-二甲基氨基丙基)-碳化二亚胺盐酸盐(edc)及n-羟基琥珀酰亚胺(nhs)激活羧甲基化的葡聚糖生物感测器芯片(cm5,biacore inc.)。将抗原用10mm醋酸钠ph 4.8稀释至5μg/ml(约0.2μm),之后以5μl/分钟的流量进行注射以获得大约10响应单位(ru)的偶联蛋白。注射抗原之后,注射1m乙醇胺以阻断未反应的基团。关于动力学测量,在25℃,以约25μl/min的流速将fab的两倍连续稀释液(0.78nm至500nm)注射在含有0.05%聚山梨酯20(tween 20tm)表面活性剂(pbst)的pbs中。通过同时拟合缔合和解离传感图,使用简单的一对一朗缪尔结合模型(评估软件3.2版)计算缔合速率(kon)和解离速率(koff)。平衡解离常数
(kd)计算为比率koff/kon。参见例如chen,y.等人,j.mol.biol.293:865-881(1999)。若通过上述表面等离子体共振测定得出缔合速率超过106m-1s-1,则可通过使用荧光淬灭技术测定缔合速率,即如在分光计诸如配备止流装置的分光光度计(aviv instruments)或8000系列slm-aminco tm分光光度计(thermospectronic)中用搅拌比色杯所测得的,在浓度渐增的抗原存在下,测量在25℃pbs ph 7.2中的20nm抗抗原抗体(fab形式)的荧光发射强度(激发=295nm;发射=340nm,16nm带通)的增加或减少。
[0299]
嵌合抗体、人源化抗体和人抗体
[0300]
在某些实施例中,本文提供的抗体是嵌合抗体。某些嵌合抗体描述于,例如,美国专利号4,816,567和morrison等人,proc.natl.acad.sci.usa,81:6851-6855(1984)中。在一个实例中,嵌合抗体包含非人可变区(例如,源自小鼠、大鼠、仓鼠、兔或非人灵长类动物(诸如猴)的可变区)和人恒定区。在另一个实例中,嵌合抗体为其中类别或亚类已经与亲本抗体的类别或亚类改变的“类别转换”抗体。嵌合抗体包括其抗原结合片段。
[0301]
在某些实施例中,嵌合抗体是人源化抗体。通常,将非人抗体人源化以减少对人的免疫原性,同时保留亲本非人抗体的特异性和亲和力。通常,人源化抗体包含一个或多个可变结构域,其中hvr,例如cdr(或其部分)源自非人抗体,而fr(或其部分)源自人抗体序列。人源化抗体任选地还将包含人恒定区的至少一部分。在一些实施例中,人源化抗体中的一些fr残基被来自非人抗体(例如,hvr残基所来源于的抗体)的相应残基取代,例如以恢复或改善抗体特异性或亲和力。
[0302]
人源化抗体及其制备方法在例如almagro and fransson,front.biosci.13:1619-1633(2008)中综述,并且进一步描述于例如riechmann等人,nature 332:323-329(1988);queen等人,proc.nat’l acad.sci.usa86:10029-10033(1989);美国专利号5,821,337、7,527,791、6,982,321和7,087,409;kashmiri等人,methods 36:25-34(2005)(描述了sdr(a-cdr)移植);padlan,mol.immunol.28:489-498(1991)(描述了“表面再塑”);dall’acqua等人,methods 36:43-60(2005)(描述了“fr改组”);以及osbourn等人,methods 36:61-68(2005)和klimka等人,br.j.cancer,83:252-260(2000)(描述了用于fr改组的“指导选择”方法)中。
[0303]
可用于人源化的人框架区包括但不限于:使用“最佳拟合”方法选择的框架区(参见例如sims等人j.immunol.151:2296(1993));来源于轻链或重链可变区的特定亚组的人抗体的共有序列的框架区(参见,例如,carter等人proc.natl.acad.sci.usa,89:4285(1992);以及presta等人j.immunol.,151:2623(1993));人成熟(体细胞突变)框架区或人种系框架区(参见,例如,almagro and fransson,front.biosci.13:1619-1633(2008));以及来源于筛选fr文库的框架区(参见,例如,baca等人,j.biol.chem.272:10678-10684(1997)和rosok等人,j.biol.chem.271:22611-22618(1996))。
[0304]
在某些实施例中,本文提供的抗体是人抗体。可以使用本领域已知的各种技术来产生人抗体。人抗体一般描述于van dijk and van de winkel,curr opin pharmacol.5:368-74(2001)和lonberg,curr opin immunol.20:450-459(2008)中。
[0305]
可以通过以下方式来制备人抗体:将免疫原施用于转基因动物,所述转基因动物已被修饰以响应于抗原激发而产生具有人可变区的完整人抗体或完整抗体。此类动物通常含有全部或部分人免疫球蛋白基因座,所述全部或部分人免疫球蛋白基因座替代内源性免
疫球蛋白基因座,或者在动物的染色体外存在或随机整合至动物的染色体中。在此类转基因小鼠中,内源性免疫球蛋白基因座通常已被灭活。关于从转基因动物获得人抗体的方法的综述,参见lonberg,nat.biotech.23:1117-1125(2005)。也参见,例如,描述xenomousetm技术的美国专利号6,075,181和6,150,584;描述技术的美国专利号5,770,429;描述k-m 技术的美国专利号7,041,870,以及描述技术的美国专利申请公开号us 2007/0061900。可以进一步修饰来自由此类动物产生的完整抗体的人可变区,例如通过与不同的人恒定区组合。
[0306]
人抗体也可以通过基于杂交瘤的方法制备。已经描述了用于产生人单克隆抗体的人骨髓瘤和小鼠-人杂交骨髓瘤细胞系。(参见例如kozbor j.immunol.,133:3001(1984);brodeur等人,monoclonal antibody production techniques and applications,第51-63页(marcel dekker,inc.,new york,1987);以及boerner等人,j.immunol.,147:86(1991)。)经由人b细胞杂交瘤技术产生的人抗体也如li等人,proc.natl.acad.sci.usa,103:3557-3562(2006)中所述。另外的方法包括例如在美国专利号7,189,826(描述了从杂交瘤细胞系产生单克隆人igm抗体)和ni,xiandai mianyixue,26(4):265-268(2006)(描述了人-人杂交瘤)中描述的那些方法。人类杂交瘤技术(trioma技术)也描述于vollmers和brandlein,histology and histopathology,20(3):927-937(2005)和vollmers和brandlein,methods and findings in experimental and clinical pharmacology,27(3):185-91(2005)中。
[0307]
人抗体还可以通过分离选自人源噬菌体展示文库的fv克隆可变结构域序列产生。然后可以将此类可变结构域序列与预期的人恒定结构域结合。从抗体文库中选择人抗体的技术描述如下。
[0308]
抗体片段
[0309]
抗体片段可以通过传统方法(诸如酶消化)或通过重组技术产生。在某些情况下,使用抗体片段而不是全抗体具有优势。片段的较小尺寸允许快速清除,并可以改善对实体瘤的进入。关于某些抗体片段的综述,参见hudson等人(2003)nat.med.9:129-134。
[0310]
已经开发了用于产生抗体片段的各种技术。传统上,这些片段通过完整抗体的蛋白水解消化而获得(参见,例如,morimoto等人,journal of biochemical and biophysical methods 24:107-117(1992)和brennan等人,science,229:81(1985))。然而,这些片段现在可以直接由重组宿主细胞产生。fab、fv和scfv抗体片段均可在大肠杆菌中表达并从大肠杆菌中分泌出来,因此可轻松生产大量这些片段。可以从以上讨论的抗体噬菌体文库中分离抗体片段。替代地,可以直接从大肠杆菌中回收fab'-sh片段并化学偶联以形成f(ab')2片段(carter等人,bio/technology 10:163-167(1992))。根据另一种方法,可以直接从重组宿主细胞培养物中分离f(ab')2片段。具有增加的体内半衰期的包含挽救受体结合表位残基的fab和f(ab')2片段描述于美国专利号5,869,046中。产生抗体片段的其它技术对熟练技术人员将是显而易见的。在某些实施例中,抗体为单链fv片段(scfv)。参见wo 93/16185;美国专利号5,571,894和5,587,458。fv和scfv是具有完整结合位点没有恒定区的仅有的种类;因此,它们可能适合于在体内使用期间减少非特异性结合。可以构建scfv融合蛋白以在scfv的氨基末端或羧基末端产生效应蛋白的融合。参见antibody engineering,ed.borrebaeck,同上。例如,该抗体片段也可以是“线性抗体”,例如美国专利
号5,641,870中所述。这样的线性抗体可以是单特异性或双特异性的。
[0311]
单结构域抗体
[0312]
在一些实施例中,本公开的抗体是单结构域抗体。单结构域抗体为包含抗体的全部或部分重链可变结构域或全部或部分轻链可变结构域的单个多肽链。在某些实施例中,单结构域抗体为人单结构域抗体(domantis,inc.,waltham,mass.;参见例如美国专利号6,248,516b1)。在一个实施例中,单结构域抗体由抗体的全部或部分重链可变结构域组成。
[0313]
抗体变体
[0314]
在一些实施例中,考虑了本文描述的抗体的氨基酸序列修饰。例如,可能期望改善抗体的结合亲和力和/或其他生物特性。抗体的氨基酸序列变体可以通过向编码抗体的核苷酸序列中引入适当的改变或通过肽合成来制备。此类修饰包括例如抗体氨基酸序列内残基的缺失和/或插入和/或取代。可以进行缺失、插入和取代的任何组合以获得最终构建体,前提条件是所述最终构建体具有所需特性。可以在形成序列时将氨基酸改变引入受试抗体的氨基酸序列中。
[0315]
取代、插入和缺失变体
[0316]
在某些实施例中,提供了具有一或多个氨基酸取代的抗体变体。用于取代突变的目的位点包括hvr和fr。表a中显示了保守性取代。下面参考氨基酸侧链类别进一步描述了更实质性的变化。可以将氨基酸取代引入目标抗体中,并对产物进行所需活性(例如保留/改善的抗原结合、降低的免疫原性,或改善的adcc或cdc)筛选。
[0317]
表a.保守性取代。
[0318][0319]
可根据共同的侧链特性将氨基酸分组:
[0320]
a.疏水性:正亮氨酸、met、ala、val、leu、ile;
[0321]
b.中性亲水性:cys、ser、thr、asn、gln;
[0322]
c.酸性:asp、glu;
[0323]
d.碱性:his、lys、arg;
[0324]
e.残基,其影响链取向:gly、pro;
[0325][0326]
f.芳香族:trp、tyr、phe。
[0327]
非保守性取代将需要用这些类别中的一个的成员交换另一类别。
[0328]
一种类型的取代变体涉及取代亲本抗体(例如,人源化抗体或人抗体)的一个或多个高可变区残基。通常,相对于亲本抗体,选为用于进一步研究的一个或多个所得变体将在某些生物学特性方面(例如,亲和力增加、免疫原性降低)有改变(例如,改善)和/或将基本上保留亲本抗体的某些生物学特性。示例性取代变体是亲和力成熟抗体,其可例如使用诸如本文所述的那些基于噬菌体展示的亲和力成熟技术方便地生成。简而言之,将一个或多
个hvr残基突变并且将变体抗体展示在噬菌体上并针对特定生物活性(例如结合亲和力)进行筛选。
[0329]
例如,可改变(例如,取代)hvr,以改善抗体亲和力。可在hvr“热点”中作出此类改变,即,由在体细胞成熟过程期间经历高频突变的密码子编码的残基(参见例如,chowdhury,methods mol.biol.207:179-196(2008))和/或sdr(a-cdr),其中对所得变体vh或vl进行结合亲和力测试。通过构建并自二级文库重新选择而实现的亲和力成熟已被例如hoogenboom等人在methods in molecular biology 178:1-37(o'brien等人编,human press,totowa,nj,(2001))中进行描述。在亲和力成熟的一些实施例中,利用多种方法(例如,易错pcr、链改组或寡核苷酸定点诱变基因)中的任一种将多样性引入选择用于成熟的可变基因中。然后创建一个二级文库。然后筛选该文库以鉴定具有所需亲和力的任何抗体变体。引入多样性的另一种方法涉及hvr定向方法,其中将若干hvr残基(例如,每次4-6个残基)随机化。参与抗原结合的hvr残基可例如使用丙氨酸扫描突变或建模来特异性地鉴定。特别是cdr-h3和cdr-l3经常成为目标。
[0330]
在某些实施例中,取代、插入或缺失可发生在一个或多个hvr内,只要此类改变基本上不降低抗体的抗原结合能力即可。例如,可在hvr中进行基本上不降低结合亲和力的保守性改变(例如,如本文提供的保守性取代)。此类改变可在hvr“热点”或sdr之外。在上文提供的变体vh和vl序列的某些实施例中,每个hvr保持不变,或包含不超过一个、两个或三个氨基酸取代。
[0331]
可用于鉴别可被靶向突变的抗体残基或区域的方法称作“丙氨酸扫描突变”,如cunningham and wells,(1989)science,244:1081-1085中所述。在此方法中,鉴别残基或一组靶残基(例如,带电残基,诸如arg、asp、his、lys和glu)并用中性或带负电的氨基酸(例如,丙氨酸或多丙氨酸)置换以确定抗体与抗原的相互作用是否受到影响。可在对初始取代展示功能敏感性的氨基酸位置引入其他取代。可替代地或另外地,利用抗原-抗体复合物的晶体结构鉴定抗体与抗原之间的接触点。可靶向或消除作为取代的候选的此类接触残基和相邻残基。可筛选变体以确定它们是否具备期望的特性。
[0332]
氨基酸序列插入包括长度范围为一个残基至含有一百个或更多个残基的多肽的氨基和/或羧基末端融合,以及一个或多个氨基酸残基的序列内插入。末端插入的实例包括具有n末端甲硫氨酰残基的抗体。抗体分子的其他插入变体包括与增加抗体的血清半衰期的酶(例如,对于adept)或多肽的抗体的n末端或c末端的融合。
[0333]
糖基化变体
[0334]
在某些方面,改变本文提供的抗体以增加或降低抗体糖基化的程度。糖基化位点向抗体的添加或缺失可通过改变氨基酸序列以产生或去除一个或多个糖基化位点而方便地实现。
[0335]
当抗体包含fc区时,附接于其上的碳水化合物可以被改变。由哺乳动物细胞产生的天然抗体通常包含具有支链的双触角寡糖,所述双触角寡糖通常通过n-连接连接于fc区的ch2结构域的asn297。参见例如wright等人tibtech 15:26-32(1997)。寡糖可包括各种碳水化合物,例如,甘露糖、n-乙酰基葡糖胺(glcnac)、半乳糖和唾液酸,以及与双触角寡糖结构的“茎”中的glcnac连接的岩藻糖。在一些实施例中,可对本公开的抗体中的寡糖进行修饰,以产生具有某些改善特性的抗体变体。
[0336]
在一个实施例中,提供了包含fc区的抗体变体,其中连接至fc区的碳水化合物结构具有减少的岩藻糖或缺乏岩藻糖,这可以改善adcc功能。具体地,本文考虑相对于在野生型cho细胞中产生的相同抗体上的岩藻糖的量,具有减少的岩藻糖的抗体。也就是说,它们的特征在于其岩藻糖的量比天然cho细胞(例如,产生天然糖基化模式的cho细胞,诸如含有天然fut8基因的cho细胞)产生的岩藻糖的量低。在某些实施例中,该抗体是一种抗体,其中其上少于约50%、40%、30%、20%、10%或5%的n-连接聚糖包含岩藻糖。例如,此类抗体中的岩藻糖的量可以为1%至80%、1%至65%、5%至65%或20%至40%。在某些实施例中,该抗体是一种抗体,其中其上的n-连接聚糖均不包含岩藻糖,即,其中该抗体完全没有岩藻糖,或没有岩藻糖或被去岩藻糖基化。岩藻糖的量通过相对于通过maldi-tof质谱测得的与asn 297连接的所有糖结构(例如,复合、杂合和高甘露糖结构)的总和计算糖链内在asn297处的岩藻糖的平均量来确定,例如,如wo 2008/077546中所述。asn297是指位于fc区中约297位的天冬酰胺残基(fc区残基的eu编号);然而,由于抗体中的微小序列变化,asn297也可以位于297位上游或下游大约
±
3个氨基酸,即在294位和300位之间。此类岩藻糖基化变体可具有改善的adcc功能。参见例如美国专利公开号us 2003/0157108(presta,l.);us 2004/0093621(kyowa hakko kogyo co.,ltd)。有关“去岩藻糖化”或“岩藻糖缺陷型”抗体变体包括:us 2003/0157108;wo 2000/61739;wo 2001/29246;us 2003/0115614;us 2002/0164328;us 2004/0093621;us 2004/0132140;us 2004/0110704;us 2004/0110282;us 2004/0109865;wo 2003/085119;wo 2003/084570;wo 2005/035586;wo 2005/035778;wo2005/053742;wo2002/031140;okazaki等人,j.mol.biol.336:1239-1249(2004);yamane-ohnuki等人,biotech.bioeng.87:614(2004)。能够产生去岩藻糖基化抗体的细胞系的实例包括蛋白岩藻糖基化缺陷的lec13 cho细胞(ripka等人arch.biochem.biophys.249:533-545(1986);美国专利申请号us 2003/0157108 a1,presta,l;和wo 2004/056312 a1,adams等人,特别是实例11),和敲除细胞系诸如α-1,6-岩藻糖基转移酶基因(fut8)敲除的cho细胞(参见例如,yamane-ohnuki等人biotech.bioeng.87:614(2004);kanda,y.等人,biotechnol.bioeng.,94(4):680-688(2006);和wo2003/085107)。
[0337]
