基于二代高通量测序的哺乳动物piRNA数据分析方法与流程
基于二代高通量测序的哺乳动物pirna数据分析方法
技术领域
1.本发明涉及一种基于二代高通量测序的哺乳动物pirna数据分析方法,属于自动化测序技术领域。
背景技术:
2.近年来,人们对pirna发生过程的相关蛋白及生物学作用做了研究,发现多种piwi亚家族蛋白能与pirna结合形成piwi
‑
pirna复合体,通过表观遗传调控参与生殖细胞形成与发育、生殖干细胞分化、性别决定等生物学功能。随后pirna成为非编码小rna研究领域的一个热点,先后在果蝇、线虫、斑马鱼、小鼠、大鼠等动物研究中发现pirna在生殖细胞及干细胞分化、种系dna完整性、性别决定、胚胎发育、免疫防御及癌症预测等方面有重要作用。
3.pirna(piwi
‑
interacting rna)是指能与piwi家族蛋白交互的非编码rnas。长度约为24nt到32nt,比mirna略长,在所有的非编码rna中,pirna数量最多。pirna的发现开拓了非编码rna研究的全新领域。
4.总体来说,由于pirna在生殖系统中起着多种重要的作用,被视为哺乳动物生殖系统发育的关键调控因子之一,随着研究的不断深入,越来越多的证据显示,pirna以多种不同机制参与精子发生相关基因的表达调控,为深入理解哺乳动物睾丸发育和精子发生的分子机制和调控网络提供了一个很好的视角。
5.而目前pirna分析方式虽然很多,没有统一标准,但是多复杂步骤繁琐,且最终结果并不怎么准确,而此方法能够简单高效的鉴定到物种的pirna簇信息并预测出对应的靶基因序列。
技术实现要素:
6.为了解决上述技术问题,本发明提供一种基于二代高通量测序的哺乳动物pirna数据分析方法,其具体技术方案如下:
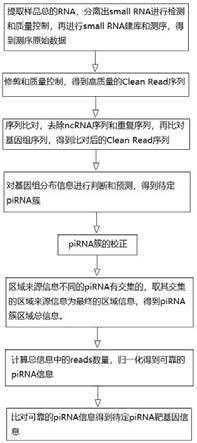
7.一种基于二代高通量测序的哺乳动物pirna数据分析方法,包括如下步骤:步骤1:对样品的总rna进行提取,并分离出small rna,对提取的small rna进行检测和质量控制,再进行small rna建库和测序,获得测序原始数据;
8.步骤2:对步骤1中得到的测序原始数据进行修剪和质量控制,得到高质量的clean read序列;
9.步骤3:将步骤2中得到的高质量的clean read序列进行序列比对,比对后去除高质量的clean read序列中各种ncrna序列和重复序列,最后比对基因组序列,得到比对后的clean read序列;
10.步骤4:将步骤3中比对后的clean read序列分成若干个指定长度,对每个长度的基因组上分布信息进行判断和预测,得到待定pirna簇;
11.步骤5:pirna簇的校正:样品是多个时,对发生区域间交叠或正负链上交叠的待定pirna簇进行鉴定,并按照取并集原则融合形成同一簇,确定最终簇的的区域来源信息,并
将该区域内对应的样品按照步骤6处理;样品是单个时,对待定pirna簇则按照步骤7处理;
12.步骤6:步骤5所得到的该区域内对应的样品中,区域来源信息不同的pirna簇有交集的,则取其交集的区域来源信息作为最终的区域信息,将所有最终的区域信息作为pirna簇区域总信息;
13.步骤7:计算pirna簇区域总信息中的reads数量,用rpm方法进行归一化处理,获得可靠的pirna信息;
14.步骤8:将步骤7中得到的可靠的pirna信息与对应物种的基因序列信息比对,然后预测与可靠的pirna信息互补的基因,通过筛选得到待定pirna靶基因信息。
15.进一步的,所述步骤1中样品指同一物种某一时期的单个取样或同一物种多个时期多个取样。
16.进一步的,所述步骤2中的修剪和质量控制具体步骤为:
17.步骤2.1:对测序原始数据对3’端和5’端进行接头序列的修剪,得到去接头small rna序列;
18.步骤2.2:修剪步骤2.1得到的去接头small rna序列的末端质量值低于20的碱基,得到高质量去接头small rna序列;
19.步骤2.3:修剪步骤2.2得到的高质量去接头small rna序列中未知碱基含量大于或等于5的序列,得到去多碱基序列;
20.步骤2.4:保留步骤2.3得到的去多碱基序列中大于等于24nt且小于等于32nt的序列,得到高质量的clean read序列。
21.进一步的,所述步骤3中序列比对时具体与mirbase数据库、rfam数据库和repbase数据库进行比对。
22.进一步的,所述步骤3中按micro rna、核糖体rna、转运rna、核内小rna、核仁小rna等ncrna以及重复序列的顺序进行比对。
