均匀分散有生理活性物质的微球及含有其的缓释制剂的制作方法
1.本发明涉及均匀分散有生理活性物质的微球及含有其的缓释制剂。本发明特别涉及均匀分散有生理活性物质的以乳酸/乙醇酸共聚物(plga)或聚乳酸(pla)为主要成分的微球及含有其的缓释制剂。
背景技术:
2.作为含有生理活性物质的药品等缓释制剂等,最近,微球或纳米球备受关注。微球通常指的是粒径为1μm至150μm左右的制剂,将小于其的小于1μm的制剂称为纳米球。它们例如可以使生理活性物质被内包在生物降解性的合成高分子或天然高分子内,局部持续释放生理活性物质,或者可以对组织进行生理活性物质的靶向等。
3.在以一定的速度缓慢释放生理活性物质的缓释微球制剂中,例如需要适当控制生物降解性高分子、生理活性物质、添加剂、溶剂等的制剂。为了使缓释微球制剂在生物体内在一定期间有效地显示药理学效果,需要适当地控制生理活性物质的初期释放量和之后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
4.决定该生理活性物质的释放速度的重要因素之一有生物降解性高分子的种类。特别就最广泛使用的乳酸/乙醇酸共聚物(polylactide-co-glycolide acid,plga)以及作为乳酸的聚合物的聚乳酸(pla)而言,根据其物理化学特性、例如作为构成成分的乳酸与乙醇酸的比例、分子量、水亲和性等而在生物体内的分解速度不同,因此能够通过它们来调整为所希望的释放期间(专利文献1)。
5.进而,在此基础上,为了抑制生理活性物质的初期释放量异常(初期爆发)、将释放期间的释放速度控制为恒定,涉及到微球的粒径和微球中的生理活性物质的分散状态。微球的粒径存在成品率的问题,但可以通过过滤等操作调节为目标粒径。但是,微球中的生理活性物质的分散状态只是被认为是均匀的而没有被确认。
6.plga或pla的微球例如可以使用液中干燥法、喷雾干燥法、喷雾冷冻干燥法、使用超临界流体工序的干燥法、双乳化法等制造。就其中最常用的制造方法而言,在生理活性物质为亲油性的情况下,是使plga或pla和生理活性物质溶解或分散在有机溶剂中,使其与溶解有聚乙烯醇(pva)的水溶液混合而乳液化,从乳液中去除溶剂的液中干燥法。
7.专利文献1中公开了利用喷雾干燥法、喷雾冷冻干燥法或使用超临界流体工序的干燥法制造包含plga等生物降解性高分子和肽药物的缓释微球的方法。但是,对于缓释微球的粒径以何种程度波动、肽药物在缓释微球中是否均匀地分散、能否得到均匀的产物,没有记载。
8.在专利文献2中公开了使用由卤代烃和药物的溶解度为0.3%(w/v)以上的非水混合性有机溶剂组成的混合溶剂,采用液中干燥法制造plga微粒的方法。虽然记载了制造例1和2中得到的微粒的粒径(中值粒径)为14和16μm,但对于微粒中的药物的分散状态没有记载。
9.在专利文献3中公开了包含药物的plga纳米粒子。这些纳米粒子主要用于对特定
组织的靶向,因此是能够穿过毛细血管的微小的孔的几十~几百nm左右的纳米粒子。但是,在专利文献3中,对于远大于这些纳米粒子的1μm以上的微球没有记载。另外,即使使用专利文献3的技术,本领域技术人员也无法制造粒径为1μm以上的微球。
10.在专利文献4中公开了通过皮下注射而释放作为黄体生成素释放激素衍生物的醋酸亮丙瑞林约1个月至几个月的制剂。在该制剂中存在粒径的粒度分布从1μm到400μm、非常宽的问题。因此,在专利文献5中,作为解决该问题的方法,提出了采用双乳化法制造在载体用高分子内封入有生理活性物质的微球的方法。但是,就实施例1~5中得到的含有醋酸亮丙瑞林的微球而言,对于粒子中的药物的分散状态没有记载。
11.在专利文献6中公开了减轻慢性疼痛至少28天(672小时)的微球。该微球包含生物降解性聚合物和局部麻醉剂(生理活性物质),局部麻醉剂的约75%释放直至约72小时,局部麻醉剂的约80~90%释放直至约120小时。由此暗示,微球中的局部麻醉剂的分布在微球中不均匀而偏向外侧。根据图2中记载的微球的截面的sem(扫描型电子显微镜)图像,无法确认局部麻醉剂的分散状态。
12.在专利文献7中公开了一种核壳结构的微球,其中,核含有固体状的阿立哌唑,包含生物降解性聚合物的壳包覆核的表面。这样,专利文献7的微球不是生理活性物质均匀地分散在微球中的微球。另外,在图5所记载的将实施例中得到的微球切断得到的截面的电子显微镜照片中,在壳中无法确认阿立哌唑的分散状态。
13.现有技术文献
14.专利文献
15.专利文献1:日本特开2005-035994号公报
16.专利文献2:日本特开2005-015476号公报
17.专利文献3:日本专利4856752号公报
18.专利文献4:日本专利2653255号公报
19.专利文献5:日本特开2014-224114号公报
20.专利文献6:日本特开2016-069378号公报
21.专利文献7:日本特表2010-531303号公报
技术实现要素:
22.发明所要解决的课题
23.就平均体积基准粒径为1μm以上且150μm以下的生物降解性高分子微球而言,如果未控制微球中的生理活性物质(有时也记作药物)的分布,则无法按照设计实现释放期间。例如,如果生理活性物质偏向微球的表面附近,则在给药后初期从微球释放大量的生理活性物质,产生初期爆发的问题。相反,如果生理活性物质偏向微球的中心部,或者如果是核壳那样的状态,则无法从初期持续地释放。因此,希望微粒的生理活性物质均匀地分散的状态。在大块的生理活性物质散布那样的分散状态下,无法从初期持续地释放。同样地,如果未进行孔隙的控制,则在生理活性物质的释放中产生同样的问题。
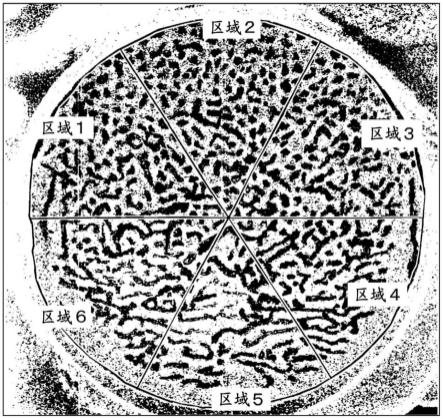
24.实际上,在使用大鼠等小动物调查药代动力学时,有时会在释放速度、释放曲线上产生大量波动,作为其原因,大多结论为大鼠等的个体差异。但是,如果微球中的生理活性物质的分散状态均匀,则能够更为改善波动的频发,通过plga的种类、分子量来控制分解速
度,也能够按照设计实现从微球的生理活性物质的释放。
25.在微球中均匀地分散生理活性物质是用于在生物体内在一定期间持续释放生理活性物质的绝对条件。但是,现状是对于均匀的分散尚未进行探索。由于微球的粒径与纳米粒子不同而较大,因此一般难以均匀化。因此,需要确认微球中的生理活性物质的分散状态。为此,制作将微球的粒子切断而成的截面观察试样,以能够确认微球中的生理活性物质的倍率以上进行电子显微镜观察,确认其分散状态是能够容易地实施、可靠的。