进一步提供了包含两分型寡糖的抗体变体,例如,其中连接至抗体的fc区的双角寡糖被glcnac两分。此类抗体变体可具有减少的岩藻糖基化和/或改善的adcc功能。此类抗体变体的实例描述于例如wo 2003/011878(jean-mairet等人);美国专利号6,602,684(umana等人);us 2005/0123546(umana等人);以及ferrara等人,biotechnology and bioengineering,93(5):851-861(2006)中。还提供了在连接于fc区的寡糖中具有至少一个半乳糖残基的抗体变体。这样的抗体变体可以具有改善的cdc功能。此类抗体变体描述于例如wo 1997/30087(patel等人);wo 1998/58964(raju,s.);以及wo 1999/22764(raju,s.)中。
[0338]
在某些实施例中,包含本文所述的fc区的抗体变体能够结合fcγriii。在某些实施例中,与包含人野生型igg1 fc区的其它相同抗体相比,包含本文所述的fc区的抗体变体在人效应细胞存在下具有adcc活性,或在人效应细胞存在下具有增加的adcc活性。
[0339]
fc区变体
[0340]
在某些实施例中,一个或多个氨基酸修饰可引入本文提供的抗体的fc区中,从而
生成fc区变体。fc区变体可包含人fc区序列(例如人igg1、igg2、igg3或igg4 fc区),其在一个或多个氨基酸位置上包含氨基酸修饰(例如取代)。
[0341]
在某些实施例中,本公开考虑了具有一些但不是全部效应子功能的抗体变体,这使其成为应用的期望候选物,其中体内的抗体的半衰期为重要的而某些效应子功能(诸如补体和adcc)为不必要的或有害的。可以进行体外和/或体内细胞毒性测定,以确认cdc和/或adcc活性的降低/消耗。例如,可以进行fc受体(fcr)结合测定以确保抗体缺乏fcγr结合(因此可能缺乏adcc活性),但是保留fcrn结合能力。介导adcc的主要细胞nk细胞仅表达fcγriii,而单核细胞表达fcγri、fcγrii和fcγriii。造血细胞上的fcr表达总结在ravetch和kinet,《免疫学年评》(annu.rev.immunol.)9:457-492(1991)的第464页的表3中。用于评估目的分子的adcc活性的体外测定的非限制性示例描述于美国专利号5,500,362(参见,例如,hellstrom,i.等人proc.nat’l.acad.sci.usa83:7059-7063(1986))和hellstrom,i等人,proc.nat’l.acad.sci.usa82:1499-1502(1985);5,821,337(参见bruggemann,m.等人,j.exp.med.166:1351-1361(1987))中。替代地,可以采用非放射性测定方法(参见,例如,用于流式细胞术的acti
tm
非放射性细胞毒性测定(celltechnology,inc.mountain view,ca);以及cytotox非放射性细胞毒性测定(promega,madison,wi))。用于此类测定的有用效应物细胞包括外周血单核细胞(pbmc)和自然杀伤(nk)细胞。替代地或另外地,所关注分子的adcc活性可以在体内评估,例如,在动物模型(诸如在clynes等人proc.nat’l.acad.sci.usa 95:652-656(1998)中所公开的)中评估。也可以进行c1q结合测定以确认抗体不能结合c1q,因此缺乏cdc活性。参见,例如,wo 2006/029879和wo 2005/100402中的c1q和c3c结合elisa。为了评估补体活化,可以执行cdc测定(参见例如gazzano-santoro等人,j.immunol.methods 202:163(1996);cragg,m.s.等人,blood 101:1045-1052(2003);以及cragg,m.s.and m.j.glennie,blood 103:2738-2743(2004))。fcrn结合和体内清除/半衰期测定也可以使用本领域已知的方法执行(参见例如petkova,s.b.等人,int'l.immunol.18(12):1759-1769(2006))。
[0342]
具有降低的效应物功能的抗体包括具有fc区残基238、265、269、270、297、327和329中一个或多个的取代的那些(美国专利号6,737,056)。此类fc突变体包括在第265、269、270、297和327位氨基酸中两个或多个处具有取代的fc突变体,包括所谓的“dana”fc突变体,其残基265和297被取代为丙氨酸(美国专利7,332,581)。
[0343]
描述了具有改善的或降低的与fcr的结合的某些抗体变体。(参见例如,美国专利号6,737,056;wo 2004/056312,以及shields等人,j.biol.chem.9(2):6591-6604(2001)。)
[0344]
在某些实施例中,抗体变体包含具有一个或多个氨基酸取代的fc区,取代改善adcc,例如,在fc区的298、333和/或334位(残基的eu编号)处的取代。在一个示例性的实施例中,该抗体在其fc区中包含以下氨基酸取代:s298a、e333a和k334a。
[0345]
在一些实施例中,例如,如美国专利号6,194,551、wo 99/51642和idusogie等人j.immunol.164:4178-4184(2000)中所述,在fc区中进行改变,导致改变(即,改善或减少)的c1q结合和/或补体依赖性细胞毒性(cdc)。
[0346]
具有延长的半衰期和改善的新生儿fc受体(fcrn)结合的抗体描述于us2005/0014934a1(hinton等人)中,该新生儿fc受体负责将母体igg转移至胎儿(guyer等人,j.immunol.117:587(1976)和kim等人,j.immunol.24:249(1994))。那些抗体包含这样的fc
区,所述fc区中具有改善fc区与fcrn的结合的一个或多个取代。此类fc变体包括在以下fc区残基中的一处或多处具有取代的fc变体:238、256、265、272、286、303、305、307、311、312、317、340、356、360、362、376、378、380、382、413、424或434,例如对fc区残基434的取代(美国专利号7,371,826)。也参见duncan&winter,nature 322:738-40(1988);美国专利号5,648,260;美国专利号5,624,821;和wo 94/29351关于fc区变体的其它实例。
[0347]
v.药物组合物和制剂
[0348]
本文还提供了例如用于治疗癌症的药物组合物和制剂,其包含抗pd-l1抗体(例如,阿特珠单抗)。在一些实施例中,药物组合物和制剂进一步包含药用载体。
[0349]
在一些实施例中,本文所述的抗pd-l1抗体(诸如阿特珠单抗)处于制剂中,该制剂包含约60mg/ml量的抗体、约20mm浓度的乙酸组氨酸、约120mm浓度的蔗糖和0.04%(w/v)浓度的聚山梨酸酯(例如聚山梨酸酯20),且制剂的ph为约5.8。在一些实施例中,本文所述的抗pd-l1抗体(诸如阿特珠单抗)处于制剂中,该制剂包含约125mg/ml量的抗体、约20mm浓度的乙酸组氨酸、约240mm浓度的蔗糖和0.02%(w/v)浓度的聚山梨酸酯(例如聚山梨酸酯20),且制剂的ph为约5.5。
[0350]
在制备目的抗体后(例如,生产可以如本文所公开地配制的抗体的技术在本文中阐述并且是本领域已知的),制备了包含它的药物制剂。在某些实施例中,待配制的抗体未经预先冻干,并且本文目标制剂是水性制剂。在某些实施例中,抗体是全长抗体。在一个实施例中,制剂中的抗体是抗体片段,诸如f(ab')2,在这种情况下,可能需要解决使用全长抗体时可能不会发生的问题(诸如将抗体剪接到fab)。制剂中存在的抗体的治疗有效量例如通过考虑所需的剂量体积和施用方式来确定。约25mg/ml至约150mg/ml、或约30mg/ml至约140mg/ml、或约35mg/ml至约130mg/ml、或约40mg/ml至约120mg/ml、或约50mg/ml至约130mg/ml、或约50mg/ml至约125mg/ml、或约50mg/ml至约120mg/ml、或约50mg/ml至约110mg/ml、或约50mg/ml至约100mg/ml、或约50mg/ml至约90mg/ml、或约50mg/ml至约80mg/ml、或约54mg/ml至约66mg/ml是制剂中的示例性抗体浓度。在一些实施例中,本文所述的抗pd-l1抗体(诸如阿特珠单抗)以约1200mg的剂量施用。
[0351]
制备在ph缓冲溶液中包含抗体的水性制剂。在一些实施例中,本公开的缓冲剂的ph在约5.0至约7.0的范围内。在某些实施例中,ph在约5.0至约6.5的范围内、ph在约5.0至约6.4的范围内、在约5.0至约6.3的范围内、ph在约5.0至约6.2的范围内、ph在约5.0至约6.1的范围内、ph在约5.5至约6.1的范围内、ph在约5.0至约6.0的范围内、ph在约5.0至约5.9的范围内、ph在约5.0至约5.8的范围内、ph在约5.1至约6.0的范围内、ph在约5.2至约6.0的范围内、ph在约5.3至约6.0、ph在约5.4至约6.0的范围内、ph在约5.5至约6.0的范围内、ph在约5.6至约6.0的范围内、ph在约5.7至约6.0的范围内、或ph在约5.8至约6.0的范围内。在一些实施例中,制剂的ph值为6.0或约6.0。在一些实施例中,制剂的ph值为5.9或约5.9。在一些实施例中,制剂的ph值为5.8或约5.8。在一些实施例中,制剂的ph值为5.7或约5.7。在一些实施例中,制剂的ph值为5.6或约5.6。在一些实施例中,制剂的ph值为5.5或约5.5。在一些实施例中,制剂的ph值为5.4或约5.4。在一些实施例中,制剂的ph值为5.3或约5.3。在一些实施例中,制剂的ph值为5.2或约5.2。将ph控制在该范围内的缓冲剂的实例包括组氨酸(例如l-组氨酸)或乙酸钠。在某些实施例中,缓冲剂包含浓度为约15mm至约25mm的乙酸组氨酸或乙酸钠。在一些具体实施例中,缓冲剂包含乙酸组氨酸或乙酸钠,其浓度为
约15mm至约25mm、约16mm至约25mm、约17mm至约25mm、约18mm至约25mm、约19mm至约25mm、约20mm至约25mm、约21mm至约25mm、约22mm至约25mm、约15mm、约16mm、约17mm、约18mm、约19mm、约20mm、约21mm、约22mm、约23mm、约24mm或约25mm。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.0。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.1。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.2。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.3。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.4。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.5。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.6。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.7。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.8。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 5.9。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 6.0。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph6.1。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 6.2。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约20mm,ph 6.3。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.2。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.3。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.4。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.5。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.6。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.7。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.8。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 5.9。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 6.0。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 6.1。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 6.2。在一实施例中,缓冲剂是乙酸组氨酸或乙酸钠,其量为约25mm,ph 6.3。
[0352]
在一些实施例中,缓冲剂的浓度为约60mm至约240mm。在一些实施例中,制剂中的蔗糖为约60mm至约230mm、约60mm至约220mm、约60mm至约210mm、约60mm至约200mm、约60mm至约190mm、60mm至约180mm、约60mm至约170mm、约60mm至约160mm、约60mm至约150mm、约60mm至约140mm、约80mm至约240mm、约90mm至约240mm、约100mm至约240mm、约110mm至约240mm、约120mm至约240mm、约130mm至约240mm、约140mm至约240mm、约150mm至约240mm、约160mm至约240mm、约170mm至约240mm、约180mm至约240mm、约190mm至约240mm、约200mm至约240mm、约80mm至约160mm、约100mm至约140mm、或约110mm至约130mm。在一些实施例中,制剂中的蔗糖为约60mm、约70mm、约80mm、约90mm、约100mm、约110mm、约120mm、约130mm、约140mm、约150mm、约160mm、约170mm、约180mm、约190mm、约200mm、约210mm、约220mm、约230mm或约240mm。
[0353]
在一些实施例中,制剂中的抗体浓度为约40mg/ml至约125mg/ml。在一些实施例中,制剂中的抗体浓度为约40mg/ml至约120mg/ml、约40mg/ml至约110mg/ml、约40mg/ml至约100mg/ml、约40mg/ml至约90mg/ml、约40mg/ml至约80mg/ml、约40mg/ml至约70mg/ml、约50mg/ml至约120mg/ml、约60mg/ml至约120mg/ml、约70mg/ml至约120mg/ml、约80mg/ml至约120mg/ml、约90mg/ml至约120mg/ml或约100mg/ml至约120mg/ml。在一些实施例中,制剂中
的抗体浓度为约60mg/ml。在一些实施例中,制剂中的抗体浓度为约65mg/ml。在一些实施例中,制剂中的抗体浓度为约70mg/ml。在一些实施例中,制剂中的抗体浓度为约75mg/ml。在一些实施例中,制剂中的抗体浓度为约80mg/ml。在一些实施例中,制剂中的抗体浓度为约85mg/ml。在一些实施例中,制剂中的抗体浓度为约90mg/ml。在一些实施例中,制剂中的抗体浓度为约95mg/ml。在一些实施例中,制剂中的抗体浓度为约100mg/ml。在一些实施例中,制剂中的抗体浓度为约110mg/ml。在一些实施例中,制剂中的抗体浓度为约125mg/ml。在一些实施例中,本文所述的抗pd-l1抗体(诸如阿特珠单抗)以约60mg/ml的浓度施用。
[0354]
在一些实施例中,将表面活性剂添加到抗体制剂中。示例性的表面活性剂包括非离子表面活性剂,诸如聚山梨酯(例如聚山梨酯20、80等)或泊洛沙姆(例如泊洛沙姆188等)。添加的表面活性剂的量应使得其减少所配制抗体的聚集和/或最小化制剂中颗粒的形成和/或减少吸附。例如,表面活性剂可以约0.001%至约0.5%(w/v)的量存在于制剂中。在一些实施例中,表面活性剂(例如聚山梨酯20)为约0.005%至约0.2%、约0.005%至约0.1%、约0.005%至约0.09%、约0.005%至约0.08%、约0.005%至约0.07%、约0.005%至约0.06%、约0.005%至约0.05%、约0.005%至约0.04%、约0.008%至约0.06%、约0.01%至约0.06%、约0.02%至约0.06%、约0.01%至约0.05%或约0.02%至约0.04%。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.005%或约0.005%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.006%或约0.006%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.007%或约0.007%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.008%或约0.008%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.009%或约0.009%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.01%或约0.01%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.02%或约0.02%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.03%或约0.03%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.04%或约0.04%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.05%或约0.05%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.06%或约0.06%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.07%或约0.07%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.08%或约0.08%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.1%或约0.1%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.2%或约0.2%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.3%或约0.3%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.4%或约0.4%的量存在于制剂中。在某些实施例中,表面活性剂(例如聚山梨酯20)以0.5%或约0.5%的量存在于制剂中。