23.进一步的,所述步骤4中的指定长度范围为24~32bp。
24.进一步的,所述步骤7中的rpm方法定义为reads per million mapped reads,即每1百万个比对上的reads中比对到pirna的reads个数。
25.进一步的,所述步骤8中筛选得到待定pirna靶基因信息时,允许2个以内的错配。
26.本发明的有益效果是:只需要提供二代small rna测序数据,便能够自动化地完成分析任务,输出详细pirna分析结果。该方法能够快速从二代small rna测序数据中快速识别和鉴定不同样品的pirna信息,快速识别物种的pirna簇区域、表达信息及靶基因调控信息,简单高效。
附图说明
27.图1是本发明的步骤流程图。
具体实施方式
28.按如图1所示的本发明步骤来操作,
29.以下表1的绵羊(ovis aries)的small rna采集样品为实施例实验对象,所示同一时期的为不同个体上取样所得的,以测得雄性绵羊(ovis aries)的生殖性状和哪些基因有
关:
30.表1
[0031][0032][0033]
步骤1:对样品的总rna进行提取,并分离出small rna,对提取的small rna进行检测和质量控制,再进行small rna建库和novaseq 6000se50(平台)测序,获得测序原始数据;
[0034]
步骤2:为了保证信息分析的准确性,需要对原始数据进行质量控制及处理,对步骤1中得到的测序原始数据进行修剪和质量控制,得到高质量的clean read序列;修剪和质量控制具体步骤为:
[0035]
步骤2.1:对测序原始数据对3’端和5’端进行接头序列的修剪,得到去接头small rna序列;接头去除的越干净,测序得到的small rna序列越正确。
[0036]
步骤2.2:修剪步骤2.1得到的去接头small rna序列的末端质量值低于20的碱基,得到高质量去接头small rna序列;高质量值small rna测序序列的单碱基准确性更高。
[0037]
步骤2.3:修剪步骤2.2得到的高质量去接头small rna序列中未知碱基含量大于或等于5的序列,得到去多碱基序列;n碱基越少,small rna测序序列在分析过程中得到的准确性越高。
[0038]
步骤2.4:保留步骤2.3得到的去多碱基序列中大于等于24nt且小于等于32nt的序列,得到高质量的clean read序列。过长过短的测序small rna序列不可能是pirna。
[0039]
原始数据质控越严格,分析得到的结果假阳性越低,越接近真实可靠的结果。步骤3:利用bowtie(v1.2.2)软件,将步骤2中得到的高质量的clean read序列与mirbase数据库、rfam数据库和repbase数据库进行比对。比对后去除高质量的clean read序列中各种
ncrna序列和重复序列,最后比对基因组序列,得到比对后的clean read序列;按micro rna(mirna)、核糖体rna(rrna)、转运rna(trna)、核内小rna(snrna)、核仁小rna(snorna)等ncrna以及重复序列(repeat)的顺序进行比对。
[0040]
步骤4:将步骤3中比对后的clean read序列分成若干个指定长度,对每个长度的基因组上分布信息进行判断和预测,得到待定pirna簇;使用protrac(v2.4.3)软件基于reads在基因组上的分布信息预测出可能的pirna簇。protrac软件使用参数:
‑
pimin 24
‑
pimax 32
‑
pdens 0.05
‑
1tor10a 0.01
‑
1tand10a 0.01
‑
distr 1
‑
90
‑
clsize 1000
‑
clsplit 0.2
‑
clhitsn 100。
[0041]
步骤5:pirna簇的校正:对发生区域间交叠或正负链上交叠的待定pirna簇进行鉴定,并按照取并集原则融合形成同一簇,确定最终簇的的区域来源信息,并将该区域内对应的样品按照步骤6处理;对未发生交叠的待定pirna簇则按照步骤7处理;
[0042]
步骤6:步骤5所得到的该区域内对应的样品中,区域来源信息不同的pirna簇有交集的,则取其交集的区域来源信息作为最终的区域信息,将所有最终的区域信息作为pirna簇区域总信息,得到如表2所示数据:
[0043]
表2
[0044][0045]
上图为pirna簇所在位置总信息,第一列为染色体位置,第二列为pirna簇在染色体上的起始位置,第三列为pirna在染色体上的终止位置,第四列为pirna簇的命名,第五列为pirna的起始和终止位置信息,第六列为pirna的来源信息,+表示来自一个正链,