26.图1是相当于专利文献4中记载的lh-rh衍生物的长期缓释型微囊的、微囊型缓释制剂利普安(注册商标)注射用1.88mg(武田药品工业株式会社制造)的电子显微镜照片。
27.本制剂中,包含从大的粒子到小的粒子的各种粒子,但选择作为其代表性的粒子的约6μm的粒子,该粒子的截面的sem(扫描型电子显微镜)图像为图2。通过确认图像中的边缘效应,另外通过sem-eds(能量色散型x射线光谱仪)可知图中的箭头所示的大的分散体为孔隙。图3是将图2的sem截面图像在图像上分割为6个(区域1~区域6),使用市售的图像处理分析软件item(tem照相机控制,图像分析软件,emsis公司制造)以像素范围3
×
3进行平均化处理后,进行强调边缘部分的处理,进行对比度的最佳化。之后,进行二值化处理,在图像处理中进行噪声除去及对比度低的粒子的强调处理后,再次以像素范围3
×
3进行平均化处理后进行强调边缘部分的处理的图像。基于图3,求出分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数,结果为1.06。通过这样实施,能够由粒子截面确认生理活性物质的分散状态。该变动系数超过0.35,是分散于粒子中的生理活性物质不均匀的分散状态,因此在本制剂中无法适当地控制释放期间的释放速度。
28.因此,本发明的课题在于提供微球,其能够适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
29.用于解决课题的手段
30.本发明人等为了解决上述课题而进行了深入研究,结果发现,通过制成微球的电子显微镜截面观察的像的分割为6个的区域中的生理活性物质的面积比率的变动系数为0.35以下的微球,生理活性物质均匀地分散于微球中,没有不均匀的孔隙,从而能够适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质,完成了本发明。即,本发明如下所述。
31.[1]本发明的第1方式是微球,其为均匀分散有生理活性物质的以乳酸/乙醇酸共聚物(plga)或聚乳酸(pla)作为主要成分的微球,其特征在于,
[0032]
所述微球的平均体积粒径为1μm~150μm以下,
[0033]
制作将所述微球切断而成的截面观察试样,以能够确认微球中的生理活性物质的倍率以上对所述截面观察试样进行电子显微镜观察,将所述电子显微镜截面观察的像分割为6个,计算出分割的各个区域的区域面积(a)与该区域所包含的所述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%),6个区域中的所计算出的所述比率的变动系数为0.35以下。
[0034]
[2]本发明的第2方式是[1]所述的微球,其中,所述生理活性物质为亲油性生理活性物质。
[0035]
[3]本发明的第3方式是[1]或[2]所述的微球,其中,分散的所述生理活性物质的
平均体积基准粒径为5nm~500nm。
[0036]
[4]本发明的第4方式是[1]~[3]中任一项所述的微球,其中,所述微球中的所述生理活性物质的含量为0.35~1.5质量%。
[0037]
[5]本发明的第5方式是含有[1]~[4]中任一项所述的微球的缓释制剂。发明的效果
[0038]
通过本发明的微球,能够适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
附图说明
[0039]
图1是利普安(注册商标)注射用1.88mg(武田药品工业株式会社制造)的sem(扫描型电子显微镜)图像。
[0040]
图2是利普安(注册商标)注射用1.88mg(武田药品工业株式会社制造)的代表性粒子的截面的sem(扫描型电子显微镜)图像。
[0041]
图3是将图2的截面图像分割为6个,为了求出所分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)而进行了二值化处理的图像。
[0042]
图4是参考例1的不含生理活性物质的微球的截面的sem图像。
[0043]
图5示出微球的分割方法的例子。(a)为在同心圆上分割为6个。(b)是在纵向上分割为6个。(c)是在圆周方向上分割为6个。
[0044]
图6-1是实施例1的微球的截面的sem图像。
[0045]
图6-2是将图6-1的截面图像分割为6个,为了求出所分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)而进行了二值化处理的图像。
[0046]
图7-1是实施例3的微球的截面的sem图像。
[0047]
图7-2是放大图7-1的截面图像并进行了二值化处理的图像。
[0048]
图8-1是实施例4的微球的截面的sem图像。
[0049]
图8-2是将图8-1的截面图像分割为6个,为了求出所分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)而进行了二值化处理的图像。
[0050]
图9是将比较例1的截面图像分割为6个,为了求出所分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)而进行了二值化处理的图像。
[0051]
图10是比较例2的微球的截面的sem图像。
[0052]
图11是比较例3的微球的截面的sem图像。
[0053]
图12是比较例4的微球的截面的sem图像。
具体实施方式
[0054]
1.微球
[0055]
本发明的微球是均匀分散有生理活性物质的以乳酸/乙醇酸共聚物(plga)或聚乳
酸(pla)作为主要成分的微球,其特征在于,所述微球的平均体积粒径为1μm~150μm以下,制作将所述微球切断而成的截面观察试样,以能够确认微球中的生理活性物质的倍率以上对所述截面观察试样进行电子显微镜观察,将所述电子显微镜截面观察到的像分割为6个,算出所分割的各个区域的区域面积(a)与该区域所包含的所述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%),6个区域中的算出的所述比率的变动系数为0.35以下。