[0355]
在一个实施例中,制剂包含上述试剂(例如抗体、缓冲液、蔗糖和/或表面活性剂),并且基本上不含一种或多种防腐剂诸如苯甲醇、苯酚、间甲酚、氯丁醇和苄索铵。在另一实施例中,制剂中可以包含防腐剂,特别是其中制剂为多剂量制剂。防腐剂的浓度可以在约0.1%至约2%的范围内,优选约0.5%至约1%。制剂中可以包含一种或多种其他药用载体、赋形剂或稳定剂,诸如《雷明顿药物科学》(remington's pharmaceutical sciences,第16版,osol,a.编撰(1980))中描述的那些,只要它们不会不利地影响制剂的所需特征即可。可
接受的载体、赋形剂或稳定剂在所采用的剂量和浓度下对受者无毒;并且包括:其他缓冲剂;助溶剂;抗氧化剂,包括抗坏血酸和蛋氨酸;螯合剂,诸如edta;金属络合物(例如锌-蛋白络合物);可生物降解的聚合物,诸如聚酯;和/或成盐抗衡离子。本文的示例性药用载体进一步包括间质药物分散剂,诸如可溶中性活性透明质酸酶糖蛋白(shasegp),例如人可溶性ph-20透明质酸酶糖蛋白,诸如rhuph20(baxter international,inc.)。某些示例性shasegp和使用方法,包括rhuph20,描述于美国专利公开号2005/0260186和2006/0104968中。在一个方面中,将shasegp与一种或多种另外的糖胺聚糖酶(诸如软骨素酶)组合。
[0356]
对于所治疗的特定适应症,本文的制剂还可以包含一种以上的蛋白质,优选具有对其它蛋白质没有不利影响的互补活性的那些蛋白质。例如,当抗体是抗pd-l1(诸如阿特珠单抗)时,可以与另一种药物(例如化疗剂和抗肿瘤剂)联合组合使用。
[0357]
本文所述的药物组合物和制剂可通过将具有所需纯度的活性成分(诸如抗体或多肽)与一种或多种任选的药用载体混合来制备(《雷明顿药学科学》(remington's pharmaceutical sciences),第16版,osol,a.编撰(1980)),以冻干制剂或水溶液的形式。可接受的载体在所采用的剂量和浓度下对受者无毒;并且包括但不限于:缓冲剂,诸如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸;防腐剂(诸如十八烷基二甲基苄基氯化铵;氯化六甲双铵;苯扎氯铵;苄索氯铵;苯酚、丁醇或苄醇;对羟基苯甲酸烷基酯,诸如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇和间甲酚);低分子量(少于约10个残基)多肽;蛋白质,诸如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,诸如聚乙烯吡咯烷酮;氨基酸,诸如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂,诸如edta;糖,诸如蔗糖、甘露醇、海藻糖或山梨糖醇;成盐抗衡离子,诸如钠;金属络合物(例如锌-蛋白络合物);和/或非离子表面活性剂,诸如聚乙二醇(peg)。本文的示例性药用载体进一步包括间质药物分散剂,诸如可溶中性活性透明质酸酶糖蛋白(shasegp),例如人可溶性ph-20透明质酸酶糖蛋白,诸如rhuph20(baxter international,inc.)。某些示例性shasegp和使用方法,包括rhuph20,描述于美国专利公开号2005/0260186和2006/0104968中。在一个方面中,将shasegp与一种或多种另外的糖胺聚糖酶(诸如软骨素酶)组合。
[0358]
示例性的冻干抗体制剂描述于美国专利号6,267,958中。水性抗体制剂包括在美国专利号6,171,586和wo2006/044908中描述的那些,后一者中的制剂包含组氨酸-乙酸盐缓冲剂。
[0359]
本文的制剂和组合物还可含有多于一种对于所治疗的特定适应症是必需的活性成分,优选是具有不会彼此不利地影响的互补活性的活性成分。此类活性成分适当地以对预期目的有效的量组合存在。
[0360]
活性成分可以包埋在例如通过凝聚技术或通过界面聚合而制备的微胶囊(例如分别为羟甲基纤维素或明胶微胶囊和聚(甲基丙烯酸甲酯)微胶囊)中;在胶体药物递送系统(例如,脂质体、白蛋白微球、微乳液、纳米粒子和纳米胶囊)中;或在粗乳液中。此类技术公开于remington’spharmaceutical sciences第16版,osol,a.编辑(1980)中。
[0361]
可以制备缓释制备物。缓释制备物的合适示例包括含有抗体的固态疏水聚合物的
半透性基质,所述基质是例如膜或微胶囊等成型制品的形式。待用于体内施用的制剂通常是无菌的。例如,无菌可以通过无菌过滤膜过滤而容易地实现。
[0362]
vi.制品或试剂盒
[0363]
本文进一步提供一种制品或试剂盒,其包含本公开的抗pd-l1抗体(例如,阿特珠单抗)和带有根据本文所述任何方法使用抗pd-l1抗体的说明书的包装插页。
[0364]
在一些实施例中,抗pd-l1抗体存在于药用载体中。在一些实施例中,抗pd-l1抗体以单位剂量提供。在一些实施例中,单位剂量为840mg。在一些实施例中,单位剂量为840mg,并且该单位剂量在14ml溶液(例如,包含药用载体)中提供。
[0365]
在一些实施例中,抗pd-l1抗体存在于容器中。合适的容器包括例如瓶、小瓶、袋子和注射器。容器可以由多种材料形成,例如玻璃、塑料(诸如聚氯乙烯、聚乙烯或聚烯烃)或金属合金(诸如不锈钢或哈氏合金)。在一些实施例中,容器容纳制剂,容器上或与容器相关的标签可以指示使用说明。制品或试剂盒还可以包括从商业和用户角度出发期望的其它材料,包括其它缓冲剂、稀释剂、过滤器、针头、注射器和带有使用说明的包装插页。在一些实施例中,制品还包括一种或多种其他试剂(例如化疗剂和抗肿瘤剂)。用于一种或多种试剂的合适容器包括例如瓶、小瓶、袋子和注射器。
[0366]
实例
[0367]
前述书面说明书被认为足以使本领域技术人员能够实施本发明。提供以下实例仅用于说明性目的并且不旨在以任何方式限制本发明的范围。事实上,除了本文中示出和描述的之外,本发明的各种修改对于根据说明书前文的本领域技术人员而言将变得显而易见,并且落入所附权利要求的范围内。
[0368]
概述
[0369]
靶向程序性死亡配体1(pd-l1)或程序性死亡-1(pd-1)的免疫检查点抑制已成为治疗多种人类癌症的重要方法,因为pd-l1在肿瘤细胞和肿瘤浸润免疫细胞上的表达可以抑制抗癌免疫应答(chen等人,(2013)immunicty doi:10.1016/j.immuni.2013.07.012)。阿特珠单抗是一种人源化、工程化的单克隆免疫球蛋白(ig)g1抗体,选择性地靶向pd-l1以阻断与其受体的相互作用,从而促进t细胞活化并重振和增强抗癌活性,同时保留pd-l2与pd-1之间的相互作用完整(chen等人,(2013)immunicty doi:10.1016/j.immuni.2013.07.012;chen等人,(2012)clin cancer res doi:10.1158/1078-0432.ccr-12-1362;herbst等人,(2014)nature doi:10.1038/nature14011)。阿特珠单抗在美国、欧洲和其他地方被批准用于治疗某些类型的局部晚期或转移性非小细胞肺癌(nsclc)和尿路上皮癌(uc),并且在美国被批准用于治疗局部晚期或转移性三阴性乳腺癌(tnbc)和广泛期小细胞肺癌(sclc)(tecentriq(阿特珠单抗)[包装插页]。south san francisco,ca:基因泰克公司;2019年。south san francisco,ca,usa:基因泰克公司;tecentriq(阿特珠单抗)[产品特性总结]welwyn garden city,uk: 罗氏注册有限公司;2018)。uc和nsclc阿特珠单抗单药治疗适应症以及nsclc和sclc阿特珠单抗联合治疗适应症首次被批准进行1200mg q3w的iv输注。
[0370]
确定可以互换使用的替代给药方案将为患者的癌症治疗提供更大的便利,特别是对于具有不同给药要求的联合方案。
[0371]
以下实施例描述了确定晚期非小细胞肺癌(nsclc)或尿路上皮癌(uc)患者中阿特
珠单抗暴露与疗效或安全性之间的暴露-应答(er)关系的研究,并确定替代给药方案。具体而言,基于来自九项临床研究的可用于二线(2l)非小细胞肺癌(nsclc)以及一线(1l)不符合进行顺铂-治疗条件的和2l转移性尿路上皮癌(uc)中的阿特珠单抗的综合临床药理学信息,以下实例提供阿特珠单抗单药治疗的药代动力学(pk)建模和模拟预测(表1a和表1b)。
[0372]
这些研究的目标是确定阿特珠单抗的疗效和安全性的er关系,并应用这些知识以及群体pk(poppk)模拟和阿特珠单抗的已知安全性特征,以鉴定替代给药方案。
[0373]
本文所述的结果表明,经批准的1200mg q3w给药方案的阿特珠单抗暴露和暴露-应答(er)关系(首次施用为历时60分钟以静脉输注施用,且然后,如果患者耐受,施用历时30分钟的后续输注)与本文公开的1680mg q4w和840q2w给药方案(首次施用为历时60分钟以静脉输注施用,且然后,如果患者耐受,施用历时30分钟的后续输注)相当。基于研究pcd4989g、研究go28915(oak)和研究go29294(imvigor211)数据的安全性分析和免疫原性数据也支持新的840mg q2w和1680mg q4w给药方案。
[0374]
[0375][0376]
1l=一线;2l=二线;2l+=二线及以上;muc=转移性尿路上皮癌;nsclc=非-小细胞肺癌;orr=总应答率;q3w=每3周;os=总存活;pk=药代动力学。
[0377]a不符合进行顺铂治疗的条件的患者
[0378]b对于随机研究(即imvigor211、poplar、oak),入组人数包括纳入阿特珠单抗组的患者
[0379]
表1b.阿特珠单抗研究总结
[0380]
[0381][0382][0383]
实例1
[0384]
阿特珠单抗单一疗法的药代动力学性质
[0385]
在本实例中,在单一疗法设置中进行的八项阿特珠单抗研究中比较了阿特珠单抗
的药代动力学(pk)特征(见表1)。关键pk特征诸如c
min
、c
max
和auc是基于使用固定的1200mg q3w剂量的临床研究计算的,并针对固定的1680mg q4w和840mg q2w剂量进行估计。重要的患者特征也作为潜在的协变量而被分析。
[0386]
阿特珠单抗pk在1至20mg/kg的阿特珠单抗剂量范围内呈线性,包括固定的1200mg阿特珠单抗剂量。阿特珠单抗pk似乎跨研究具有可比性,如在相同剂量水平下观察到的相似的周期1中c
max
和c
min
所示(表2)。
[0387]
表2.pcd4989g、jo28944、imvigor210、imvigor211、birch、poplar、fir和oak的周期1中的阿特珠单抗血清pk参数的汇总统计
[0388][0389][0390]
方法
[0391]
软件
[0392]
在一些实施例中,在本实例和本文提供的所有其他实例中,使用了以下软件工具和方法。使用r 3.4.3版和综合r档案网络包执行数据集准备、探索、可视化和分析,包括描述性统计。使用具有交互作用的一阶条件估计算法进行非线性混合效应建模(非线性混合效应建模工具[nonmem]7.3版;icon development solutions,ellicott city,md,usa)(beal等人,(2011)nonmem用户指南。(1989-2011))用于个体pk参数的贝叶斯估计。逻辑回
归使用r中的广义线性模型函数和族“二项式”(方差=二项式;链接=logit)。蒙特卡罗pk模拟是使用nonmem 7.3版实现的,要评估的模拟数据集是使用r创建的。
[0393]
poppk模型
[0394]
首先基于两项临床研究的i期数据(“i期poppk模型”)评估了阿特珠单抗的群体pk(poppk):研究pcd4989g和研究jo28944。随后,i期poppk模型分别对uc和nsclc进行了外部验证,分别使用在imvigor210和imvigor211中收集的用于uc的pk数据和在birch、poplar、fir和oak中收集的用于nsclc的数据。
[0395]
分析中使用的数据
[0396]
对于i期poppk模型,在来自研究pcd4989g和jo28944的472例患者的4563个样品中评估了阿特珠单抗在血清中的药代动力学。
[0397]
poppk模型使用来自以下各项的阿特珠单抗血清pk样品进行外部验证:来自imvigor210的423例患者(占接受治疗的429例患者的98.6%)的1251个样品;来自birch、poplar和fir的920例患者(占接受治疗的938例患者的98.1%)的3891个样品;来自oak的596例患者(占接受治疗的608例患者的98%)的2754个样品;以及来自imvigor211的455例患者(占接受治疗的467例患者的97%)的1939个样品。
[0398]
基本群体pk模型
[0399]
对于i期poppk模型,使用具有nonmem 7版本7.3(icon,maryland)的具有交互作用的一阶条件估计方法的非线性混合效应方法来开发基本poppk模型。几个候选模型拟合pk数据。评估了各种残差omega矩阵模型(块:考虑iiv之间的相关性;对角线:相互独立的iiv)。使用michaelis-menten模型评估药代动力学的非线性。
[0400]
协变量的选择
[0401]
对于i期poppk模型,一旦基本模型最终确定,就对协变量对于主要pk参数的潜在影响进行评估。
[0402]
在第一步中,针对分析中包含的协变量绘制由群体基础pk模型生成的pk参数的随机效应,以定性地评估相关性的程度。散点图用于检查连续变量的影响,箱线图用于检查分类变量的影响。
[0403]
在第二步中,正式的协变量分析涉及一种前向加性包容(forward additive inclusion)和后向消元的逐步方法,其中结构模型用作基线,并且协变量模型变得越来越复杂。在每个模型估计之后,评估协变量以查看哪个导致了目标函数值(ofv)的最大改进大于阈值(一个自由度的
△
ofv》-6.64,显著性水平为p《0.01)。该协变量被添加到结构参数的回归模型中,并对模型进行估计。重复这个过程,直至所有显著的影响都被考虑在内。然后,以后向删除的相反方向重复该过程,以消除协变量对于参数的影响,这些协变量的去除导致拟合优度的最小降低小于阈值(一个自由度的
△
ofv》+10.83,两个自由度的该值为13.8,显著性水平为p《0.001)。
[0404]
探讨了以下协变量:性别;年龄;体重(bw);东部肿瘤合作组(ecog)体能状态;肿瘤负荷;肝转移、脑转移、内脏转移的存在和转移部位的数量、肝功能(ast、alt、白蛋白、胆红素);肾功能(肌酐清除率、估计的肾小球滤过率(egfr));治疗中出现的抗药抗体(ada)。
[0405]
在通过前向选择方法和后向消元方法选择具有统计学意义的人口统计学或病理生理学协变量后,评估了其他协变量:制剂(f01对f03)、pd-l1状态(ic评分和tc评分)、种
族、地区、肿瘤类型(尿路上皮癌与其他和nsclc与其他)。
[0406]
外部验证:尿路上皮癌
[0407]
使用i期poppk模型基于在imvigor210和imvigor211中观察到的阿特珠单抗浓度-时间曲线推导出个体pk估计值。在nonmem 7版本7.3(icon,maryland)中,非线性混合效应建模方法与贝叶斯事-后估计(maxeval=0)一起使用。
[0408]
基于i期poppk模型进行预测校正的视觉预测检查(pcvpc),并将imvigor210和imvigor211中观察到的峰值(c
max
)和谷值(c
min
)与相应的预测分布进行比较。获得了imvigor210和imvigor211患者水平随机效应的个体估计,并将其对基线协变量作图,以评估i期poppk模型是否充分捕获了imvigor210和imvigor211中的协变量效应。
[0409]
外部验证:非小细胞肺癌
[0410]
使用i期poppk模型基于在birch、poplar、fir和oak中观察到的阿特珠单抗浓度-时间曲线推导出个体pk估计值。在nonmem 7版本7.3(icon,maryland)中,非线性混合效应建模方法与贝叶斯事-后估计(maxeval=0)一起使用。
[0411]
基于i期poppk模型进行pcvpc,并将birch、poplar、fir和oak中观察到的峰值(c
max
)和谷值(c
min
)与相应的预测分布进行比较。获得了birch、poplar、fir和oak患者水平随机效应的个体估计,并将其对基线协变量作图,以评估i期poppk模型是否充分捕获了birch、poplar、fir和oak中的协变量效应。
[0412]
结果
[0413]
i期poppk模型概述
[0414]
非房室分析(nca)表明≥1mg/kg的剂量显示与剂量-成比例的药代动力学。
[0415]
对于i期poppk模型,跨pcd4989g和jo28944两项研究(剂量范围:1-20mg/kg q3w,包括固定的1200mg q3w阿特珠单抗剂量)的阿特珠单抗血清药代动力学通过具有一级消除的线性双-房室处置模型描述。对于白蛋白为40g/l的男性患者,估计的典型群体药物总清除率(cl)为0.200l/天,中央房室(v1)的典型分布容积为3.28l。
[0416]
稳态条件下的典型分布容积(v
ss
)和终末t
1/2
估计值分别为6.9l和27天。根据当前群体中的模拟,在以下q3w周期的中位数(范围)后达到90%的稳态:针对c
min
、c
max
和auc,分别为3个周期(1-6)、2个周期(1-4)和3个周期(1-5)。cl、v1和外周房室中的分布容积(v2)的个体间变异性(iiv)分别估计为29%、18%和34%。
[0417]
可通过poppk模型鉴定的统计上显著的参数-协变量关系在图1中提供。表3提供了最终的poppk参数。
[0418]
表3.阿特珠单抗的最终群体药代动力学模型参数估计。
[0419][0420][0421]
在ada阳性的患者中,cl估计比没有ada的患者高16%。在女性中,v1和v2的分布容积将分别比男性低13%和27%。对于极值,没有协变量引起相对于典型pk模型参数的超过27%的变化。
[0422]
在多次给药1200mg阿特珠单抗q3w后,poppk模型估计c