‑
表示来自一个负链,=表示多个来源。
[0046]
步骤7:计算pirna簇区域总信息中的reads数量,用rpm(reads per million mapped reads)方法对pirna簇区域覆盖的测序reads的数量进行归一化处理,获得可靠的pirna信息;rpm方法定义为reads per million mapped reads,即每1百万个比对上的reads中比对到pirna的每个碱基上的reads个数。以便不同样品处理之间进行比较分析。
[0047]
步骤8:将步骤7中得到的可靠的pirna信息与对应物种的基因序列信息比对,然后预测与可靠的pirna信息互补的基因,通过筛选得到待定pirna靶基因信息,采用bowtie(v1.2.2)软件,允许2个以内的错配。预测与pirna互补的基因,然后通过筛选得到可能的pirna靶基因信息。得到多个数据,其中如下表3所示数据为一部分由pirna得出的影响雄性绵羊(ovis aries)生殖的靶基因序列信息:
[0048]
最后得出所要测性状对应的靶基因信息如下表3:
[0049]
表3
[0050][0051]
第一列为来源的pirna簇;第二列:靶基因信息;第三列:来源的样本信息。
[0052]
得出tssk3影响精子的发育,igf2bp1影响雄性动物生殖机能的维持,pld4在精子获能和受精方面发挥作用。
[0053]
其中tssk3为影响精子发育的靶基因,其序列具体为:
[0054]
>s16_001029363_x2
[0055]
gcaagtcctggcattcagccgagatgcccaga
[0056]
>s16_000785157_x2
[0057]
gccccccattcagcacacagtcaaa
[0058]
>s16_001066195_x2
[0059]
agtcaaagacatcccctccttcag
[0060]
igf2bp1为影响雄性动物生殖机能维持的靶基因,其序列具体为:
[0061]
>s10_000165807_x2
[0062]
atgcttgctgctggaaattcaccttaaa
[0063]
>s10_000260804_x2
[0064]
ttctgggggtctacagaggatcgtca
[0065]
>s10_000080794_x4
[0066]
ttctgggggtctacagaggatcgtca
[0067]
>s10_000078781_x4
[0068]
gggtctacagaggatcgtcatgggaaga
[0069]
>s10_000065664_x4
[0070]
cagaggatcgtcatgggaagaaagtga
[0071]
>s10_000112082_x3
[0072]
aacccgtccccagttacattcccaaa
[0073]
>s10_000054393_x5
[0074]
ggtccactgttagaatcaatcatatca
[0075]
>s10_000272383_x2
[0076]
actttaaaatatctcaacagcacca
[0077]
pld4在精子获能和受精方面发挥作用,其序列具体为:
[0078]
>s13_000331156_x6
[0079]
tgcgtgtctgagactttctgcagagaa
[0080]
>s13_000806933_x3
[0081]
tgcgtgtctgagactttctgcagagaa
[0082]
>s13_001592107_x2
[0083]
tgcgtgtctgagactttctgcagagaaa
[0084]
>s13_000890089_x2
[0085]
taatgaagcagcggagtaagagaagatgc
[0086]
>s13_000110868_x21
[0087]
ttagaaaactgatgacttcaaacctatga
[0088]
>s13_000077360_x23
[0089]
tagaaaactgatgacttcaaacctatgaga
[0090]
由此可得本发明的方法和装置只需要提供二代small rna测序数据,便能够快速从二代small rna测序数据中识别和鉴定不同样品的pirna信息,快速识别物种的pirna簇区域、表达信息及靶基因调控信息,能够自动化地完成分析任务,输出详细的可能的pirna靶基因分析结果,相比市面上其他同类产品简单高效。
[0091]
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1