[0056]
若生理活性物质偏析于表层或中心部、或者存在粗大粒子、凝聚体或大的孔隙等,则上述比率的变动系数变大。如果上述比率的变动系数为0.35以下,则生理活性物质的分散均匀。本发明涉及的微球在将上述微球的截面分割为6个的各区域内,生理活性物质所占的面积相对于各区域面积的比率的变动系数为0.35以下,优选为0.25以下,更优选为0.20以下。
[0057]
通过本发明的微球,能够适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
[0058]
<微球的截面的观察>
[0059]
以下叙述微球中的生理活性物质的分散状态的确认方法。
[0060]
可以通过以能够明确地确认分散的生理活性物质的微粒的倍率利用电子显微镜观察微球的截面来实施。电子显微镜有将透射电子作为信息源的透射型电子显微镜(tem)和检测二次电子(反射电子)的扫描型电子显微镜(sem)等。根据观察的试样进行选择,但就纳米球等而言,透射型电子显微镜能够观察微细结构。具体地,首先将金、铂、铂钯合金等的薄膜涂覆于微球。在实施例中,使用锇进行涂覆。接着,利用液氮使微球冷冻。冷冻后,采用fib(聚焦离子束:focused ion beam)制作截面。即,使用fib装置,将聚焦的离子束照射试样,切出试样内部的所希望位置的结构,由此制造微球的截面观察试样。分散于plga或pla的母材中的生理活性物质的微粒的优选粒径为数10nm~数100nm,但也存在几μm的情况。以能够确认该大小的分散微粒的电子显微镜的观察倍率观察微球的整个截面。通常,电子显微镜的观察倍率为2500倍至几十万倍左右以上。另外,在使用无法观察整个微球的高倍率的情况下,可以将观察到的部分连接起来而观察整体。
[0061]
在将截面图像分割为6个时,例如,如图5的a~c所示,可以以同心圆状分割为6个,也可以在纵向或横向上分割为6个,也可以在通过中心的圆周方向上分割为6个。优选采取显著地显示生理活性物质的偏析状态的分割方法。例如,在以同心圆状分割为6个的情况下(图5a),必须从微球的截面的最大直径的中心点起将半径进行6等分而分割成同心圆状。在纵向或横向上进行分割的情况下(图5b),必须等间隔地分割为6个。必须在与上述最大直径平行或垂直的任一方向上分割为6个。在通过中心的圆周方向上分割为6个的情况下(图5c),必须以上述最大直径的中心点为中心每隔60度分割为6个。虽然也根据生理活性物质的构成元素而不同,但能够使用eds(能量色散型x射线光谱仪)对微球的截面进行元素分析,也能够确认是否为孔隙。在孔隙的情况,不能累计到生理活性物质的面积中。在没有eds检测元素的情况下,可以使用四氧化钌、四氧化锇、磷钨酸、醋酸铀酰、碘等对截面观察试样进行染色。另外,通过与未加入生理活性物质的微球的比较,可以确定生理活性物质。在电子显微镜截面观察图像中难以得到对比度的情况下,染色法是有效的。上述为一例,也可以用树脂包埋试样,或者在微球的截面制作中使用切片机。
[0062]
另外,就截面积的计算而言,对方法没有特别限制,优选使用市售的图像处理分析
软件。作为市售的图像处理分析软件,可以使用image-proplus(media cybernetics公司制造)、item(tem照相机控制、图像分析软件、emsis公司制造)等多种软件。
[0063]
<plga和pla>
[0064]
plga是具有来源于乳酸的结构单元和来源于乙醇酸的结构单元的乳酸/乙醇酸共聚物。pla是乳酸的聚合物。plga可以包含聚丙交酯(polylactide,pla)、聚乙交酯(polyglycolide,pga)等其他生物降解性高分子。本说明书中记载的plga是作为一例而记载的物质,并不限于所记载的物质。
[0065]
作为plga中的来源于乳酸的结构单元(l)与来源于乙醇酸的结构单元(g)的摩尔比率(l:g),没有特别限制,可以根据目的适当选择,优选为1:99~99:1,更优选为25:75~99:1,进一步优选为30:70~90:10,特别优选为50:50~85:15。另外,也可以仅使用pla。为了实现生理活性物质的均匀的分散状态,该摩尔比率的选择是重要的,plga或pla的分子量的选择也同样重要。
[0066]
本发明的微球中使用的plga例如可以通过以离子交换树脂作为催化剂将乳酸和乙醇酸在弱的减压下加热、缩聚来制造。此时,也可以使用丙交酯来代替乳酸。plga可以是市售品。作为市售品,例如可以购自富士胶片和光纯药工业(株)、多木化学(株)、evonic rohm gmbh公司、merck公司、sigma-aldrich公司等。
[0067]
本发明的微球中的plga或pla的含量没有特别限制,可以根据目的适当选择,优选为1质量%以上,更进一步优选为30质量%以上且95质量%以下,特别优选为50质量%以上且90质量%以下。
[0068]
<微球>
[0069]
在本发明的微球中包含plga或pla和生理活性物质。进而根据需要,含有分散剂、其他成分。在微球中,在plga或pla的母材中分散有生理活性物质、分散剂、其他成分等。
[0070]
[生理活性物质]
[0071]
作为本发明的微球中所包含的生理活性物质,没有特别限制,可以根据目的适当选择,例如可列举医药化合物、功能性食品化合物、功能性化妆品化合物等。包含医药化合物的微球例如可适合用作缓释医药制剂。在生理活性物质中包含亲油性生理活性物质和亲水性生理活性物质中的任一种。作为优选的生理活性物质,可列举亲油性生理活性物质。亲油性生理活性物质意指例如水/辛醇分配系数的logp值为3以上的物质,亲油性生理活性物质中不含的生理活性物质被分类为亲水性生理活性物质。水/辛醇分配系数可以依据jisz7260-107(2000)烧瓶振荡法进行测定。生理活性物质只要是缓释制剂所希望的物质就没有特别限制,可以根据目的适当选择。生理活性物质中也包括盐、水合物等的任一形态。
[0072]