min
、c
max
和auc的几何平均累积比分别为2.75、1.46和1.91倍。在研究pcd4989g中,从nca估计的几何平均累积比的范围分别为2.07至2.39和1.21至1.41,分别针对c
min
和c
max
,与poppk模型估计一致。观察到的累积程度与基于poppk报告的27天q3w给药的t
1/2
预测的程度非常一致。
[0423]
poppk模型估计c
min
、c
max
和auc的几何平均累积比在多次给药840mg阿特珠单抗q2w后分别为3.05、1.84和2.54倍,并且在多次给药1680mg阿特珠单抗q4w后分别为1.88、1.35和1.72倍。
[0424]
进行敏感性分析以检查统计学上显著的协变量对于稳态阿特珠单抗暴露(稳态时血清浓度时间曲线下面积[auc
ss
]、稳态时最大观察血清浓度[c
max,ss
]和稳态时观察到的最低血清浓度[c
min,ss
])的影响。图2示出了每个协变量(在连续协变量的第10个和第90个百分
位数之间变化)对1200mg剂量q3w后阿特珠单抗稳态暴露的独立影响。
[0425]
总体而言,与男性相比,女性的暴露程度略高。
[0426]
白蛋白低的患者往往暴露量较低,对c
min,ss
的影响较大。
[0427]
基线肿瘤负荷和治疗-中出现的阳性ada对本分析中研究的剂量范围(即1至20mg/kg的阿特珠单抗q3w,或固定的1200mg剂量q3w)的影响较小。
[0428]
总体而言,与典型患者相比,没有协变量效应引起超过30%的暴露变化(典型患者是男性,治疗后-出现ada阴性,体重77kg,白蛋白水平为40g/l,肿瘤负荷为63mm),以最低极端体重(即第10个百分位数)进行评估时的bw除外。bw低于54kg的患者的auc
ss
、c
max,ss
或c
min,ss
分别比典型患者高32%、28%、40%。
[0429]
预计这些协变量效应均不会导致c
min,ss
低于6μg/ml的目标血清浓度。在下面提供的er评估(例如,实例2-3)中描述了对这些对阿特珠单抗药代动力学的相对中等影响的临床意义(如果有)的进一步评估。
[0430]
根据年龄范围为21-89岁(n=472)且中位年龄为62岁的患者,年龄未被鉴定为影响阿特珠单抗药代动力学的显著协变量。在《65岁患者(n=274)、65-75岁之间的患者(n=152)和》75岁(n=46)患者中,未观察到阿特珠单抗的药代动力学具有临床意义的差异。不需要根据年龄调整剂量。
[0431]
与具有正常(egfr大于或等于90ml/min/1.73m2;n=140)肾功能的患者相比,在肾功能轻度受损(egfr 60至89ml/min/1.73m2;n=208)或中度受损(egfr 30至59ml/min/1.73m2;n=116)的患者中,未观察到阿特珠单抗的cl具有临床重要的差异。很少患者有严重的肾功能受损(egfr 15至29ml/min/1.73m2;n=8)。
[0432]
在轻度肝受损(胆红素≤uln和ast》uln或胆红素》1.0至1.5
×
uln和任何ast;n=71)和正常肝功能(胆红素和ast小于或等于uln;n=401)的患者之间,阿特珠单抗的cl没有临床重要的差异。没有中度或重度肝功能受损患者的数据。
[0433]
未发现ecog体能状态或转移(部位数量;脑、肝或内脏转移)影响阿特珠单抗的药代动力学。在对最终模型中显著的人口统计学和病理生理学协变量效应进行调整后,对患者水平随机效应的图形探索显示,制剂既不影响阿特珠单抗的药代动力学,也不影响免疫细胞或肿瘤细胞中的pd-l1表达。与其他肿瘤类型的患者相比,uc或nsclc患者没有表现出任何具有不同pk参数的趋势。
[0434]
尿路上皮癌poppk模型的外部验证
[0435]
对于外部验证,使用来自imvigor210和imvigor211的实际给药历史以及i期poppk模型模拟了来自imvigor210和imvigor211的pk数据(1000次重复)。imvigor210和imvigor211的阿特珠单抗数据的预测校正视觉预测检查(pcvpc)分别提供在图3a和图3b中。
[0436]
imvigor210和imvigor211的pcvpc表明,所有周期的观察到的c
max
和c
min
的中位数、第95个和第5个百分位数通常被很好地捕获,除了观察到的周期1c
max
的第95个和第5个百分位数比相应的预测百分位数略窄。在多次给药时,似乎没有出现对阿特珠单抗暴露数据高估或低估的一致趋势。pcvpc表明,i期poppk模型足以预测来自imvigor210和imvigor211的所有患者的阿特珠单抗pk数据。使用i期poppk模型进行事后估计,以获得来自imvigor210和imvigor211的患者的个体随机效应和pk参数。imvigor210和imvigor211数据中的协变量
效应与i期poppk模型中确定的一致;似乎没有任何新的协变量效应是之前在i期poppk模型中未发现的。
[0437]
nsclc poppk模型的外部验证
[0438]
类似地,使用来自birch、poplar、fir和oak的实际给药历史以及i期poppk模型模拟了来自birch、poplar、fir和oak的pk数据(1000次重复)。birch、poplar和fir的阿特珠单抗合并数据的pcvpc以及单独的oak分别显示在图4a和图4b中。
[0439]
所有患者的pcvpc(birch、poplar和fir研究相结合,以及单独的oak)表明,所有周期的观察到的c
max
和c
min
的中位数、第95个和第5个百分位数通常被很好地捕获。在多次给药时,似乎没有出现对阿特珠单抗暴露高估或低估的一致趋势。通过研究,pcvpc表明,i期poppk模型足以预测birch(所有群组)以及fir(所有群组)和oak中的阿特珠单抗pk数据。观察到poplar出现负的群体-水平预测和残差的趋势,但这种趋势在个体预测和残差中得到解决,这表明i期poppk模型允许在所有研究中对个体参数进行可靠和稳健的贝叶斯估计。使用i期poppk模型进行事后估计,以从纳入birch、fir、poplar和oak的患者中获得个体随机效应和pk参数。birch、fir、poplar和oak数据中的协变量效应通常与i期poppk模型中鉴定的那些一致。尽管poplar中存在更快cl和更大v1的趋势,但poplar中的暴露仅受这些效应的中度影响(即,auc、c
max
和c
min
通常与birch、fir和oak估计值相差20%以内)。cl的随机效应与bw之间的关系以负相关系数为特征,表明nsclc患者中的这种关系可能不像i期poppk模型所暗示的那样夸大。在birch、fir、poplar和oak中未发现新的意外协变量效应。在nsclc患者的birch、fir、poplar和oak中获得的组合阿特珠单抗pk数据与i期poppk模型估计值一致。
[0440]
内在因素对阿特珠单抗pk影响的汇总
[0441]
尚未在老年患者中进行过阿特珠单抗的专门研究。在poppk分析中,根据年龄为21至89岁(n=472)且中位年龄为62岁的患者,年龄未被鉴定为影响阿特珠单抗药代动力学的显著协变量。在《65岁患者(n=274)、65-75岁之间的患者(n=152)和》75岁(n=46)患者中,未观察到阿特珠单抗的药代动力学具有临床重要的差异。不需要根据年龄调整剂量。尚未在儿科患者中完成对阿特珠单抗的专门研究。
[0442]
在poppk分析中,基于包括276名男性(58.5%)和196名女性(41.5%)的数据集,性别被鉴定为v1和v2的统计学上显著的协变量,但不是cl的。在女性中,v1和v2的容积分别比男性低13%和27%。对于典型的女性患者(体重标准化为77kg),与典型的男性患者相比,阿特珠单抗的auc
ss
、c
max,ss
或c
min,ss
增加不到10%。
[0443]
在对最终poppk模型中的协变量效应进行调整后,种族(亚裔n=17、黑人n=15和白人n=375)不是阿特珠单抗药代动力学的显著协变量,并且与阿特珠单抗cl没有临床相关性。
[0444]
尚未在肾功能受损的患者中进行正式的pk研究。基于poppk分析,与具有正常(egfr大于或等于90ml/min/1.73m2;n=140)肾功能的患者相比,在肾功能轻度受损(egfr 60至89ml/min/1.73m2;n=208)或中度受损(egfr 30至59ml/min/1.73m2;n=116)的患者中,未观察到阿特珠单抗的cl具有临床重要的差异。很少患者有严重的肾功能受损(egfr 15至29ml/min/1.73m2;n=8)。不需要根据与肾功能相关的协变量调整剂量。
[0445]
尚未在肝功能受损的患者中进行正式的pk研究。基于poppk分析,在轻度肝受损
(胆红素≤uln和ast》uln或胆红素》1.0至1.5
×
uln和任何ast;n=71)和正常肝功能(胆红素和ast小于或等于uln;n=401)的患者之间,阿特珠单抗的cl没有临床重要的差异。轻度肝功能受损的患者无需调整剂量。没有中度或重度肝功能受损患者的数据。
[0446]
基于poppk分析,未发现ecog体能状态或转移(部位数量;脑、肝或内脏转移)影响阿特珠单抗的药代动力学。白蛋白和肿瘤负荷被鉴定为cl的统计学上显著的协变量。当以这些协变量分布的极值(即第10个和第90个百分位数)进行评估时,这些协变量都不会导致典型患者的auc
ss
、c
max,ss
或c
min,ss
变化超过30%。在调整最终poppk模型中的协变量效应后,肿瘤浸润免疫细胞(ic评分)或肿瘤细胞(tc评分)中的pd-l1表达均不影响阿特珠单抗的药代动力学。uc或nsclc患者没有表现出任何与其他肿瘤类型患者具有不同pk参数的趋势。
[0447]
外在因素对阿特珠单抗pk影响的汇总
[0448]
在poppk分析中,药物产品/制剂的变化对阿特珠单抗的药代动力学没有影响。尚未进行pk药物-药物相互作用研究。
[0449]
在对最终poppk模型中的协变量效应进行调整后,地区(日本与西班牙与法国与英国与美国)对阿特珠单抗的药代动力学不是一个显著的协变量,并且它与阿特珠单抗cl没有临床相关性。
[0450]
实例2
[0451]
阿特珠单抗在尿路上皮癌和非小细胞肺癌中的暴露-疗效关系
[0452]
进行了暴露-应答(er)分析,以针对每种适应症分别评估独立(uc或nsclc)以及合并的(uc和nsclc)患者人群的临床疗效与阿特珠单抗暴露之间的可能关系。
[0453]
方法
[0454]
合并的er分析概述
[0455]
与药代动力学(pk)度量相比,评估了客观应答率、总存活和不良事件,如下所述。
[0456]
进行er分析以告知pk度量与orr、os、3至5级ae和基于周期1数据在先前临床研究中评估的aesi终点之间的任何关系,以最小化由于与基线预后因素混杂而导致的潜在偏倚(yang等人,(2013)j clin pharmacol doi:10.1177/0091270012445206;wang等人,(2014)clin pharmacol ther doi:10.1038/clpt.2014.24)以及对阿特珠单抗和其他检查点抑制剂观察到的清除率随时间的变化(tecentriq(阿特珠单抗)[包装插页]。south san francisco,ca:基因泰克公司;2019年。south san francisco,ca,usa:基因泰克公司;bi等人,(2019)ann oncol doi:10.1093/annonc/mdz037;bajaj等人,(2017)cpt pharmacometrics syst pharmacol doi:10.1002/psp4.12143;li等人,(2017)j pharmacokinet pharmacodyn doi:10.1007/s10928-017-9528-y;liu等人,(2017)clin pharmacol ther doi:10.1002/cpt.656;wang等人,(2017)clin pharmacol ther doi:10.1002/cpt.628)。这些分析是使用来自经阿特珠单抗治疗的nsclc或uc患者(来自pcd4989g、oak和imvigor211)的合并数据进行的,这些患者的暴露数据可用,但如下所述,总存活(os)除外。使用周期1最大血清浓度(c
max
)、c
min
和浓度-时间曲线下面积(auc;时间0-21天)进行探索性er分析,如所推荐的(liu等人,(2017)clin pharmacol ther doi:10.1002/cpt.656)以最小化先前针对抗pd-1和抗pd-l1药物观察到的应答依赖性时变清除率的影响(li等人,(2017)j pharmacokinet pharmacodyn doi:10.1007/s10928-017-9528-y)。auc(时间0-21天)、c
max
和c
min
在周期1基于仅使用周期1数据和先前开发的poppk模
型估计的个体pk参数得出(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587)。评估的疗效终点是研究人员评估的确认的实体瘤应答评估标准1.1版(recist 1.1)客观应答率(orr;所有研究的次要终点)和os(oak和imvigor211的主要终点)。orr分析使用来自pcd4989g、oak(前850例随机化患者)和imvigor211中阿特珠单抗治疗的nsclc或uc患者的数据,而os分析仅使用来自oak(前850例随机化患者)和imvigor211的数据。评估的安全性终点包括根据美国国家癌症研究所不良事件通用术语标准第4版和医学词典第20.1版的3至5级不良事件(ae)(pcd4989g中的主要终点,也在oak和imvigor211中进行评估)和特别关注的ae(aesi;在所有研究中评估)。aesi为提示自身免疫性疾病的病症,先前已定义(petrylak等人,(2018)jama oncol doi:10.1001/jamaoncol.2017.5440)。
[0457]
orr和ae作为二元终点(是/否)进行评估,并使用逻辑回归将其对作为连续变量的暴露进行研究。报告了每个逻辑回归的wald检验p值,以及针对暴露四分位数计算的比例/频率及其95%ci。对于os数据,为了减轻患者基线信息与阿特珠单抗清除率和暴露之间的混杂因素,进行了tgi-os建模(bruno等人,(2014)clin pharmacol ther doi:10.1038/clpt.2014.4;claret等人,(2018)clin cancer res doi:10.1158/1078-0432.ccr-17-3662)。为了可以在该分析中进行评估(tgi可评估),患者需要进行≥1次治疗后最长直径(sld)评估。使用kaplan-meier和cox回归分析探索了个体基线预后因素和tgi度量(根据recist 1.1在靶标病灶的sld双指数纵向模型中估计的肿瘤收缩率和肿瘤生长率)对os的影响,以及建立参数多元回归tgi-os模型。通过模拟其在描述os分布和风险比(hr)的能力方面与不同亚组(特别是根据暴露四分位数分组)中的对照进行比较,验证了最终的tgi-os模型。对于hr模拟,对照患者的tgi度量估计和基线协变量取自先前的分析(claret等人,(2018)clin cancer res doi:10.1158/1078-0432.ccr-17-3662;bruno等人,(2018)j clin oncol doi:10.1200/jco.2018.36.5_suppl.62)。在调整了与预后因素的混杂后,在最终的多变量模型上测试了暴露度量。如果合适,将“肿瘤类型”因素并入模型中。
[0458]
尿路上皮癌的er分析和os建模
[0459]
在imvigor210和imvigor211这两项研究中,对muc患者的阿特珠单抗暴露-疗效关系进行了单独评估。在这两项研究中,周期1暴露度量用于适应先前使用抗pd-1抗体和抗pd-l1抗体观察到的清除率的轻微时间和应答依赖性的变化。对于imvigor210,主要终点客观应答率(orr)被用作疗效度量。对于imvigor211,orr和主要终点os用于暴露-疗效评估。
[0460]
阿特珠单抗暴露度量(auc、c
max
和c
min
)在周期1从基于个体pk参数的模拟pk曲线推导出来。阿特珠单抗auc
ss
计算为起始剂量/cl。
[0461]
orr的特征在于应答者状态(是/否)。应答者的比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个相关性,进行逻辑回归并报告逻辑回归中暴露效应对应答概率的wald检验p值。
[0462]
为了减轻患者预后因素与阿特珠单抗清除率和暴露之间的混杂,进行了肿瘤生长抑制-总存活(tgi-os)建模(疾病建模)。患者水平的肿瘤生长抑制(tgi)度量使用来自stein等人(2011)clin cancer res 18:907-917先前描述的纵向肿瘤大小模型的参数估计值进行估计,并由与可评估的患者适配的claret等人(2013)j clin oncol 31:2110-2114实现。通过来自tgi模型的事后经验贝叶斯估计来估计以个体患者的生长率常数(kg)为特征的生长率。
[0463]
多变量参数os模型是使用kg和其他协变量开发的。“完整”os模型的构建首先包括来自单变量分析的所有显著协变量(cox,p《0.05),然后使用p《0.01的截止值进行后向逐步消元。评估os模型在imvigor211中模拟观察到的os分布和风险比(hr)的能力。(stein等人,(2011)clin cancer res 18:907-917;claret等人,(2013)j clin oncol 31:2110-2114)。
[0464]
nsclc的er分析和os建模
[0465]
根据birch的实体瘤应答评估标准(recist)v1.1的独立审查机构(irf)评估的orr,以及根据poplar和oak的recist v1.1的os和研究人员评估的orr,均被考虑在暴露-疗效评估中。根据recist v1.1的irf评估orr为birch的主要终点,而os为poplar和oak的主要终点。对于birch,暴露-疗效评估中的分析群体为二线及以上患者(2l+)tc2/3或ic2/3nsclc,代表群组2和3中的意向治疗人群。对于poplar和oak,暴露-疗效评估中的分析群体为pd-l1未选择的nsclc患者群体(即所有参与者)。针对er分别分析了来自birch的根据recist v1.1的irf评估的orr和来自poplar和oak的根据recist v1.1的研究者评估的orr。
[0466]
疗效终点orr的特征在于应答者状态(是/否)。频率比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个相关性,进行逻辑回归并报告逻辑回归中暴露效应的wald检验p值。
[0467]
p(orr)~暴露
[0468]
其中,p(orr)为客观应答的概率,暴露为阿特珠单抗暴露度量。
[0469]
为了减轻患者预后因素与阿特珠单抗清除率和暴露之间的混杂,进行了tgi-os建模(疾病建模)。患者水平的tgi度量使用来自如stein等人(2011)clin cancer res 18:907-917先前描述的纵向肿瘤大小模型的参数估计值进行估计,并由与可评估的患者适配的claret等人(2013)j clin oncol 31:2110-2114实现。通过来自tgi模型的事后经验贝叶斯估计来估计以个体患者的kg为特征的生长率。
[0470]
多变量参数os模型是使用具有kg和其他协变量的回归分析开发的。“完整”os模型的构建首先包括来自单变量分析的所有显著协变量(cox,p《0.05),然后使用p《0.01的截止值进行后向逐步消元。评估os模型在poplar和oak中模拟观察到的os分布和hr的能力。然后模拟该模型以表征由于kg对os的影响导致的(非混杂)er(stein等人,(2011)clin cancer res 18:907-917;claret等人,(2013)j clin oncol31:2110-2114)。
[0471]
合并的(uc和nsclc)er分析和os建模
[0472]
在pcd4989g、imvigor211和oak研究中对muc或nsclc患者的合并分析中评估了阿特珠单抗暴露-疗效关系。考虑用于暴露-应答分析的疗效终点在研究pcd4989g、imvigor211和oak中是所有经阿特珠单抗治疗的muc和nsclc患者的orr(研究者使用recist v1.1评估),而在研究imvigor211和oak中是所有经阿特珠单抗治疗的muc和nsclc患者的os。周期1暴露度量用于适应先前针对抗pd1和pd-l1抗体观察到的清除率的轻微时间和应答依赖性的变化。
[0473]
疗效终点orr的特征在于应答者状态(是/否)。应答者的比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个相关性,进行逻辑回归并报告逻辑回归中暴露效应的wald检验p值。
[0474]
为了减轻患者预后因素与阿特珠单抗清除率和暴露之间的混杂,进行了tgi-os建模(疾病建模)。患者水平的tgi度量使用来自如stein等人(2011)clin cancer res 18:
907-917先前描述的与可评估的患者适配的纵向肿瘤大小模型的参数估计值进行估计,并由claret等人(2013)j clin oncol 31:2110-2114实现。通过来自tgi模型的事后经验贝叶斯估计来估计以个体患者的kg为特征的生长率。
[0475]
多变量参数os模型是使用kg和其他协变量开发的。“完整”os模型的构建首先包括来自单变量分析的所有显著协变量(cox,p《0.05),然后使用p《0.01的截止值进行后向逐步消元。评估os模型在imvigor211和oak中模拟观察到的os分布和hr的能力(stein等人,(2011)clin cancer res 18:907-917;claret等人,(2013)j clin oncol 31:2110-2114)。
[0476]
阿特珠单抗暴露度量(auc、c
max
和c
min
)在周期1从基于个体pk参数的模拟pk曲线推导出来。
[0477]
结果
[0478]
尿路上皮癌er分析和os建模结果
[0479]
考虑到imvigor210(群组1和2)中使用阿特珠单抗1200mg q3w治疗的患者的任何暴露度量的情况下,应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系。对于imvigor210中接受阿特珠单抗1200mg q3w的患者的orr与周期1auc、周期1c
min
和auc
ss
之间的关系,患有1l不符合进行顺铂治疗条件的尿路上皮癌的患者的这些关系提供于图5a-5c中,而2l尿路上皮癌患者的这些关系提供于图6a-6c中。
[0480]
类似地,对于imvigor211中的患者,在阿特珠单抗1200mg q3w后没有发现与orr有统计学上显著的er关系(周期1auc)(图7)。最初,使用单变量分析鉴定了与os的统计学上显著的er关系。然而,在最终多变量模型上测试时,暴露(auc周期1)不再显著(p》0.01)(p=0.0812),表明多变量os模型针对auc-os关系中的混杂进行了调整单变量分析。tgi度量log(kg)或log(ks)无一与auc周期1显著相关。
[0481]
与用poppk模型(见实例1)鉴定的统计学上显著的协变量相关的阿特珠单抗暴露的任何变化预计无一具有临床意义或需要调整剂量。因此,与典型患者相比,在施用阿特珠单抗1200mg q3w固定剂量后,在体重极值(即第90个百分位数)下评估时,预计阿特珠单抗暴露量的减少没有临床意义或不需要根据bw调整剂量。
[0482]
非小细胞肺癌er分析和os建模结果
[0483]
对于在birch和oak中接受阿特珠单抗1200mg q3w治疗的患者,应答概率与阿特珠单抗暴露之间存在统计学上显著的er关系,其中至少考虑了一项暴露度量。
[0484]
对于birch和oak,在与阿特珠单抗暴露应答概率增加趋势相关的暴露度量中,与auc
ss
相关的p值(分别为p=0.0005343和p《0.0003)最低。对于birch,周期1c
min
、周期1auc、auc
ss
和体重的逻辑回归分别提供于图8a-8d中。对于oak,周期1c
min
、周期1auc、auc
ss
和体重的逻辑回归分别提供于图9a-9d中。
[0485]
对于在poplar中接受阿特珠单抗1200mg q3w治疗的患者,应答概率与阿特珠单抗暴露之间不存在统计学上显著的er关系,其中考虑了任何暴露度量。周期1c
min
、auc周期1和auc
ss
的逻辑回归分别提供于图10a-10c中。对poplar中的2l/3l tc2/3或ic2/3nsclc患者进行了敏感性分析,这进一步表明应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系。
[0486]
在poplar和oak的暴露-疗效评估中也考虑了基于模型的os评估。对于poplar和oak,kg的对数(logkg)和一定范围的患者预后因素解释了阿特珠单抗对os的影响。
[0487]
具体来说,对于poplar多变量os模型,转移部位的数量、白蛋白水平和logkg解释了阿特珠单抗对os的影响。kg的对数与阿特珠单抗aucss相关。多变量os模型用于根据logkg上的er推断os上的er。模拟了在每组aucss三分位数中比较阿特珠单抗与多西他赛os的hr。在跨aucss三分位数和多西他赛组校正预后因素(转移部位的数量和白蛋白水平)不平衡后对os模型的模拟表明,所有患者都将从阿特珠单抗治疗中受益(hr估计值[95%预测区间]在低暴露患者[第1个三分位数]中为=0.859[0.820,0.906];在高暴露患者[第3个三分位数]中为0.614[0.556,0.681])(图11a)。
[0488]
具体而言,对于oak多变量os模型,最长直径(bsld)、白蛋白水平、ecog体能状态》0、乳酸脱氢酶(ldh)水平和logkg的基线总和解释了阿特珠单抗对os的影响。logkg与阿特珠单抗aucss相关。多变量os模型用于根据logkg上的er推断os上的er。模拟了在每组aucss三分位数中比较阿特珠单抗与多西他赛os的hr。在跨aucss三分位数和多西他赛组校正预后因素(基线bsld、白蛋白、ecog体能状态和ldh水平)不平衡后对os模型的模拟表明,所有患者都将从阿特珠单抗治疗中受益(hr估计值[95%预测区间]在低暴露患者[第1个三分位数]中为=0.870[0.831,0.908];在高暴露患者[第3个三分位数]中为0.624[0.582,0.670])(图11b)。
[0489]
在birch中,aucss的er关系模拟表明,对于aucss中位数和第25个百分位数的患者,orr(估计[预测间隔])分别从0.16(0.13,0.20)降至0.13(0.10,0.17)。鉴于重叠的置信区间(ci)、orr的小幅下降以及在这种治疗设置中通过orr衡量的疗效与os之间缺乏相关性,因此认为orr的这种变化不太可能具有临床意义。此外,由于使用抗pd-1和pd-l1抑制剂观察到清除率的时间和应答依赖性的降低,因此在暴露-应答分析中使用aucss作为暴露度量可能会高估暴露与orr之间的潜在关系。
[0490]
在oak中,aucss的er关系模拟表明,对于aucss中位数和第25个百分位数的患者,orr(估计[预测间隔])分别从0.13(0.10,0.16)降至0.10(0.07,0.14)。鉴于重叠的ci、orr的小幅下降以及在这种治疗设置中通过orr衡量的疗效与os之间缺乏相关性,因此认为orr的这种变化也不太可能具有临床意义。在poplar中,与orr之间没有统计学上显著的er关系。
[0491]
由于i期poppk模型中没有与aucss降低》25%相关的单一效应(即bw、性别、ada、白蛋白和肿瘤负荷),因此对于birch(图8c)或oak(图9c),预计与用poppk模型鉴定的统计学上显著的协变量相关的aucss的变化无一将会超过在aucss的第25个百分位处的orr变化或在阿特珠单抗暴露的最低三分位数处的os的hr。与uc一样,预计与这些通过poppk模型确定的具有统计学意义的协变量相关的阿特珠单抗暴露的倍数-变化无一将会具有临床意义或需要调整剂量。
[0492]
因此,在使用阿特珠单抗1200mg q3w固定剂量后,与典型患者相比,在极端体重值下评估时阿特珠单抗暴露量的降低(即auc
ss
降低21%)被认为不太可能需要调整剂量或根据bw进行调整。对于birch(图8d)和oak(图9d),orr与bw没有统计学上显著关系的观察结果进一步支持选择1200mg q3w的阿特珠单抗固定剂量。模拟表明,对那些在固定1200mg阿特珠单抗剂量后处于阿特珠单抗暴露最低四分位数的患者施用基于体重的15mg/kg阿特珠单抗剂量不会改善这些患者的orr。对1200-mg q3w固定剂量阿特珠单抗的进一步支持来自oak,其中按bw四分位数绘制的os的kaplan-meier图(图12)表明,体重较重的患者与体重较
轻的患者具有相似的os。
[0493]
合并的(nsclc和uc)er分析和os建模结果
[0494]
在暴露-功效评估中评估了在pcd4989g、imvigor211和oak中使用阿特珠单抗治疗的患者的muc和nsclc中的orr。群体包含muc和nsclc患者(1042例接受阿特珠单抗治疗的患者有暴露数据)。在分析群体中,根据recist v1.1的orr(确认的cr和pr的比例;研究人员评估的)为15.7%(1042例有暴露数据的患者中有164例应答者)。muc(15.9%,n=541例患者)和nsclc(15.6%,n=501例患者)的orr没有差异,因此,肿瘤类型不包括在逻辑回归模型中。
[0495]
如表4和图13a-13b所示,在应答概率与阿特珠单抗暴露之间没有统计学上显著的er关系,其中考虑了任何暴露度量(auc周期1、c
max
周期1和c
min
周期1)。
[0496]
表4.合并的muc和nsclc患者中应答概率与暴露的逻辑回归结果汇总。
[0497][0498]
为了减轻预后因素与阿特珠单抗清除率和暴露之间的混杂,开发了多变量os模型来解释基线预后因素和tgi度量,如概述。oak nsclc患者(n=425例意向治疗[itt]患者中388tgi可评估[91%])的中位os为467天(95%ci,402-508天),而imvigor211 uc患者(n=467例itt患者的382tgi可评估[82%])为344天(95%ci,290-383天)。由于与nsclc患者相比,muc患者的中位os较短,因此将肿瘤类型并入多变量模型中。在770例tgi可评估患者中,764例有暴露数据。
[0499]
log(肿瘤生长率[kg])和基线预后因素(诸如ecog体能状态》0、基线肿瘤大小、白蛋白水平、乳酸脱氢酶、碱性磷酸酶、pd-l1状态和肿瘤类型)的个体估计是os的强独立预测因子(表5)。值得注意的是,在最终模型中考虑基线协变量后,在最终模型上进行测试时,周期1阿特珠单抗暴露(周期1的auc、c
min
或c
max
)不再显著(p》0.01)。
[0500]
表5.在以muc肿瘤类型为因子的oak和imvigor211中,最终多变量os模型的参数估计。
[0501]
[0502][0503]
即使暴露不在模型中,该模型在通过每种肿瘤类型的暴露四分位数模拟os分布和hr方面也表现良好。预测的和观察到的os数据的比较提供在图14a-14b和图15a-15b中。在调整基线协变量(固定为中值)后,通过auc四分位数的hr模拟也说明了阿特珠单抗的平坦er关系图16a-16b。
[0504]
实例3
[0505]
阿特珠单抗在尿路上皮癌和非小细胞肺癌中的暴露-安全性关系
[0506]
进行了暴露安全性分析,以分别评估每个适应症(uc或nsclc)以及合并的(uc和nsclc)患者群体的安全性终点与阿特珠单抗暴露之间的可能关系。
[0507]
方法
[0508]
尿路上皮癌
[0509]
分析了来自研究pcd4989g(uc群组)、imvigor210(群组1和群组2)和imvigor211(阿特珠单抗组)的3至5级不良事件(aeg35)和特别关注的不良事件(aesi)的暴露-安全性关系。安全性终点以频率为特征(是/否)。频率比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个这样的相关性,进行逻辑回归并报告逻辑回归中暴露效应的wald检验p值。
[0510]
p(ae)~暴露
[0511]
其中p(ae)为不良事件的概率(即aeg35或aesi),而暴露为阿特珠单抗暴露度量。阿特珠单抗暴露度量(auc、c
max
和c
min
)在周期1从基于个体pk参数的模拟pk曲线推导出来。
[0512]
非小细胞肺癌
[0513]
来自birch、poplar、fir和pcd4989g研究(nsclc群组)的合并数据以及oak的单独数据中的aeg35和aesi用于暴露-安全性分析。这些安全性终点以频率为特征(是/否)。频率比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个这样的相关性,进行逻辑回归并报告逻辑回归中暴露效应的wald检验p-值。
[0514]
p(ae)~暴露
[0515]
其中p(ae)为不良事件的概率(即aeg35或aesi),而暴露为阿特珠单抗暴露度量。阿特珠单抗暴露度量(auc、c
max
和c
min
)在周期1从基于个体pk参数的模拟pk曲线推导出来。
[0516]
合并分析
[0517]
uc和nsclc中阿特珠单抗的暴露-安全性关系的合并分析如上文和实例2中的“合并er分析概述”部分所述进行。
[0518]
对研究pcd4989g、imvigor211和oak中所有经阿特珠单抗治疗的muc和nsclc患者的2至5级不良事件(aeg25)、3至5级不良事件(aeg35)和特别关注的不良事件(aesi)进行了暴露与安全性的关系分析。安全性终点以频率为特征(是/否)。频率比例和95%ci是针对等效数量的个体(例如四分位数)的暴露区间计算的。对于每个这样的相关性,进行逻辑回归并报告逻辑回归中暴露效应的wald检验p值。
[0519]
p(ae)~暴露
[0520]
其中p(ae)为不良事件的概率(即aeg25、aeg35或aesi),而暴露为阿特珠单抗暴露度量。阿特珠单抗暴露度量(auc、c
max
和c
min
)在周期1从基于个体pk参数的模拟pk曲线推导出来。
[0521]
结果
[0522]
尿路上皮癌
[0523]
对aeg35发生率的分析未显示与研究的任何暴露度量的任何统计学上显著的er关系,包括pcd4989g和imvigor210中的uc患者的组合分析中的周期1auc(图17a)、c
max
(图17b)或auc
ss
(图17c),或者在研究imvigor211的独立分析中的周期1auc(图18a)或c
max
(图18b)。
[0524]
类似地,对aesi发生率的分析未显示与研究的任何暴露度量的任何统计学上显著的er关系,包括pcd4989g和imvigor210中的uc患者的组合分析中的周期1auc(图19a)、周期1c
max
(图19b)或auc
ss
(图19c),或者在研究imvigor211的独立分析中的周期1auc(图20a)或周期1c
max
(图20b)。
[0525]
非小细胞肺癌
[0526]
对aeg35发生率的分析未显示与研究的任何暴露度量的任何统计学上显著的正er关系,包括pcd4989g、birch、poplar和fir中的nsclc患者的组合分析中的周期1auc(图21a)、周期1c
max
(图21b)和auc
ss
(图21c),或者在oak的独立分析中的周期1auc(图22a)、周期1c
max
(图22b)或auc
ss
(图22c)。
[0527]
对pcd4989g、birch、poplar和fir中nsclc患者的合并分析的aesi发生率的分析未显示与周期1auc(图23a)或c
max
(图23b)有任何统计学上显著的er关系,但是确实与auc
ss
具有统计学上显著的关系(图23c)。对于oak,对aesi发生率的分析未显示与研究的任何暴露度量的任何统计学上显著的er关系,包括周期1auc(图24a)、周期1c
max
(图24b)或auc
ss
(图24c)。
[0528]
对于来自birch、poplar、fir和pcd4989g(nsclc群组)研究的合并数据,aesi包括许多不同的事件;评估了发生最多的aesi(见于15例或更多患者)与auc
ss
的关系。虽然研究结果表明aesi的可能性略有增加,但这种增加不被认为具有临床意义或需要调整剂量。在oak中未观察到有关aesi的这一发现。aesi的显著性与阿特珠单抗er的auc
ss
在oak与早期合并研究数据之间的差异的原因尚不清楚。还应注意,如下详述,aesi合并研究数据中鉴定的er趋势未被视为具有临床意义。
[0529]
对于来自birch、poplar、fir和pcd4989g(nsclc群组)研究的合并数据,对于具有auc
ss
中位数和第90个百分位数的患者,对auc
ss
的逻辑回归模型的模拟表明aesi的概率(估
计[预测区间])从0.18(0.16,0.21)分别增加到0.22(0.18,0.26)。对于合并的研究数据,预计aesi的这种增加没有临床意义或不需要调整剂量。在i期poppk模型鉴定的统计学上显著的协变量中,模拟表明,阿特珠单抗auc
ss
的最大正估计变化为》32%,并且与体重的极值(即10%百分位数)相关。由于没有与auc
ss
的》32%变化相关的单一效应,因此预计与poppk模型鉴定的统计学上显著的协变量相关的auc
ss
变化无一将会具有临床意义或需要调整剂量。与典型患者相比,在施用阿特珠单抗1200mg q3w固定剂量后,在体重极值(即第10个百分位数)下评估时,预计auc
ss
的提高没有临床意义或不需要根据bw调整剂量。
[0530]
合并(nsclc和uc)分析