在本发明的微球中生理活性物质均匀地分散。通过采取该结构,能够适当地控制生理活性物质的初期释放量和之后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。生理活性物质在微球中均匀地分散能够用生理活性物质相对于微球总量的含量进行控制。作为生理活性物质的优选含量,根据生理活性物质而变化,相对于微球总量,例如可列举0.1~3质量%,优选列举0.3~2质量%,更优选列举0.35~1.5质量%,进一步优选列举0.5~1.25质量%。
[0073]
分散的生理活性物质的微粒的平均体积基准粒径优选为5nm~500nm,更优选列举10nm~400nm,进一步优选列举20nm~200nm。
[0074]
[分散剂]
[0075]
为了分散生理活性物质,可以使用分散剂。作为分散剂,可以是低分子量的分散剂,也可以是高分子量的分散剂聚合物。低分子量的分散剂意指重均分子量小于15000的化合物,高分子量的分散剂聚合物意指在1个以上的单体之间包含重复的共价键、重均分子量为15000以上的化合物。
[0076]
作为低分子量的分散剂,只要是医药化合物、功能性食品化合物、功能性化妆品化合物等中可容许的分散剂,就没有特别限制,可以根据目的适当选择。具体地,可列举脂质类、糖类、环糊精类、氨基酸类、有机酸类、其他成分等。它们可以单独使用1种,也可以并用2种以上。
[0077]
作为脂质类,没有特别限制,可以根据目的适当选择,例如可列举中链或长链的甘油单酯、甘油二酯或甘油三酯、磷脂、植物油(例如大豆油、鳄梨油、角鲨烯油、芝麻油、橄榄油、玉米油、菜籽油、红花油、葵花籽油等)、鱼油、调味油、水不溶性维生素、脂肪酸以及它们的混合物、它们的衍生物等。它们可以单独使用1种,也可以并用2种以上。
[0078]
作为糖类,没有特别限制,可以根据目的适当选择,例如,可列举葡萄糖、甘露糖、艾杜糖、半乳糖、岩藻糖、核糖、木糖、乳糖、蔗糖、麦芽糖、海藻糖、松二糖、棉子糖、麦芽三糖、阿卡波糖、水溶性纤维素、合成纤维素、糖醇、甘油、山梨糖醇、乳糖醇、麦芽糖醇、甘露醇、木糖醇、赤藓糖醇、或多元醇、或者它们的衍生物等。它们可以单独使用1种,也可以并用2种以上。
[0079]
作为其他成分,没有特别限制,可以根据目的适当选择,优选以往可以用于医药的成分。
[0080]
<平均体积基准粒径>
[0081]
本发明的微球的平均体积基准粒径为1μm以上且150μm以下,优选为10μm以上且100μm以下,更优选为20μm以上且75μm以下。平均体积基准粒径可以使用激光衍射式粒度分布测定装置进行测定。在本发明中,如果平均体积基准粒径超过150μm,则由于微球内的生理活性物质的分散不均匀性会产生初期爆发的问题,容易凝聚、沉降,后工序的处理也变得困难。如果小于1μm,则显著产生初期爆发的问题。
[0082]
通过本发明的微球,能够适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
[0083]
2.缓释制剂
[0084]
使用本发明的微球,可以制备含有微球的缓释制剂。通过本发明的缓释制剂,可以适当地控制生理活性物质的初期释放量和其后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质,有效地显示出药理学效果。
[0085]
本发明的缓释制剂可以作为注射剂和埋入剂,或者作为经皮剂,容易地直接给药至肌肉内、皮下、血管、脏器、关节腔、肿瘤等病灶等。也可以作为其他各种制剂形态进行给药。例如,为了将本发明的缓释制剂制成注射剂,与分散剂(tween80、hco-60、羧甲基纤维素、海藻酸钠等)、保存剂(尼泊金甲酯、尼泊金丙酯等)、等渗剂(氯化钠、甘露醇、山梨糖醇、葡萄糖等)等一起制成水性悬浮剂,或者与大豆油、芝麻油、玉米油等植物油一起分散而制成油性悬浮剂,从而制成缓释注射剂。
[0086]
3.微球的制造方法
[0087]
<微球的制造工序>
[0088]
本发明的微球的制造方法至少包括粒子形成工序,进而根据需要,还可以包括过滤灭菌工序、良溶剂除去工序、其他工序等。
[0089]
[粒子形成工序]
[0090]
粒子形成工序优选使用在日本特开2009-132871号公报或日本特开2011-189348号公报中记载的在能够接近或远离的相对配置的、至少一方相对于另一方相对旋转的多个处理用面之间进行粒子化处理的粒子化处理装置。粒子形成工序例如通过如下方式实施:使用上述的粒子化处理装置,将plga或pla和生理活性物质溶解或分散在plga或pla的良溶剂中而得到的plga或pla和生理活性物质的溶液与包含plga或pla的不良溶剂的溶液连续投入,制造乳化粒子,从所制造的粒子中除去良溶剂,来使本发明的微球析出。在此,“分散”包括:使生理活性物质以固体的状态分散在plga或pla的良溶剂中;使生理活性物质在plga或pla的良溶剂中乳化;形成包含亲水性生理活性物质的水性溶液和plga或pla的良溶剂的w/o型乳液等。
[0091]
作为plga或pla和生理活性物质的溶液,只要是plga或pla和生理活性物质溶解或分散在plga或pla的良溶剂中的溶液,就没有特别限制,可以根据目的适当选择。作为良溶剂,没有特别限制,可以根据目的适当选择,例如,可列举:卤代脂肪族烃、脂肪族酯、醇、酮、醚、乙腈等。作为卤代脂肪族烃,例如,可列举二氯甲烷、氯仿、四氯化碳、氯乙烷、2,2,2-三氯乙烷等。作为脂肪族酯,例如,可列举乙酸乙酯、乙酸丙酯、乙酸丁酯等。作为醇,例如可列举苄醇、苯基醇、正丁醇等在水中的溶解度低的醇。作为酮,例如,可列举碳原子数3~6的酮(例如丙酮、甲乙酮、甲基异丁基酮、环己酮等)等。作为醚,例如,可列举碳原子数2~6的醚(例如二甲基醚、甲基乙基醚、二乙基醚等)等。从生理活性物质的含量的观点、防止初期爆发的目的出发,优选选择在水中的溶解度低的溶剂。作为优选的良溶剂,可列举卤代脂肪族烃、酮及它们的混合溶剂,更优选列举二氯甲烷、丙酮及它们的混合溶剂。另外,它们可以单独使用1种,也可以并用2种以上。通过改变溶剂种类、混合量,能够控制粒径。
[0092]
良溶剂意指plga或pla的溶解度大的溶剂,不良溶剂意指plga或pla的溶解度小或不溶解的溶剂。就良溶剂和不良溶剂而言,分别选择使得微球中的生理活性物质不发生偏析或不产生粗大粒子、凝聚体的溶剂。另外,良溶剂和不良溶剂例如可以用在25℃下的溶剂100g中能溶解的plga或pla的质量来规定。本发明中,良溶剂优选为溶解0.1g以上plga或pla的溶剂,更优选列举0.2g以上,进一步优选列举0.5g以上。不良溶剂优选为仅溶解0.05g以下的plga或pla的溶剂,更优选列举0.02g以下,进一步优选列举0.01g以下。作为不良溶剂,没有特别限制,可以根据目的适当选择,优选水。