[0531]
对所有具有暴露数据的局部晚期或转移性nsclc或uc患者(n=1228)进行了合并的阿特珠单抗暴露-安全性分析。
[0532]
1228例患者中的209例(17.0%)和298例(24.3%)分别发生了≥3级ae和aesi。与uc相比,nsclc患者的ae频率相似(≥3级ae为14.9%对19.6%;aesi为24.6%对23.9%);因此,逻辑回归模型中不包括肿瘤类型。
[0533]
在研究pcd4989g、imvigor211和oak中,对所有经阿特珠单抗治疗的muc和nsclc患者的aeg35(≥3级ae)发生率的分析未显示与研究的任何周期1暴露度量有任何统计学上显著的er关系,包括周期1auc)(图25a)或c
max
(图26a)。
[0534]
类似地,在研究pcd4989g、imvigor211和oak中,对所有经阿特珠单抗治疗的muc和nsclc患者的aesi发生率的分析未显示与研究的任何周期1暴露度量有任何统计学上显著的er关系,包括周期1auc(图25b)或c
max
(图26b)。
[0535]
实例4
[0536]
观察到的阿特珠单抗暴露与预测的840mg q2w和1680mg q4w暴露的比较
[0537]
实例1-3的汇总
[0538]
如上所述,对于批准的1200mg q3w给药方案,阿特珠单抗表现出被认为没有临床意义的er趋势或被转移性uc或nsclc患者的疗效和安全性预后因素混杂的er趋势。就uc和nsclc两者的疗效的er而言,未观察到与orr或os有临床意义的er关系(参见实例2)。这表明通过批准的1200mg q3w给药方案实现的暴露处于er曲线的平坦或平台部分。
[0539]
因此,只要任何新的给药方案达到已批准的1200-mg q3w给药方案的预期范围内,就不会对应答产生影响。重要的是,840mg q2w和1680mg q4w给药方案预计将落入该暴露范围内。
[0540]
就uc和nsclc两者的安全性的er而言,在10mg/kg q3w至20mg/kg q3w的剂量范围内,包括1200mg固定剂量q3w方案,未观察到对阿特珠单抗的安全性具有临床意义的er(参见实例3)。840mg q2w、1200mg q3w和1680mg q4w的固定剂量方案在标准化为80kg bw时分别等效于10.5mg/kg q2w、15mg/kg q3w和21mg/kg q4w。任何新的阿特珠单抗给药方案提供在针对高达20mg/kg q3w(首次人体剂量范围研究pcd4989g中施用的最高剂量,通常耐受性良好)的剂量范围观察到的范围内的暴露,预计该给药方案将表现出与之前观察到的类似的暴露-安全性关系。840-mg q2w和1680-mg q4w给药方案预计落入针对批准的1200-mg q3w给药方案和20mg/kg q3w观察到的暴露范围内(参见实例6)。应该注意的是,在剂量范围研究pcd4989g中没有确定最大耐受剂量(mtd)。
[0541]
在本实例中,基于前述实例中描述的poppk模型,针对840mg q2w、1200mg q3w、
1680mg q4w和20mg/kg q3w给药方案预测了虚拟患者的pk曲线。阿特珠单抗暴露度量随后从模拟的pk曲线推导。
[0542]
方法
[0543]
先前开发的阿特珠单抗的群体pk模型(参见前述实例)用于针对以下给药方案预测虚拟患者在周期1和稳态时的个体pk曲线:840mg q2w、1200mg q3w、1680mg q4w和20mg/kg q3w。
[0544]
阿特珠单抗暴露度量(周期1和稳态时的c
max
、c
trough
和auc)从模拟的个体pk曲线推导,并针对每个给药方案跨个体汇总。为了比较涉及不同给药间隔(每2、3或4周)的几种给药方案,还推导了周期1和稳态时的每周auc。计算每个给药方案的每周auc,
ss
与20mg/kg q3w(首次人体剂量范围研究pcd4989g中施用的最高剂量)的每周auc,
ss
的几何平均值的差异。
[0545]
为了模拟不同阿特珠单抗方案(840mg q2w、1200mg q3w、1680mg每4周[q4w]和20mg/kg q3w)的pk参数,使用之前使用pcd4989g数据开发的包括协变量效应的阿特珠单抗poppk模型进行了蒙特卡罗模拟(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587)以获得在周期1和稳态时的虚拟个体pk曲线。在用于pk模拟的poppk模型中,发现体重、白蛋白、肿瘤负荷、治疗中出现的抗药抗体(ada)状态和性别对阿特珠单抗pk具有统计学上显著的影响。每个方案模拟500例患者的单个重复。在控制流中提供了种子编号以确保模拟的可再现性。随机效应是从先前估计的分布中采样的,并且不考虑单个预测的残差。假设每个给药方案的虚拟患者具有1:1的男性:女性比(男性体重85kg,女性体重64kg,这是用于开发poppk模型的1期数据库中的中位数体重)。其他影响阿特珠单抗pk参数的协变量设置为分类协变量的中位数或最常见类别:白蛋白水平为40g/l,基线肿瘤大小为63mm,对于抗药抗体(ada)为阴性。模拟了四种给药方案:1200mg q3w、20mg/kg q3w(即男性1700mg和女性1280mg)、840mg q2w和1680mg q4w。为了评估固定剂量方案后体重对暴露的影响,每个体重四分位数有500例具有中位白蛋白水平、基线肿瘤大小和ada阴性的虚拟患者被分配了840mg q2w或1680mg q4w的剂量。1期患者群体的体重分布按四分位数区分如下:36.5至63.7、63.7至77.0、77.0至90.9和90.9至168.0kg。假设截断正态分布,在每个四分位数中对500个个体体重进行采样。为了保持性别和体重之间的相关性,女性的比例设置为第一个四分位数为80%,第二个四分位数为50%,第三个四分位数为25%,最后一个四分位数为10%,如在用于开发poppk模型的1期数据库中观察到的。
[0546]
阿特珠单抗暴露度量(周期1:auc[使用梯形法计算;时间0-21天]、c
max
和c
min
;稳态:auc[剂量/清除率]、c
max
和c
min
)从模拟的个体pk曲线推导,并针对每个给药方案跨个体汇总。为了比较涉及不同给药间隔(每2、3或4周)的几种给药方案,还推导了稳态每周auc数据。
[0547]
结果
[0548]
将840mg每2周(q2w)和1680mg每4周(q4w)方案的群体pk模拟暴露与1200mg每3周(q3w)和最大评估剂量(mad;20mg/kg q3w)的批准方案进行比较。
[0549]
下表5b和表6分别提供了周期1和稳-态时所有可用研究中poppk估计暴露的汇总。
[0550]
表5b.使用poppk模型(pk可评估群体)预测的周期1中1200mg q3w阿特珠单抗暴露度量的汇总统计(几何平均值,%cv)。
[0551][0552]
表6.使用poppk模型(pk可评估群体)在稳态时1200mg q3w阿特珠单抗暴露度量的汇总统计(几何平均值,%cv)。
[0553][0554]
poppk预测的4种给药方案(840-mg q2w、1200-mg q3w、1680-mg q4w和20-mg/kg q3w)的模拟阿特珠单抗暴露曲线(浓度-时间曲线)显示在图27中。显示了28天期间的曲线,显示2个剂量的1200mg q3w、20mg/kg q3w和840mg q2w;以及1个剂量的1680mg q4w。与每个给药方案相关的相应暴露度量的汇总(在周期1和稳态时的预测c
max
和c
min
值)在表7中给出。
[0555]
表7.针对各种方案模拟的阿特珠单抗暴露的汇总统计(500例患者的几何平均值[90%ci])。
[0556][0557]
表8中给出了每周预测的周期1auc和auc
ss
。
[0558]
表8.针对各种方案模拟的阿特珠单抗暴露的汇总统计(500例患者的几何平均值[90%ci])。
[0559][0560]
与1200mg q3w给药方案的预测c
min
相比,840-mg q2w给药方案的预测c
min
浓度在周期1时低13%并且在稳态时高16%。然而,在周期1时和稳态时,840mg q2w方案的预测c
min
值仍然比c
min
靶标浓度(6μg/ml(deng等人,(2016)mabs doi:10.1080/19420862.2015.1136043))高至少10-倍(》10倍)。在周期1和稳态时,840mg q2w给药方案的预测c
max
低于1200mg q3w给药方案的预测c
max
。
[0561]
与1200mg q3w给药方案的预测c
min
相比,1680mg q4w给药方案(就80kg患者而言,等效于21mg/kg q4w)的预测c
min
在周期1时高低14%并且在稳态时低6%。然而,在周期1时和稳态时,1680-mg q4w方案的预测c
min
值仍然比c
min
靶标浓度(6μg/ml)高至少10倍(》10倍)。
[0562]
1680-mg q4w方案的预测c
max
在周期1时和稳态时分别比相对于20mg/kg给药方案的预测几何平均c
max
高12%和高0.8%,并且与针对pcd4989g中20mg/kg q3w给药方案观察到的暴露一致(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587;center for drug evaluation and research(2016)bla 761034clinical pharmacology review atezolizumab,可在网址www[dot]accessdata[dot]fda[dot]gov/drugsatfda_docs/nda/2016/761034orig1s000clinpharmr.pdf获得)。1680mg q4w方案在周期1和稳态时的c
max
预测第90个百分位数分别为754μg/ml和1037μg/ml。尽管存在周期1的c
max
比20-mg/kg给药方案更高的趋势,但1680-mg q4w给药方案的预测暴露仍处于研究pcd4989g中针对20mg/kg q3w给药方案观察到的暴露范围内(图28)。
[0563]
稳态时840mg q2w和1680mg q4w方案的预测每周auc分别比模拟的1200mg q3w方案高3.5%和4.8%。
[0564]
在考虑固定剂量方案时,由于清除率和体积受阿特珠单抗poppk模型中体重的影响(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587),与体重较重的患者相比,体重较低的患者预计会表现出更高的阿特珠单抗暴露。为了进一步评估q2w和q4w方案,对于840mg q2w和1680mg q4w的剂量水平,通过体重的四分位数模拟c
min
或c
max
(表9)。
[0565]
对于1680mg q4w方案,最低体重四分位数(《63.7kg,大多数女性)的预测c
max
值在周期1和稳态时分别为692和950μg/ml,这处于针对1200mg q3w和20mg/kg q3w观察到的c
max
值范围内(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587;center for drug evaluation and research(2016)bla 761034clinical pharmacology review atezolizumab,可在网址www[dot]accessdata[dot]fda[dot]gov/drugsatfda_docs/nda/2016/761034orig1s000clinpharmr.pdf获得)。对于840mg q2w方案,最高体重四分位数(》90.9kg,大多数男性)的预测c
min
值在周期1和稳态时分别为58和158μg/ml,这处于针对1200mg q3w观察到的c
min
值范围内并且高于6μg/ml的c
min
靶标浓度。
[0566]
如上所述,采用1680mg q4w方案的最低体重患者的预测c
max
值处于研究pcd4989g中观察到的20mg/kg q3w给药方案的c
max
值范围内(图28)。
[0567]
表9.按体重四分位数模拟阿特珠单抗c
max
和c
min
值。
[0568][0569]
总之,1680mg q4w和840mg q2w方案预计与批准的1200mg q3w方案具有相当的疗效(例如orr和os)和安全性。由于840mg q2w和1680mg q4w方案的预测暴露量(c
min
)超过靶标浓度(6μg/ml)并且处于批准的1200mg q3w方案的c
min
值范围内,因此在以1200mg q3w给药的nsclc或uc患者中,阿特珠单抗暴露与orr或os之间没有临床意义的er关系(参见实例2),与批准的1200mg q3w方案相比,使用840mg q2w或1680mg q4w方案预计对于应答没有影响。
[0570]
类似地,由于840mg q2w和1680mg q4w方案的预测c
max
值处于通常耐受性良好的最大评估剂量20mg/kg的c
max
值范围内,并且在给药1200mg q3w或20mg/kg的nsclc或uc患者中,阿特珠单抗暴露与等级≥3的ae或aesi之间没有临床意义的er关系(参见实例3),预计
840mg q2w和1680mg q4w方案具有类似于批准的1200mg q3w方案的安全性概况。对以下患者安全性概况的详细评估进一步支持了这一点:(1)接受20mg/kg q3w给药方案与1200mg q3w给药方案的患者,(2)低bw的患者,(3)c
max
高于1680mg q4w方案的预测第90个百分位的患者,(4)c
max
高于1680mg q4w的预测平均值的患者(参见实例6-9)。
[0571]
实例5
[0572]
在tnbc中验证poppk预测的840mg q2w暴露
[0573]
在本实例中,3期impassion130(nct02425891)数据用于验证840mg q2w的pk模拟。
[0574]
材料和方法
[0575]
基于先前的1期poppk模型(外部评估)执行预测校正的视觉预测检查(pcvpc)。使用1期poppk模型基于在impassion130中观察到的阿特珠单抗浓度-时间曲线推导出个体pk参数估计值。使用实际给药和患者协变量(体重、性别、ada状态、白蛋白水平和肿瘤负荷)和1期poppk模型模拟了impassion130中经阿特珠单抗治疗的患者的pk数据(1000次重复)。将impassion130中观察到的阿特珠单抗峰值(c
max
)和谷值(c
min
)浓度与相应的预测分布进行比较。
[0576]
结果
[0577]
作为1期poppk模型的外部评估并确认840mg q2w pk模拟,来自impassion130研究的阿特珠单抗加白蛋白结合型紫杉醇q2w组的pk基于基线患者协变量(pcvpc)进行模拟。四百四十三例(共445例)接受阿特珠单抗治疗的患者具有可评估的血清样品用于pk分析,总共2232个样品。结果如图29所示。剂量1和稳态暴露度量与基于1期poppk模型为840mg q2w给药方案预测的度量相似。对于poppk模型,观察到在长期施用(剂量2、4、6、14和30+)后阿特珠单抗暴露数据(谷值)的中位数和第五个百分位数有低估的趋势,这与阿特珠单抗的时间依赖性清除率一致(tecentriq(阿特珠单抗)[包装插页]。south san francisco,ca:基因泰克公司;2019年。south san francisco,ca,usa:基因泰克公司)。
[0578]
实例6
[0579]
来自研究pcd4989g的临床安全性数据汇总,包括20mg/kg q3w(研究pcd4989g中测试的最高剂量)
[0580]
20mg/kg q3w剂量提供的临床暴露范围类似于1680mg q4w固定剂量给药方案的预测稳态最大或c
max
浓度759μg/ml。在20mg/kg剂量水平未观察到剂量限制性毒性,并且报告的ae的发生率和强度并未显示出依赖于剂量。因此,尚未确定最大耐受剂量。
[0581]
在本实例中,分析了研究pcd4989g中阿特珠单抗的安全性。
[0582]
对c
max
高于或低于针对1680mg剂量的预测c
max
的患者的不良事件的分析
[0583]
在来自pcd4989g研究的640例可安全评估的患者中,82例患者被鉴定为在任何时间观察到的c
max
高于759μg/ml;其中62例患者来自20mg/kg剂量组。然后将这组82例患者的观察到的安全性与在研究pcd4989g中观察到的c
max
≤759μg/ml的其余558例患者进行比较(表10)。
[0584]
表10.pcd4989g研究中患者的总体安全性概况。
[0585][0586]
总体而言,在研究pcd4989g中,观察到的c
max
》759μg/ml的82例患者和观察到的c
max
≤759μg/ml的558例患者的安全性概况似乎与阿特珠单抗单一疗法或基线疾病的已知风险相当并一致。
[0587]
例如,在常见的ae(≥20%的患者)中,c
max
》759μg/ml的患者和c
max
≤759μg/ml的患者中的大多数相似,包括疲劳、发热、恶心、腹泻、便秘、呼吸困难和食欲下降。在c
max
》759μg/ml的患者和c
max
≤759μg/ml的患者中报告的更高比例的ae(差异≥5%)是疲劳、寒战、流感样疾病、恶心、咳嗽、呼吸困难、排痰性咳嗽、咯血、肺炎、肌肉骨骼疼痛、食欲下降、皮肤干燥、上呼吸道感染和鼻窦炎。这些事件的严重程度大多为1级或2级,但一例恶心和五例呼吸困难报告为3或4级。这些事件被认为预期会在研究治疗或潜在疾病中发生。
[0588]
根据研究人员的评估,c
max
》759μg/ml的患者比c
max
≤759μg/ml的患者经历了更多的研究治疗相关ae(75.6%对69.7%)。大多数最常见的治疗相关ae(≥10%的患者)在c
max
》759μg/ml的患者和
cmax
≤759μg/ml的患者中相似。
[0589]
对c
max
高于或低于针对1680mg剂量的预测c
max
的患者的严重不良事件的分析
[0590]cmax
≤759μg/ml患者中出现严重ae(sae)的患者比例(43.0%)高于c
max
》759μg/ml的患者(35.4%),并且c
max
≤759μg/ml患者中3-4级sae的比例(33.7%)也高于c
max 759μg/ml的患者(25.6%)。两个亚组中报告的常见sae(≥2%的患者)包括呼吸困难(2.4%对3.9%)和发热(3.7%对2.9%)。c
max
≤759μg/ml的亚组中感染和胃肠道疾病的发生频率高于c
max
》759μg/ml的亚组,但是,没有确定个体首选术语(pt)来解释所指出的差异。
[0591]cmax
》759μg/ml的患者没有致命的ae;在c
max
≤759μg/ml的患者中有10起致命ae(1.7%)。10起致命事件包括以下项:呼吸衰竭、肺炎、肺动脉高压、败血症、头部损伤、过量(酒精和吗啡)、急性心肌梗塞、肝衰竭、肝血肿和死亡(原因不明)。
[0592]
在c
max
》759μg/ml的患者中,2例(2.4%)患者报告了导致研究药物停用的ae,这低于c