[0093]
plga或pla和生理活性物质的溶液中的plga或pla的含量可以根据良溶剂、根据目标微球的粒径、另外以使得生理活性物质均匀地分散在微球中的方式而改变。plga或pla的含量例如可列举1~30质量%,优选列举3~20质量%,更优选列举5~15质量%。plga或pla溶液中的生理活性物质的含量可以根据目的、药理效果等、另外以使得微球中的生理活性物质均匀地分散的方式而适当地改变。
[0094]
为了进一步确保制造的微球的稳定性,可以在不良溶剂中加入稳定剂。作为稳定剂,没有特别限制,可以根据目的适当选择。例如,可列举聚乙烯醇(pva)、聚乙烯吡咯烷酮(pvp)、羧甲基纤维素(cmc)、羟丙基纤维素(hpc)、羟丙基甲基纤维素(hpmc)、卵磷脂、聚山
梨酯80等,优选聚乙烯醇(pva)。另外,添加的稳定剂的浓度优选列举0.01~20质量%,更优选为5质量%以下。作为优选的不良溶剂,例如,可列举pva水溶液等。
[0095]
希望使用使棒状、板状、螺旋桨状等各种形状的搅拌器在槽内旋转的装置、具备相对于搅拌器相对旋转的筛网的装置等、对流体施加剪切力等,从而实现均质的混合的旋转式分散机等制备装置来制备plga或pla和生理活性物质的溶液以及包含不良溶剂的溶液。作为旋转式分散机的优选例,可以应用日本专利第5147091号中公开的搅拌机。为了使微球中的生理活性物质均匀地分散,需要将plga或pla溶液与不良溶剂完全混合。为了完全混合,需要以至少分子水平的均匀化为目标,混合不充分成为不均匀的分散状态的原因。
[0096]
旋转式分散机可以以间歇式也可以以连续式进行。在以连续式进行的情况下,可以连续地进行对于搅拌槽的流体的供给和排出,也可以不使用搅拌槽而使用连续式的混合机进行,可以使用公知的搅拌机、搅拌手段适当地控制搅拌能量。应予说明,关于搅拌能量,在本技术人的日本特开平04-114725号公报中详细叙述。对本发明中的搅拌的方法没有特别限定,可以使用各种剪切式、摩擦式、高压喷射式、超声波式等的搅拌机、溶解机、乳化机、分散机、均化器等来实施。作为一例,可列举ultra-turrax(ika制造)、polytron(kinematica制造)、tk均质混合器(primix制造)、ebara milder(荏原制作所制造)、tk homomic line flow(primix制造)、胶体磨(神钢环境solutions制造)、thrasher(日本coke工业制造)、trigonal湿式微粉碎机(三井三池化工机制造)、cavitron(eurotec制造)、fine flow mill(太平洋机工制造)等连续式乳化机、clearmix(m-technique制造)、clearmix dissolver(m-technique制造)等间歇式或连续两用乳化机。另外,搅拌处理希望使用具备高速旋转的搅拌叶片、在搅拌叶片的外侧具备筛网、流体从筛网的开口成为射流而排出的搅拌机,特别希望使用上述的clearmix(m-technique制造)、clearmix dissolver(m-technique制造)。
[0097]
在上述的粒子化处理装置中,通过调整旋转的处理用面的停止时的接触面压力,能够控制plga或pla微粒的粒径及粒径分布。本发明人的实验的结果为,接触面压力优选为20g/cm2~250g/cm2。在接触面压力低于20g/cm2的情况下,薄膜不稳定,粒径分布变宽。明确了如果接触面压力高于250g/cm2,则目标的粒径的调整变得困难。更优选列举50g/cm2~200g/cm2,进一步优选列举80g/cm2~150g/cm2。
[0098]
优选防止plga或pla和生理活性物质的溶液与包含不良溶剂的溶液通过相接而形成的各个微球聚结。作为防止其聚结的方法,优选在溶液排出液回收罐中预先放入包含不良溶剂的溶液,缓慢搅拌。通过进行搅拌,能够更为抑制微球的聚结。就搅拌而言,优选旋转式分散机,希望为clearmix dissolver(m-technique制造)。只要能够使整体缓慢地流动,则并无特别限定。如果搅拌强,则plga或pla的乳化粒子被破坏,分布宽度变宽,生理活性物质的分散状态有可能崩溃。
[0099]
在生理活性物质为亲油性生理活性物质的情况下,可以根据上述的说明,适合地实施粒子形成工序,从而可以制造微球。在生理活性物质为亲水性生理活性物质的情况下,可以通过使用例如分散剂使亲水性生理活性物质分散于plga或pla的良溶剂中,同样地实施粒子形成工序,制造微球。
[0100]
另外,在生理活性物质为亲水性生理活性物质的情况下,也可以通过将亲水性生理活性物质与根据需要的稳定剂一起溶解在水等水性溶剂中,与将plga或pla溶解在plga
或pla的良溶剂中而成的溶液一起混合而制备的w/o型乳液用作plga或pla和生理活性物质的溶液,使用上述的粒子化处理装置,实施上述的粒子形成工序。在w/o型乳液的制备中,可以使用间歇振荡法、使用螺旋桨型搅拌机、涡轮型搅拌机的采用混合机的方法、胶体磨法、均化器法、超声波照射法。使用上述的粒子化处理装置,连续投入作为该w/o型乳液的plga或pla和生理活性物质的溶液和包含plga或pla的不良溶剂的溶液,作为w/o/w乳液制造乳化粒子,从所制造的粒子中除去良溶剂,由此使本发明的微球析出来实施。可以直接使用该微球,但也可以进一步加入赋形剂(甘露醇、山梨糖醇、乳糖、葡萄糖等),再分散后,进行冷冻干燥或喷雾干燥从而固体化。该固体化的微球在使用时,可以通过添加注射用蒸馏水或适当的分散介质,来得到更稳定的缓释注射剂。
[0101]
[过滤灭菌工序]
[0102]
根据需要,在粒子形成工序之前,优选对制备的plga或pla和生理活性物质的溶液以及包含不良溶剂的溶液进行无菌过滤。包含不良溶剂的溶液可以使用亲水性过滤器进行过滤灭菌、plga或pla和生理活性物质的溶液可以使用疏水性过滤器进行过滤灭菌。用于过滤的过滤器的孔径优选为0.1μm~0.45μm,更优选为0.2μm。
[0103]
作为上述的过滤灭菌过滤器,没有特别限制,可以根据目的适当选择。例如,为了对包含不良溶剂的溶液进行灭菌过滤,可列举聚偏氟乙烯(pvdf)、聚醚砜等亲水性过滤器。为了对plga或pla和生理活性物质的溶液进行过滤灭菌,可列举聚四氟乙烯(ptfe)等疏水性过滤器。并不限定于在此记载的材质,需要根据药物的吸附、溶剂种类来进行选定。
[0104]
[良溶剂除去工序]
[0105]
在良溶剂除去工序中,从包含plga或pla及生理活性物质的上述乳化粒子中除去良溶剂。就良溶剂除去工序而言,只要能够在微球中的生理活性物质均匀地分散的状态下从含有上述乳化粒子的液体中除去上述良溶剂,就没有特别限制,可以根据目的适当选择。例如,可列举通过一边搅拌一边加热上述液体、使氮气等气体流向上述液体的液面、以及使上述液体减压的至少任一种方式使上述良溶剂蒸发而从上述液体中除去的方法等,优选列举使氮气等气体流向上述液体的液面的方式。为了维持微球中的生理活性物质均匀地分散的状态,大多优选迅速除去良溶剂,但有时缓慢除去为宜。作为除去良溶剂的时间,例如,可列举30分钟~12小时,优选列举1~10小时,更优选列举1~5小时。
[0106]
另外,除去良溶剂时的温度根据良溶剂的种类而不同。需要从良溶剂的沸点附近的高温至低温,进行微球的截面观察,在适当的温度下实施。在除去良溶剂时,在plga或pla的浓度低的情况下,粒子的体积变化大,此时经常看到好不容易分散的生理活性物质凝聚的情况,因此需要注意。
[0107]
[其他工序]
[0108]
作为其他工序,例如,可列举溶剂组成制备、分级工序、粒子清洗工序等。通常,在分级工序中进行粗粉去除、细粉去除,但本发明中制造的粒子基本上不必进行分级工序。但是,为了慎重起见,也可以包括分级工序。
[0109]
通过上述的制造方法,能够制造微球的粒径为1μm~150μm以下、微球中的生理活性物质为均匀的分散状态的微球。即,能够制造如下微球:制作将微球的粒子切断而成的截面观察试样,以能够确认微球中的生理活性物质的倍率以上进行电子显微镜观察,将截面图像分割为6个,分割的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截
面积的总和(s)的比率为(s/a)
×
100(%),分割为6个的区域内的生理活性物质所占的面积比率的变动系数为0.35以下。
[0110]
实施例
[0111]
以下,对本发明的实施例进行说明,但本发明不受这些实施例的任何限定。
[0112]
(参考例1)
[0113]
在参考例1中,制造不含生理活性物质的微球(plga微粒)。使用参考例1的微球作为指标,通过sem图像观察实施例及比较例的微球的截面,由此在以下确认实施例及比较例的微球中的生理活性物质的分散状态。
[0114]
<plga溶液和pva水溶液的制备>
[0115]
以乳酸/乙醇酸共聚物(resomer rg504,evonik制造)成为13质量%的方式加入二氯甲烷(关东化学制造),使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到plga溶液。然后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。以聚乙烯醇(pva,eg-40p,日本合成化学工业制造)成为1.5质量%的方式加入离子交换水,使用高速旋转式分散机clearmix(m-technique制造)使其溶解,得到pva水溶液。然后,用亲水性pvdf膜滤器(φ47,merck制造)进行过滤。在回收plga乳化粒子的罐中预先放入pva水溶液,以液面移动的程度进行搅拌。
[0116]
<微球(plga微粒)的制备>
[0117]
作为粒子形成工序,使用日本特开2011-189348号公报中记载的粒子化处理装置将制备的plga溶液和pva水溶液混合。在此,日本特开2011-189348号公报中记载的粒子化处理装置是该公报的图25中记载的装置,其中第2导入口d20是围绕作为环状圆盘的处理用面2的中央的开口的同心圆状的圆环形状,圆盘直径为75mm。具体地,将制备的pva水溶液从第1导入部d1以0.02mpag、65ml/分钟、30℃导入处理用面1、2间,一边使处理用部10以2000rpm进行旋转,一边将所制备的plga溶液从第2导入部d2以0.65mpag、20ml/分钟、30℃导入处理用面1、2间,将pva水溶液和plga溶液在强制薄膜中混合,在处理用面1、2间制造包含二氯甲烷的plga乳化粒子。使处理用面1、2间的包含plga乳化粒子的流体(以下称为plga乳化粒子分散液)从粒子化处理装置的处理用面1、2间排出。将排出的plga乳化粒子分散液回收至回收罐。
[0118]
接着,作为脱溶剂工序,一边使用clearmix dissolver(m-technique制造)以圆周速度4.7m/秒对上述排出液进行搅拌,一边使氩气流动以向液面喷射,用3.5小时除去二氯甲烷,得到包含plga微粒的悬浮液(plga微粒悬浮液)。得到的plga微粒的平均体积基准粒径为34.0μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图4)。
[0119]
如图4所示,确认在上述的fib截面没有粒子状的块和孔隙。另外,可知参考例1的微球的截面能够用作实施例和比较例的微球的截面的观察中的指标。
[0120]
(实施例1)
[0121]
<plga和生理活性物质的溶液和pva水溶液的制备>
[0122]
以乳酸/乙醇酸共聚物(resomer rg752h,evonik制造)成为10质量%、作为生理活性物质的姜黄素(富士胶片和光纯药制造,和光特级)成为0.5质量%的方式,加入二氯甲烷(关东化学制造)64.5质量%和丙酮(关东化学制造)25质量%,使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到了plga和生理活性物质的溶液。之
后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。pva水溶液与参考例1同样地制备。在回收plga和生理活性物质的乳化粒子的罐中预先放入pva水溶液,以液面移动的程度进行搅拌。