max
≤759μg/ml患者报告的频率(28,5.0%)。在c
max
》759μg/ml患者组中导致研究药物停用的两起ae为血液胆红素增加和结肠炎,这是阿特珠单抗的已知ae。
[0593]
基于观察到的c
max
》759μg/ml患者的安全性数据分析,预计1680mg q4w剂量的阿特珠单抗具有良好的耐受性,安全性可控。
[0594]
实例7
[0595]
基于来自研究pcd4989g、imvigor211和oak的阿特珠单抗治疗组的安全性分析比较
[0596]
方法
[0597]
分析群体
[0598]
该分析内的安全性群体包括来自研究pcd4989g、imvigor211和oak的患者,这些患者接受了至少一剂阿特珠单抗,根据实际接受的治疗将患者分配到治疗组。以下治疗组和亚组用于安全性分析:
[0599]
研究pcd4989g:
[0600]
ο“pcd4989g 20mg/kg”(n=146):研究pcd4989g中接受阿特珠单抗20mg/kg iv q3w剂量的患者。
[0601]
ο“pcd4989g 1200mg”(n=210):研究pcd4989g中接受阿特珠单抗1200mg iv q3w剂量的患者。
[0602]
按bw分组的研究pcd4989g亚组:
[0603]
ο“最低四分位数bw pcd4989g 20mg/kg”(n=37):在研究pcd4989g中给药20mg/kg阿特珠单抗的患者,其bw处于该群组中bw分布的最低四分位数中。
[0604]
ο“上3个四分位数bw pcd4989g 20mg/kg”(n=109):具有可用于该剂量群组的bw的剩余患者。
[0605]
按周期1观察到的c
max
值分组的研究pcd4989g亚组
[0606]
ο“pcd4989g 20mg/kg》90%-分位数c
max”(n=4):在研究pcd4989g中给药20mg/kg阿特珠单抗的患者在周期1中的c
max
值高于1680mg阿特珠单抗iv的预测c
max
的第90个百分位数。
[0607]
ο“pcd4989g 20mg/kg≤90%-分位数c
max”(n=134):在研究pcd4989g中给药20mg/kg阿特珠单抗的患者在周期1中的c
max
值高达1680mg阿特珠单抗iv的预测c
max
的第90个百分位数。
[0608]
ο“pcd4989g 20mg/kg》平均c
max”(n=40):在研究pcd4989g中给药20mg/kg阿特珠单抗的患者在周期1中的c
max
值高于1680mg阿特珠单抗iv的预测c
max
的均值。
[0609]
ο“pcd4989g 20mg/kg≤平均c
max”(n=98):在研究pcd4989g中给药20mg/kg阿特珠单抗的患者在周期1中的c
max
值高达1680mg阿特珠单抗iv的预测c
max
的均值。
[0610]
如上所述的研究pcd4989g 20mg/kg亚组,但使用患者的周期1模型预测的c
max
值而不是观察到的c
max
值
[0611]
研究go28915(oak;n=609):研究go28915中接受阿特珠单抗1200mg iv q3w剂量的患者。
[0612]
研究go29294(imvigor211;n=459):研究go29294中接受阿特珠单抗1200mg iv q3w剂量的患者。
[0613]
安全性参数
[0614]
研究pcd4989g、imvigor211和oak的ae术语使用药事管理医学辞典(medical dictionary for regulatory activities,meddra 20.1版)编码为首选术语。根据美国国家癌症研究所不良事件通用术语标准4.0版(nci ctcae v4.0)标准对ae严重程度进行分级。
[0615]
出于此分析的目的,使用meddra-标准化smq、发起人定义的不良事件分组术语(aegt)和高级术语(hlt)的一组综合定义,用于从ae临床医学概念数据库中识别鉴定出特别关注的ae(aesi)。医学概念包括与阿特珠单抗相关的重要已鉴定风险以及其他免疫检查点抑制剂报告的潜在风险和类别效应。
[0616]
对需要使用皮质类固醇治疗的aesi进行了单独分析。这些ae是使用以下标准鉴定的:
[0617]
·
ae术语属于特别关注的ae分组
[0618]
·
全身性皮质类固醇的开始日期是在ae发生日期当天或之后最多30天
[0619]
·
全身性皮质类固醇的开始日期在ae解决日期之前
[0620]
皮质类固醇是根据标准药物篮子确定的。全身使用被定义为不具有以下任何施用途径的任何药物:耳部(耳)、膀胱内、玻璃体内、鼻、眼、呼吸(吸入)、局部或阴道。
[0621]
为了捕捉潜在的输注相关反应(irr),对在阿特珠单抗输注期间或之后24小时内发生的ae进行了分析。
[0622]
结果
[0623]
安全性概况概述
[0624]
如图30中所示,阿特珠单抗以20mg/kg q3w剂量给药的总体安全性与以1200mg q3w固定剂量给药时观察到的相似。观察到的发生率跨治疗组存在一些差异,与其他治疗组相比,研究pcd4989g 20mg/kg的aesi和irr(输注后24小时内的ae)发生率更高。对于aesi,更频繁地观察到免疫介导的皮疹以及肝功能测试异常,而对于irr,20mg/kg治疗组的发生率较高主要是由于关节痛、皮疹和寒战事件较多。
[0625]
常见的ae
[0626]
对于所有治疗组,相似比例的患者经历了至少一次任何级别的ae(99.3%pcd4989g 20mg/kg对96.7%pcd4989g 1200mg对94.4%oak对95.9%imvigor211)。
[0627]
20mg/kg和1200mg治疗组中最常见的ae相似。与任何1200mg治疗组相比,20mg/kg组中有≥10%差异的患者是呼吸困难、恶心和呕吐的全身症状。其中,与所有1200mg治疗组相比,在20mg/kg群组中观察到的唯一发生率更高的事件是呼吸困难(pcd4989g(20mg/kg,n=146)中为32.9%;18%为pcd4989g(1200mg,n=210);oak(1200mg,n=609)中为19.5%;imvigor211(1200mg,n=459)中为15.0%)。个体ae发生率的这些发现被认为是继发于潜在疾病,不太可能是由于20mg/kg群组中的潜在暴露。
[0628]
按强度分类的ae
[0629]
imvigor211中出现至少一次≥3级ae的患者比例(59.5%)高于其他治疗组(49.3%pcd4989g 20mg/kg对55.2%pcd4989g 1200mg对40.2%oak)。
[0630]
观察到的贫血(5.5%pcd4989g 20mg/kg(n=146)对5.7%pcd4989g1200mg(n=210)对2.3%oak 1200mg(n=609)对10.2%imvigor211 1200mg(n=459))和尿路感染(0.7%pcd4989g 20mg/kg(n=146)对1.4%pcd4989g 1200mg(n=210)对0.2%oak 1200mg(n=609)对5.7%imvigor211 1200mg(n=459))的发生率有≥5%的跨治疗组差异。在imvigor211中,贫血和尿路感染的报告频率更高,这与在膀胱癌群体中通常观察到的情况一致。
[0631]
严重ae
[0632]
总体而言,在所有治疗组中,经历至少一起sae的患者比例相似,但oak中的发生率较低(42.5%pcd4989g 20mg/kg对44.3%pcd4989g1200mg对33.5%oak对45.5%imvigor211)。与1200mg治疗组相比,20mg/kg组中有≥2%差异的是呼吸困难、腹痛、胸腔积液和骨痛的pt。其中,与任何1200mg治疗组相比,在20mg/kg群组中观察到的唯一发生率更高的事件是呼吸困难(6.2%pcd4989g 20mg/kg(n=146);3.8%pcd4989g 1200mg(n=210);2.1%oak 1200mg(n=609);1.5%imvigor211 1200mg(n=459))。个体ae发生率的这一发现被认为是继发于潜在疾病,不太可能是由于20mg/kg群组中的潜在暴露。
[0633]
导致退出的ae
[0634]
20mg/kg治疗组中导致退出的ae发生率为4.8%,而pcd4989g 1200mg为4.3%,oak中为8.2%,imvigor211中为8.1%。
[0635]
在20mg/kg群组中有7例患者因以下事件而停用阿特珠单抗:心力衰竭、死亡、虚弱、疾病进展、膀胱癌、缺氧和呼吸衰竭。
[0636]
特别关注的ae
[0637]
在所有治疗组中,与1200mg治疗组(36.2%pcd4989g 1200mg对32.7%oak对33.8%imvigor211)相比,20mg/kg治疗组中至少有一起aesi的患者比例(47.3%)更高。
[0638]
所有治疗组中最常报告的事件是免疫介导的皮疹(17.1%pcd4989g20mg/kg对6.7%pcd4989g 1200mg对9.7%oak对11.3%imvigor211)和肝功能测试升高(alt升高[6.2%对10.5%对5.7%对4.1%],ast增加[6.2%对11.4%对6.2%对4.4%])。
[0639]
20mg/kg治疗组中aesi发生率较高的主要原因是免疫-介导的皮疹事件较多,大多数为1-2级。其他aesi的发生率和类型在治疗组之间相似。
[0640]
所有治疗组之间接受糖皮质激素来治疗aesi的患者的比例相似(9.6%pcd4989g 20mg/kg对9.5%pcd4989g 1200mg对9.2%oak对9.2%imvigor211)。
[0641]
最常见的(任何治疗组中》2%的患者)需要使用皮质类固醇的aesi包括肺炎(2.7%对1.4%对1.0%对1.1%)、alt增加(0%对2.9%对1.0%对0.4%),以及ast增加(0%对2.9%对0.8%对0.7%)。
[0642]
输注后24小时内发生的ae
[0643]
20mg/kg治疗组在输注后24小时内经历至少一起ae的患者比例(83.6%)高于1200mg治疗组(68.6%pcd4989g 1200mg对70.4%oak对67.5%imvigor211)。
[0644]
20mg/kg治疗组发生率较高的主要原因是更多的关节痛事件(9.6%pcd4989g 20mg/kg(n=146);4.8%pcd4989g 1200mg(n=210);4.4%oak 1200mg(n=609);3.3%imvigor211 1200mg(n=459))、皮疹事件(6.8%pcd4989g 20mg/kg(n=146);1.4%;3.6%oak 1200mg(n=609);2.6%imvigor211 1200mg(n=459))和寒战事件(5.5%pcd4989g 20mg/kg(n=146);1.0%pcd4989g 1200mg(n=210);1.6%oak 1200mg(n=609);2.0%imvigor211 1200mg(n=459))。所有事件均报告为1-2级。输注后24小时内发生的其他ae的发生率和类型在治疗组之间大体相似。
[0645]
24小时内ae发生率较高可能是由于数据捕获方法:在研究pcd4989g中,与irr相关的事件被捕获为单起ae,而研究oak和imvigor211捕获了irr的诊断而不是单起ae。此外,在输注后24小时内报告的最常见ae主要是已知在该患者群体中发生的全身症状(例如,食欲下降、疲劳、虚弱)。irr为阿特珠单抗和其他单克隆抗体的已知风险。虽然关节痛、皮疹和寒
战可能是通常与irr发展相关的症状集群的一部分,但这些全身症状也可能与并发疾病或潜在疾病一起出现。此外,在所有亚组中,这些ae也在输注后的24-小时窗口之外报告。因此,认为irr的发展与20mg/kg治疗组无关。
[0646]
实例8
[0647]
按周期1期间的c
max
分组的研究pcd4989g 20mg/kg中的患者亚组:低于或高于针对1680mg剂量的预测c
max
的90%-分位数值
[0648]
pcd4989g 20mg/kg治疗组中,在周期1中观察到的c
max
值》针对1680mg剂量的预测c
max
值的90%-分位数的患者数量非常少(n=4),因此没有从这些分析中得出数据解释或结论。
[0649]
然而,在pcd4989g 20mg/kg中观察到的》90%-分位数c
max
亚组中四例患者的≥3级ae的描述性安全性信息如下所示:
[0650]
·
患者a在第81天死于恶性肿瘤进展,报告为5级事件。该患者也有肝转移史,并且在第64天经历了血液胆红素增加的4级ae,并且在第70天经历了alt和ast增加的3级ae。
[0651]
·
患者b在第43天报告了高血压的3级ae,在第923天报告了病理性骨折的3级ae。
[0652]
·
患者c分别在第44、93和102天报告了国际标准化比增加、疲劳和呼吸困难的3级ae。
[0653]
·
患者d在第145天死于恶性肿瘤进展,报告为5级ae。
[0654]
总体而言,使用观察到的c
max
的pcd4989g 20mg/kg周期1c
max
亚组分析的结果与使用模型预测的c
max
的结果非常相似(表11)。
[0655]
表11.接受阿特珠单抗20mg/kg iv q3w,按周期1期间观察到或建模得到的c
max
区分的患者中的不良事件的总体汇总(低于/高于针对1680mg阿特珠单抗iv预测的90%-分位数c
max
)(阿特珠单抗治疗的安全性可评估患者)。
[0656][0657]
实例9
[0658]
按周期1期间的c
max
分组的研究pcd4989g 20mg/kg中的患者亚组:低于或高于针对1680mg剂量的预测c
max
的均值
[0659]
在本实例中,分析了研究pcd4989g中的患者亚组的安全性。
[0660]
材料和方法
[0661]
总结了患者亚组的ae频率:(1)来自pcd4989g,接受阿特珠单抗20mg/kg q3w,基于与针对1680-mg q4w方案预测的c
max
相关的c
max
值;以及(2)来自pcd4989g和oak,基于体重四分位数(最低四分位数vs四分位数2-4)。在这些分析中,还规定了aesi是否需要使用皮质类固醇。
[0662]
结果
[0663]
表12提供了pcd4989g中20mg/kg q3w阿特珠单抗治疗患者的安全性汇总,其中在周期1期间观察到的c
max
相对于1680mg q4w方案的平均预测c
max
。研究pcd4989g 20mg/kg患者亚组的总体安全性概况,在周期1期间观察到的c
max
≤与》针对1680mg剂量的预测c
max
平均值的患者之间大体是相似的(表12)。一般而言,这些组之间的ae频率相似。在基于pcd4989g患者的建模得到的c
max
(即,通过poppk模型评估的个体预测)的组中,获得了相对于1680-mg q4w方案的预测c
max
均值的相似结果。
[0664]
总体而言,pcd4989g 20mg/kg在周期1期间观察到的c
max
的结果与pcd4989g 20mg/
kg在周期1期间建模得到的c
max
相似。
[0665]
表12.接受阿特珠单抗20mg/kg iv q3w(pcd4989g),按周期1期间观察到或建模得到的c
max
区分的患者中的不良事件的总体汇总(低于/高于针对1680mg阿特珠单抗iv预测的平均c
max
)(阿特珠单抗治疗的安全性可评估患者)。
[0666][0667]
对于两个治疗亚组,相似比例的患者经历了至少一起任何级别的ae(在观察到的≤平均c
max
亚组中为99.0%,而在观察到的》平均c
max
亚组中为100.0%)。发生率差异≥10%的任何级别的ae是食欲下降(在》平均c
max
亚组中更常见)和贫血(在≤平均c
max
亚组中更常见)。
[0668]
在观察到的≤平均c
max
亚组中经历了至少一起≥3级ae的患者比例(53.1%)高于观察到的》平均c
max
亚组(35.0%)。
[0669]
pt报告的最常见(任一治疗组中》5%的患者)的≥3级ae为呼吸困难、贫血和疲劳(表13)。在》平均c
max
亚组中,没有发生率较高(≥5%)的≥3级ae;在≤平均c
max
亚组中比在观察到的》平均c
max
亚组中更常见的事件是呼吸困难和贫血。
[0670]
表13.任何亚组中》5%的患者(经阿特珠单抗治疗的安全性可评估患者)报告的≥3级ae
[0671][0672]
对周期1c
max
低于或高于针对1680mg剂量的预测c
max
的患者的严重不良事件的分析
[0673]
对于两个治疗亚组,相似比例的患者经历了至少一起sae(观察到的≤平均c
max
亚组中为43.9%,而观察到的》平均c
max
亚组中为37.5%)。呼吸困难在≤平均c
max
亚组中比在观察到的》平均c
max
亚组中更常见(表14)。
[0674]
表14.任何亚组中≥5%的患者(经阿特珠单抗治疗的安全性可评估患者)报告的严重不良事件。
[0675][0676]
对导致周期1c
max
低于或高于针对1680mg剂量的预测c
max
的患者退出的不良事件的分析
[0677]
总体而言,很少有患者因ae而停用阿特珠单抗(观察到的≤平均c
max
亚组中的5.1%对观察到的》平均c
max
亚组中的2.5%)。在单个患者中报告了导致退出的事件。在≤平均c
max
亚组中的5例患者因心力衰竭、虚弱、死亡、疾病进展、缺氧和呼吸衰竭而退出。在》平均c
max
亚组中的一例患者因疾病进展而退出。
[0678]
对周期1c
max
低于或高于针对1680mg剂量的预测c
max
的患者的特别关注不良事件的分析
[0679]
总体而言,两个亚组中相似比例的患者至少经历了一起aesi(观察到的≤平均c
max
亚组中为48.0%,而观察到的》平均c
max
亚组中为45.0%)。免疫介导的皮疹(19.4%对12.5%)和肝功能测试异常(alt增加7.1%对5.0%;ast增加6.1%对7.5%)是两个亚组中最常报告的aesi。
[0680]
总体而言,两个亚组中相似比例的患者接受了针对aesi的皮质类固醇(观察到的≤平均c
max
亚组中为8.2%,而观察到的》平均c
max
亚组中为10.0%)。最常报告的需要使用皮质类固醇的aesi是肺炎(每个亚组2例患者)和皮疹(2例患者对0例患者)。
[0681]
对周期1c
max
低于或高于针对1680mg剂量的预测c
max
平均值的患者在输注后24小时内发生的不良事件的分析
[0682]
在观察到的》平均c
max
亚组中在输注后24小时内经历ae的患者比例(95.0%)高于观察到的≤平均c
max
亚组(79.6%)。
[0683]
在观察到的》平均c
max
亚组中更频繁发生(≥5%)的事件是恶心、虚弱和腹泻(表15)。
[0684]
表15.在任何亚组中,》10%的患者(经阿特珠单抗治疗的安全性可评估患者)报告的在输注后24小时内发生的常见不良事件。
[0685][0686]
按剂量组区分的安全性
[0687]
观察到的安全性数据通过暴露亚组评估。
[0688]