[0123]
<微球的制备>
[0124]
作为粒子形成工序,与参考例1同样地,使用日本特开2011-189348号公报中记载的粒子化处理装置混合所制备的plga和生理活性物质的溶液与pva水溶液。具体地,将制备的pva水溶液从第1导入部d1以0.0mpag、50ml/分钟、25℃导入处理用面1、2间,一边使处理用部10以5000rpm旋转,一边将制备的plga和生理活性物质的溶液从第2导入部d2以0.3mpag、16ml/分钟、25℃导入处理用面1、2间,将pva水溶液与plga和生理活性物质的溶液在强制薄膜中混合,在处理用面1、2间制造包含二氯甲烷的plga和生理活性物质的乳化粒子。使处理用面1、2间的包含plga和生理活性物质的乳化粒子的流体(以下称为plga和生理活性物质的乳化粒子分散液)从粒子化处理装置的处理用面1、2间排出。将用于捕获排出的plga和生理活性物质的乳化粒子分散液的plga和生理活性物质的乳化粒子分散液回收至保持为0.03mpag压力的回收罐。
[0125]
接着,作为脱溶剂工序,一边使用clearmix dissolver(m-technique制造)以圆周速度4.7m/秒对上述排出液进行搅拌,一边使氩气流动以向液面喷射,用3.5小时除去二氯甲烷和丙酮,得到包含微球的悬浮液(微球悬浮液)。得到的微球的平均体积基准粒径为7.5μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图6-1)。使用item(tem照相机控制,图像分析软件,emsis公司制造)以像素范围3
×
3对得到的sem图像的粒子截面进行平均化处理后,进行强调边缘部分的处理,进行对比度的最佳化。然后,进行二值化处理,在图像处理中进行噪声除去及对比度低的粒子的强调处理后,再次以像素范围3
×
3进行平均化处理后进行强调边缘部分的处理。在图6-2中示出在图像上将粒子截面以最大直径的中心点为中心每隔60度分割为6个(区域1~区域6)所得的图像。
[0126]
图6-2的姜黄素粒子的fib截面中的分割得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.161。
[0127]
(实施例2)
[0128]
除了使用聚乳酸(resomer r202h)代替乳酸/乙醇酸共聚物(resomer rg504,evonik制造)以外,与实施例1同样地制备包含微球的悬浮液。得到的微球的平均体积基准粒径为7.3μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像。
[0129]
用与实施例1相同的方法对所观察的sem图像进行图像分析,结果分割得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.214。
[0130]
(实施例3)
[0131]
以乳酸/乙醇酸共聚物(resomer rg504,evonik制造)成为13质量%、作为生理活性物质的孕酮(sigma-aldrich制造)成为1.0质量%的方式,加入二氯甲烷(关东化学制造),使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到plga和生理活性物质的溶液。之后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。pva水溶液与参考例1同样地制备。在回收plga和生理活性物质的乳化粒子的罐中预先放入pva水溶
液,以液面移动的程度进行搅拌。
[0132]
《微球的制备》
[0133]
作为粒子形成工序,与参考例1同样地,使用日本特开2011-189348号公报中记载的粒子化处理装置混合所制备的plga和生理活性物质的溶液与pva水溶液。具体地,将制备的pva水溶液从第1导入部d1以0.0mpag、50ml/分钟、30℃导入处理用面1、2间,一边使处理用部10以1700rpm旋转,一边将制备的plga和生理活性物质的溶液从第2导入部d2以0.35mpag、16ml/分钟、30℃导入处理用面1、2间,将pva水溶液与plga和生理活性物质的溶液在强制薄膜中混合,在处理用面1、2间制造包含二氯甲烷的plga和生理活性物质的乳化粒子。使处理用面1、2间的包含plga和生理活性物质的乳化粒子的流体(以下称为plga和生理活性物质的乳化粒子分散液)从粒子化处理装置的处理用面1、2间排出。将用于捕获排出的plga和生理活性物质的乳化粒子分散液的plga和生理活性物质的乳化粒子分散液回收至保持为0.02mpag压力的回收罐。
[0134]
用与实施例1和2相同的方法进行脱溶剂工序,得到的微球的平均体积基准粒径为34.8μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图7-1)。
[0135]
用与实施例1和2相同的方法对观察到的sem图像进行图像分析。将放大并进行二值化处理后的sem截面示于图7-2。以最大直径的中心点为中心每隔60度分割为6个得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.056。
[0136]
(实施例4)
[0137]
以乳酸/乙醇酸共聚物(resomer rg504,evonik制造)成为13质量%、作为生理活性物质的普罗布考(富士胶片和光纯药制造,生物化学用)成为1.0质量%的方式,加入二氯甲烷(关东化学制造),使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到plga和生理活性物质的溶液。之后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。pva水溶液与参考例1同样地制备。在回收plga和生理活性物质的乳化粒子的罐中预先放入pva水溶液,以液面移动的程度进行搅拌。
[0138]
用与实施例1~3相同的方法进行脱溶剂工序,得到的微球的平均体积基准粒径为32.5μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图8-1)。