表16提供了按剂量组区分阿特珠单抗暴露的pcd4989g的汇总。在10mg/kg q3w至20mg/kg q3w的剂量范围和1200mg q3w组中,中位治疗持续时间为2.07至9.48个月,中位剂量数为4至14.5。
[0689]
表16.按剂量组区分的阿特珠单抗暴露:来自pcd4989g的经阿特珠单抗治疗的患者。
[0690][0691]
表17提供了按剂量组区分的pcd4989g患者的安全性汇总。15mg/kg q3w、20mg/kg q3w和1200mg q3w组的总体安全性概况是一致的。相对于其他剂量组,10mg/kg q3w剂量组的患者表现出严重不良事件(ae)和治疗相关ae的频率增加。这可能是由于,相对于其他剂量组,该剂量组的安全性随访时间更长且患者数量更少。
[0692]
表17.按剂量组区分的ae总结:来自pcd4989g的经阿特珠单抗治疗的患者。
[0693][0694][0695]
按体重区分的安全性
[0696]
观察到的安全性数据通过暴露和体重亚组评估。
[0697]
表18提供了按体重区分的pcd4989g和oak患者的安全性汇总。pcd4989g中20mg/kg治疗组的中位体重为78.2kg(q1-q3,63.7-93.0kg),并且总体安全性概况在最低(n=37)与上3个(n=109)体重四分位数的患者之间是相似的。在最低体重四分位数亚组中观察到更高的3至5级ae发生率(48.7%对37.3%),这是由于3级ae(38.8%对27.8%)。对3级ae的评估未鉴定任何单起ae首选术语,亚组之间的差异≥2%。亚组间差异≥5%的严重ae包括疲劳和虚弱(两者均常见于恶性肿瘤)以及肺炎和心脏压塞(已知的胸癌并发症),所有此类事件都很少发生。在体重最低的亚组中,只有虚弱和呼吸系统并发症导致研究治疗停止;对于其他事件,未对研究治疗采取任何行动。为了在更大的患者群组中评估体重的影响,还分析了来自oak(1200mg q3w给药)的ae数据。体重中位数为71.0kg(q1-q3,59.5-82.2kg)。未在最低(n=152)与高3(n=442)体重四分位数之间观察到差异。
[0698]
表18.按体重区分的ae汇总:来自pcd4989g和oak的经阿特珠单抗治疗的患者。
[0699][0700][0701]
实例10
[0702]
免疫原性分析
[0703]
在研究pcd4989g、jo28944、imvigor210、imvigor211、birch、poplar、fir和oak中评估了阿特珠单抗的免疫原性。
[0704]
对于研究pcd4989g中20mg/kg q3w vs oak中1200mg q3w vs imvigor 211中1200mg q3w的基线后治疗中-突发ada发生率的分析显示,20mg/kg剂量的治疗中-突发ada发生率没有明显增加(表19)。
[0705]
表19.针对以下q3w给药的基线后治疗中突发ada发生率:pcd4989g中20mg/kg,oak和imvigor 211中1200mg。
[0706][0707]
ada血清样品中存在阿特珠单抗会干扰ada检测。在验证实验中,ada测定能够在
200μg/ml阿特珠单抗存在的情况下检测到500ng/ml替代阳性对照抗-阿特珠单抗抗体。以下百分比的基线后ada样品具有低于200μg/ml的阿特珠单抗浓度,这是基于替代阳性对照的ada检测的药物耐受水平:研究pcd4989g 80.2%、imvigor210 86.0%、imvigor21188.2%、birch 82.8%、poplar 89.6%、fir 86.9%和oak 81.9%。
[0708]
免疫原性数据高度依赖于所用测试方法的灵敏度和特异性。此外,在测试方法中观察到的阳性结果的发生率可能受到多种因素的影响,包括样品收集的时间、药物干扰、伴随用药和潜在疾病。因此,将阿特珠单抗抗体发生率与其他产品抗体发生率进行比较可能会产生误导。
[0709]
治疗中突发ada的存在对uc患者中阿特珠单抗药代动力学的影响
[0710]
尽管治疗中突发ada阳性的发生率(在研究pcd4989g、jo28944、imvigor210和imvigor211中为16.7%至41.9%),但nca分析表明ada阳性对剂量为10至20mg/kg,包括固定剂量1200mg q3w的阿特珠单抗暴露的影响较小。poppk分析还表明,治疗中突发ada的存在对阿特珠单抗暴露的影响较小。与ada-阴性患者相比,ada-阳性患者的阿特珠单抗清除率有相对较小增加,为16%(例如,参见实例1)。在所有研究中,对于接受阿特珠单抗剂量≥10mg/kg的患者,在ada-阳性患者中c
min
维持在超过6μg/ml的靶标血清浓度。
[0711]
治疗中突发ada的存在对nsclc患者中阿特珠单抗药代动力学的影响
[0712]
跨不同的临床研究,尽管ada-阳性亚组中存在较低c
min
值的趋势,但治疗中突发ada阳性似乎并未对阿特珠单抗浓度和药代动力学产生重大影响。poppk模型确定ada-阳性亚组的药物清除率比ada-阴性患者高16%,这解释了ada-阳性患者中暴露降低的趋势(例如,参见实例1)。在所有研究中,对于≥10mg/kg的剂量,在ada-阳性患者中,c
min
良好地保持为超过6μg/ml的靶标血清浓度。
[0713]
治疗中突发ada的存在对uc患者中阿特珠单抗疗效的影响
[0714]
对于跨针对uc的研究pcd4989g、imvigor210和imvigor211的orr的概述并未证明治疗中突发ada阳性始终与较低的orr相关。对imvigor211的分析显示,所有患者或ic1/2/3或ic2/3组的ada阳性与ada-阴性患者之间没有临床相关差异,其中结果测量值(os、pfs、orr和dor)的95%ci重叠。
[0715]
治疗中突发ada的存在对nsclc患者中阿特珠单抗疗效的影响
[0716]
ada-阳性与ada阴性患者之间的orr通常具有可比性,并且在存在数值差异的情况下,95%ci重叠,并且orr没有跨研究一致的增加或减少。总体而言,治疗中突发ada对基于orr的疗效没有明显影响,并且ada阴性和ada-阳性患者的置信区间重叠。
[0717]
总体而言,在ada阳性患者与ada阴性患者之间未观察到临床相关差异。poplar的os不具判断力;与ada阴性患者相比,ada阳性患者的poplar中位pfs在数值上更高,但pfs的95%ci重叠。对于oak研究,尽管与ada阳性患者相比,ada阴性患者的中位os、标志性os率和中位pfs在数值上更高,但这些结果测量的95%ci重叠。
[0718]
治疗中突发ada的存在对阿特珠单抗安全性的影响
[0719]
在所有患者群体中,治疗中-突发ada(治疗诱导和增强)的基线后发生率为42.5%(540/1272),这与所有uc群体(41.9%[161/384])和所有nsclc群体中的观察结果(42.7%[379/888])一致。
[0720]
无论基线后ada状态如何(阴性或阳性),所有级别ae、5级ae、导致治疗停止的ae、
导致剂量中断的ae和aesi的发生率相似。在3-4级ae中观察到一些数量差异(ada阴性患者中为38.4%vs ada阳性患者为44.3%),这主要是由ada阳性患者胃肠道疾病soc中报告的ae造成的(5.7%vs.8.5%),但无法鉴定个体pt来解释这种差异。ada阳性患者(40.2%)的sae发生率高于ada阴性患者(33.5%),但这种差异并非由任何特定soc或单起ae首选术语造成。
[0721]
在所有患者群体中,ada阳性和ada阴性患者的超敏反应和irr(meddra ae pt)发生率较低且一致。18例患者(1.4%)报告了超敏反应事件:8例ada阴性患者(1.1%)和10例ada阳性患者(1.9%)。20例患者(1.6%)发生输注相关反应:11例ada阴性患者(1.5%)和9例ada阳性患者(1.7%)。
[0722]
实例11
[0723]
使用预测的阿特珠单抗1680mg q4w固定剂量评估毒理学安全性边际
[0724]
1680-mg q4w给药方案代表1mg/kg的剂量,或比之前向患者施用的最高剂量高5%的以mg/kg计的剂量。如在前面的实例中所指出的,针对1680mg q4w的周期1和稳态时的预测c
min
低于针对20mg/kg q3w的预测值。在周期1和稳态时的预测c
max
分别比20mg/kg q3w给药方案高12%和0.8%。鉴于针对1680mg q4w的预测c
max
较高,对阿特珠单抗毒理学边际进行了重新评估。
[0725]
使用食蟹猴重复给药毒性研究中的最高耐受剂量50mg/kg和当前1200mg q3w剂量水平的人类pk参数评估840mg q2w和1680mg q4w方案的毒理学安全性边际(图31)。使用以下方法计算阿特珠单抗的安全系数:
[0726]
·
基于暴露auc:分别在重复给药食蟹猴毒理学研究(aucanimal/auchuman)中比较在建议的临床剂量下的预测auc与在最高耐受50mg/kg剂量水平下计算的auc。在食蟹猴的26周重复给药毒性研究(研究13-3278)中,动物每周以最高耐受剂量50mg/kg给药(即,与患者的q3w方案相比更为频繁)。因此,在3周的时间段内(与患者的q3w给药方案相匹配),猴子接受了150mg/kg的总剂量(即,50mg/kg每周一次
×
3周)。使用150mg/kg的总剂量和3.7ml/天/kg的猴子cl值,计算出猴子的auc为40,500天
·
μg/ml(即,150mg/kg除以3.7ml/天/kg)。将计算出的40,500天
·
μg/ml的猴子暴露与6,409天
·
μg/ml的人稳态暴露(来自1200mg给予q3w,研究pcd4989g)进行比较,得出6倍的安全性边际(即40,500除以6,409)。使用模拟临床auc对840mg q2w和1680mgq4w方案进行了类似的计算(图31)。
[0727]
·
基于浓度c
max
:将研究pcd4989g中报告的1200mg q3w方案的c
max
或提议的840mg q2w和1680-mg q4w方案的模拟临床c
max
分别与在重复剂量食蟹猴研究中的最高耐受剂量50mg/kg下观察到的该值进行比较(c
max
动物/c
max
人类)(图31)。对食蟹猴静脉注射(iv)27次剂量为50mg/kg的阿特珠单抗后的c
max
为3,680μg/ml。
[0728]
如上所示,基于暴露和浓度分析,阿特珠单抗在食蟹猴中的药代动力学和毒代动力学提供了足够的安全性边际,以支持840mg q2w和1680mg q4w临床给药方案。
[0729]
实例12
[0730]
1200mg q3w、840mg q2w和1680mg q4w给药方案的可互换性
[0731]
已批准的阿特珠单抗1200mg q3w给药方案的有效性和安全性已经例如在2l nsclc、2l muc和/或1l不符合使用顺铂进行治疗的条件的muc患者中确立。为了在患者护理中提供更大的便利和灵活性,本文提供了840mg q2w和1680-mg q4w作为iv输注的给药方
案。这些新的给药方案旨在与阿特珠单抗1200mg q3w给药方案互换。
[0732]
已根据前述实例中所述的八项临床研究对可用的阿特珠单抗单药治疗pk和er数据对uc和nsclc进行了评估。主要发现包括:
[0733]
·
当将阿特珠单抗作为单一疗法向muc或nsclc患者施用时,未鉴定出具有临床意义的暴露-功效或暴露-安全行关系。
[0734]
·
基于840mg q2w和1680mg q4w给药方案的基于模型的模拟,预测暴露处于使用1200mg q3w阿特珠单抗观察到的暴露范围内。840-mg q2w和1680-mg q4w给药方案在周期1和稳态时的预测c
min
浓度高于6μg/ml的靶标c
min
浓度。
[0735]
·
对阿特珠单抗的ada的总体治疗中突发发生率对于pk、疗效或安全性没有临床意义的影响。使用20mg/kg剂量,治疗中突发ada的发生率没有明显增加。
[0736]
基于来自研究pcd4989g、oak和imvigor211的安全性数据:
[0737]
·
观察到c
max
》759μg/ml(即针对阿特珠单抗1680mg q4w的预期c
max
)的患者对给药方案耐受良好,与c
max
≤759μg/ml的患者相比,未发现安全性差异。
[0738]
·
接受20-mg/kg q3w给药方案和1200-mg q3w给药方案的患者的总体安全性概况相似。
[0739]
·
具有较低或较高bw的患者在安全性概况中没有观察到有意义的差异。
[0740]
已开发出一种新的阿特珠单抗840mg呈现方式,以支持阿特珠单抗840mg q2w和1680mg q4w给药方案。这些其他给药方案利用了新的840mg呈现方式(一瓶840mg阿特珠单抗用于840mg q2w计划;两瓶840mg阿特珠单抗用于1680mg q4w计划)。阿特珠单抗的制剂(即1200-mg和840mg两种呈现方式均为活性物质浓度60mg/ml的相同强度)不变,并且赋形剂和新包装材料的成分不变。
[0741]
根据pk建模和模拟、er评估、安全性分析和免疫原性数据的结果,预计在nsclc和uc中,在提议的阿特珠单抗剂量840mg q2w和1680mg q4w与目前批准的剂量1200mg q3w之间在暴露、疗效和安全性方面不会有临床意义的差异。
[0742]
基于现有证据,可以合理地得出结论,1200mg q3w、840mg q2w和1680mg q4w给药方案认为可以互换。此处使用“可互换”是指任何阿特珠单抗给药方案都可以替代另一种给药方案,具体给药方案的选择可以基于患者特定因素,诸如阿特珠单抗给药与患者护理的其他方面的协调。
[0743]
结论
[0744]
这项研究的结果支持840mg q2w、1200mg q3w和1680mg q4w阿特珠单抗给药方案的互换使用,因为预计它们将表现出相当的疗效和安全性,同时为患者提供更大的治疗灵活性和便利性。
[0745]
提议的840mg q2w和1680-mg q4w给药方案的总体获益/风险特征与目前批准的1200-mg q3w给药方案相当,后者已被认为在nsclc和uc患者中具有积极效果。除了1200-mg q3w给药方案之外,新的840mg q2w和1680-mg q4w给药方案为患者护理提供了更大的灵活性和便利性,例如,通过减轻治疗负担和提高生活质量,以及提高治疗设施资源利用率。
[0746]
以上提供的结果表明,在安全性或有效性方面没有观察到显著的er关系。针对840mg q2w和1680mg q4w的预测暴露量与1200mg q3w和mad相当,并且与来自impassion130的观察到的pk数据一致。观察到的安全性在c
max
高于和低于针对1680mg q4w的预测c
max
的患
者之间以及在体重处于最低和上3个四分位数的患者之间相似。
[0747]
简而言之,所有使用q3w给药频率评估的剂量水平的数据,包括1200mg q3w和20mg/kg q3w(1期研究pcd4989g中的mad),表明没有临床意义的暴露-疗效或暴露-安全性关系。这些数据表明,如果新的给药方案达到针对1200mg q3w或20mg/kg q3w观察到的暴露范围内的暴露,则不太可能影响疗效或安全性。pk模拟表明,新的给药方案840mg q2w和1680mg q4w预计可实现与目前批准的方案1200mg q3w大致相当的暴露,并且处于来自1200mg q3w和20mg/kg剂量水平的观察到的暴露范围内。对c
max
高于和低于1680mg q4w方案的预测c
max
的患者观察到的安全性概况的进一步表征也支持,1680mg q4w方案的安全性概况预计与q3w方案的临床经验相似。
[0748]
1680mg q4w给药方案的pk模拟也表明与当前批准的1200mg q3w方案的总体暴露相当,而预测的稳态c
min
比当前批准的方案低6%;这个浓度也超过了靶标浓度。当与20mg/kg剂量相比时,预计周期1和稳态时的几何平均c
max
(分别为12%和0.8%)有小幅增加;然而,1680mg q4w方案的预测c
max
处于在1期研究pcd4989g中观察到的范围内。此外,来自pcd4989g的以20mg/kg q3w治疗的患者均具有相当的安全性,无论他们的c
max
是高于还是低于针对1680mg q4w方案的预测周期1值。
[0749]
类似于1200mg q3w方案的观察结果(stroh等人,(2017)clin pharmacol ther doi:10.1002/cpt.587),体重对暴露的影响预计对840mg q2w或1680mg q4w方案没有临床意义,因为低体重和高体重患者的预测暴露均处于从1200mg q3w和20mg/kg剂量水平观察到的暴露范围内。这些结果也得到了pcd4989和oak研究按体重区分的安全性分析的进一步支持,这表明观察到的总体安全性在体重处于最低和上3个四分位数的患者之间大体相似。
[0750]
维持蛋白质治疗剂的c
min
水平被认为不仅可以提供最一致的疾病控制,而且可以最小化ada发展的可能性。来自tnf抑制剂研究的临床数据表明,与持续存在相同水平的相同蛋白质相比,间歇性地暴露于蛋白质治疗剂(即暴露后完全清除,然后重新暴露)更有可能诱导免疫应答。840mg q2w和1680q4w方案的预测c
min
水平远远超过靶标浓度(6μg/ml),并且处于批准的1200mg q3w方案的c
min
值范围内。因此,预计840mg q2w或1680mg q4w方案不会导致完全清除和再暴露周期,从而导致比批准的1200mg q3w方案更高的免疫原性率。
[0751]
以较低频率给药方案(即1680mg q4w)施用阿特珠单抗的能力为患者、护理人员和医疗保健提供者提供了更大的灵活性和便利性。由于阿特珠单抗为静脉内施用,与更频繁给药的方案相比,1680mg q4w给药方案可能会减少接受治疗所需的时间(例如,访问治疗中心的次数)。此外,在整个治疗过程中切换方案的能力也将提供更大的灵活性,因为可以匹配给药方案以满足每个患者不断变化的需求。
[0752]
鉴于预测暴露处于观察到的暴露量范围内并且没有临床意义的er关系,840mg q2w和1680mg q4w的阿特珠单抗方案预计具有与批准的1200mg q3w方案相当的疗效和安全性。此外,由于阿特珠单抗pk在适应症之间以及与评估的各种药物(包括但不限于化疗、抗肿瘤药物和酪氨酸激酶抑制剂)联合使用时一致,因此这些结果适用于阿特珠单抗作为单一疗法或联合使用。
[0753]
总之,840mg q2w和1680mg q4w的阿特珠单抗方案预计与批准的1200mg q3w方案具有相当的疗效和安全性,支持它们的互换使用并为患者提供更大的灵活性。
[0754]
因此,本文提供的分析支持840mg q2w、1200mg q3w和1680mg q4w的阿特珠单抗给
药方案的互换使用,为患者在阿特珠单抗治疗期间提供更大的灵活性和便利性。这些数据有助于fda扩大针对某些类型癌症的阿特珠单抗给药方案(tecentriq(阿特珠单抗)[包装插页]。south san francisco,ca:基因泰克公司;2019年。south san francisco,ca,usa:基因泰克公司)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1