[0139]
用与实施例1~3相同的方法对观察到的sem图像进行图像分析。将二值化处理后的sem截面示于图8-2。以最大直径的中心点为中心每隔60度分割为6个得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.235。
[0140]
(比较例1)
[0141]
与实施例4同样地,作为粒子形成工序,制备plga和生理活性物质的乳化粒子分散液。接着,作为脱溶剂工序,使用clearmix dissolver(m-technique制造),一边以圆周速度4.7m/秒搅拌回收的排出液,一边在大气中用42小时除去二氯甲烷,得到包含微球的悬浮液(微球悬浮液)。得到的微球的平均体积基准粒径为31.8μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像。
[0142]
用与实施例1~3相同的方法对观察到的sem图像进行图像分析。将二值化处理后的sem截面示于图9。从最大直径的中心点起将半径进行6等分,以同心圆状分割为6个得到
的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.468。
[0143]
(比较例2)
[0144]
以与实施例4相同的配方,将plga和药物的溶解变为采用螺旋桨式搅拌机(three-one motor制造)搅拌10分钟,以与实施例4相同的条件进行粒子形成工序和脱溶剂工序,制备包含微球的悬浮液。得到的微球的平均体积基准粒径为29.8μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图10)。
[0145]
利用与实施例1~4相同的方法对所观察的sem图像进行图像分析,以最大直径的中心点为中心每隔60度分割为6个得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.357。
[0146]
(比较例3)
[0147]
以乳酸/乙醇酸共聚物(resomer rg504,evonik制造)成为5.0质量%、作为生理活性物质的姜黄素(富士胶片和光纯药制造,和光特级)成为0.25质量%的方式,加入二氯甲烷(关东化学制造)69.75质量%和丙酮(关东化学制造)25质量%,使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到了plga和生理活性物质的溶液。之后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。pva水溶液与参考例1同样地制备。在回收plga和生理活性物质的乳化粒子的罐中预先放入pva水溶液,以液面移动的程度进行搅拌。
[0148]
以与实施例1相同的条件进行粒子形成工序和脱溶剂工序,制备包含微球的悬浮液。得到的微球的平均体积基准粒径为6.8μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图11)。
[0149]
利用与实施例1~4相同的方法对所观察到的sem图像进行图像分析,以最大直径的中心点为中心每隔60度分割为6个得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为0.386。
[0150]
(比较例4)
[0151]
以乳酸/乙醇酸共聚物(resomer rg504,evonik制造)成为5.0质量%、作为生理活性物质的孕酮(sigma-aldrich制造)成为0.3质量%的方式,加入二氯甲烷(关东化学制造)69.5质量%和丙酮(关东化学制造)25质量%,使用高速旋转式分散机clearmix dissolver(m-technique制造)使其溶解,得到plga和生理活性物质的溶液。之后,用0.2μm的通气过滤器(φ62,merck制造)进行过滤。pva水溶液与参考例1同样地制备。在回收plga和生理活性物质的乳化粒子的罐中预先放入pva水溶液,以液面移动的程度进行搅拌。
[0152]
以与实施例3相同的条件进行粒子形成工序和脱溶剂工序,得到包含微球的悬浮液。得到的微球的平均体积基准粒径为21.6μm,采用液氮冷冻代表性的粒子后,制作fib截面,观察sem图像(图12)。
[0153]
利用与实施例1~4相同的方法对所观察的sem图像进行图像分析,以最大直径的中心点为中心每隔60度分割为6个得到的各个区域的区域面积(a)与该区域所包含的上述生理活性物质的截面积的总和(s)的比率(s/a)
×
100(%)的变动系数为1.094。
[0154]
在表1中示出了实施例1~4、比较例1~4以及参考例1(仅有plga)的条件的一部分。
[0155]
[表1]
[0156][0157]
在表2中示出了实施例1~4和比较例1~4和利普安的代表性粒径的微球各区域中的面积比率%、标准偏差、平均值和变动系数(cv值)。
[0158]
[表2]
[0159][0160]
由表1和表2可知,在实施例1和实施例2中,无论使用plga和pla中的哪一个,粒子内的各区域的生理活性物质所占的面积比率的变动系数都为0.35以下,粒子内的生理活性物质的分布均匀。在比较例3中,虽然粒子精制工序和脱溶剂工序为与实施例1相同的条件,但如果降低plga浓度,则粒子内的均匀性降低,各区域的生理活性物质所占的面积比率的变动系数增大为0.386。
[0161]
在实施例4和比较例1中,由于干燥条件和干燥时间的不同,生理活性物质的微粒的粒径发生变化,在干燥时间长的情况下,分布于粒子内的生理活性物质的均匀性降低,各区域的生理活性物质所占的面积比率的变动系数增大为0.468。在与实施例相比降低了plga和药物的浓度的比较例3和4中,干燥时的收缩率变大,粒子内的生理活性物质产生偏差,各区域的生理活性物质所占的面积比率的变动系数超过0.35。
[0162]
产业上的可利用性
[0163]
根据本发明,能够提供微球,其能够适当地控制生理活性物质的初期释放量和其
后的释放期间的释放速度,在生物体内在一定期间持续释放生理活性物质。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1