一种基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法
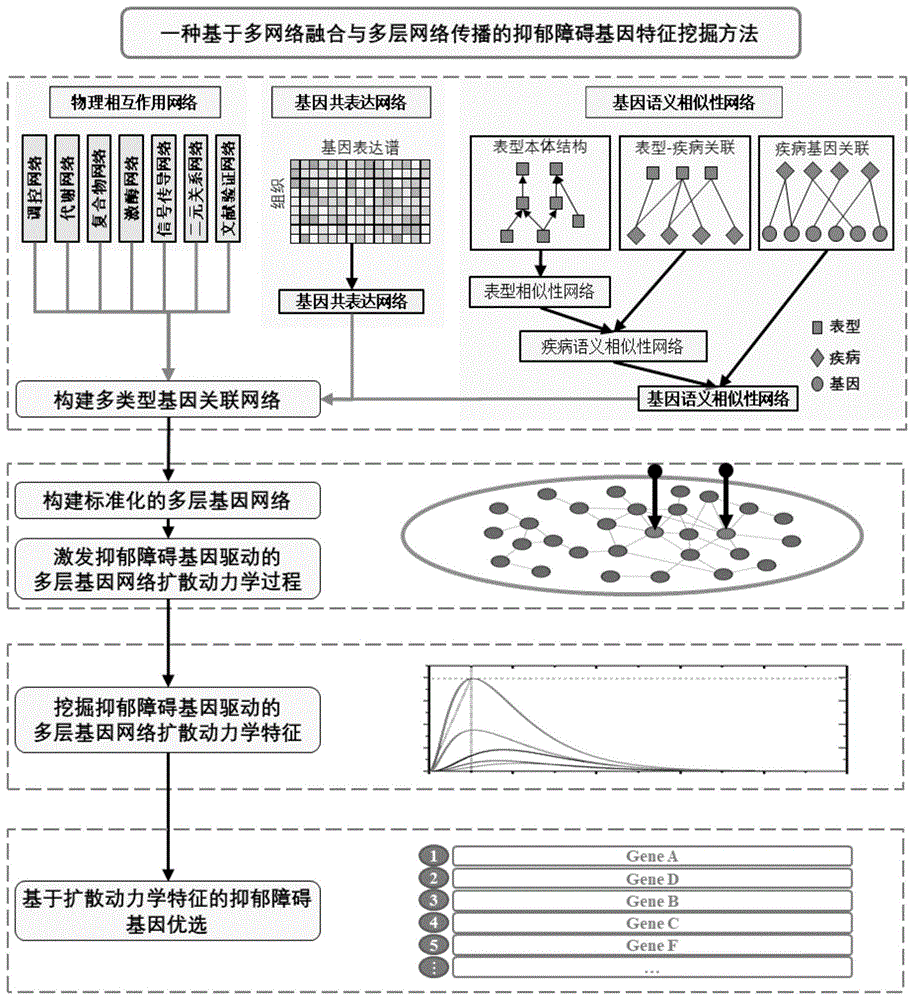
本发明属于生物信息学分析领域,涉及基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法。
背景技术:
抑郁症已成为当今社会的一种常见病,严重影响患者的生活质量,抑郁症与相关基因的功能失调密切相关,但全基因组关联研究等传统方法通常难以对抑郁症基因进行精确定位。由于生物医学实验成本高、时间周期长,因此通过开发计算方法挖掘抑郁障碍基因特征,从而识别抑郁障碍相关基因对于抑郁障碍的机制研究、预防、诊断和治疗都非常的重要。
随着蛋白质相互作用网络等生物分子网络数据快速累积,基于生物分子网络的基因特征挖掘算法成为遗传关联研究的前沿。相同疾病的致病基因,由于其功能上的相关性,在生物分子网络中趋向于聚集,因此基于网络传播的算法成为一种有效策略,例如,带重启的随机游走,异构网络随机游走,多重网络随机游走等;基于现代高通量实验技术的快速发展,各种不同类型的生物分子网络数据的快速增长,这些生物分子网络数据的有效利用将有助于更有效的挖掘抑郁障碍基因特征;传统的基于聚合网络随机游走的算法可以减少网络不完全性的影响,但它可能失去单个网络的结构特征;独立数据源的排位聚合方法利用了单个网络的特性,但忽略了不同类型网络/层之间的关系;多重图框架考虑了不同类型的网络,但网络层之间的相互作用太强烈。异构网络有助于整合多源异构关联数据,但如何从异构网络中提取有用信息来应对特定的疾病仍是一个挑战;因此,如何有效融合这些不同类型的生物分子网络,如何从这些多样化的生物分子网络中挖掘有效的抑郁障碍等疾病基因特征,从而更有效的识别抑郁障碍相关基因,仍是待研究的重要问题。
技术实现要素:
(一)要解决的技术问题
基于此,本发明公开了一种基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法(后文简称为ddsm),该方法能有效融合不同类型的生物分子网络,并从各种多样化的生物分子网络中挖掘有效的抑郁障碍基因特征,从而更有效的识别抑郁障碍相关基因,经过本发明实验可知,该方法的网络建模和动力学激发过程都是特别适合于抑郁障碍相关基因特征的挖掘。
(二)技术方案
本发明公开了一种基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法,该基因特征挖掘方法包括以下步骤:
步骤1:将多种类型的生物数据建模转换成基因关联网络:计算人类基因表达谱的皮尔逊系数,获取每个基因最相似的k个邻居,构建稀疏化的k近邻基因共表达网络;通过疾病相似性网络和疾病基因关联,计算基因语义相似性网络;
步骤2:将所述基因关联网络中的k近邻基因共表达网络、基因语义相似性网络和多类型的蛋白质物理相互作用网络中对应相同基因的节点相互连接,构建多层基因网络的邻接矩阵,实现多网络融合;通过多层基因网络的邻接矩阵的列归一化,计算多层基因网络的扩散矩阵;
步骤3:依据抑郁障碍基因驱动的多层基因网络扩散动力学方程,将抑郁障碍的已知致病基因作为扩散源,激发抑郁障碍基因驱动的多层基因网络扩散动力学过程,计算多层基因网络中每个节点的状态在扩散过程中的扩散特征。
步骤4:通过计算多层基因网络中对应相同基因的网络节点的所述扩散特征的几何均值,获得综合的抑郁障碍基因特征;依据综合的抑郁障碍基因特征优选抑郁障碍相关基因。
进一步的,步骤1中具体包括:
步骤1.1:获取抑郁障碍及各种相关疾病已知的致病基因数据、疾病表型关联注释数据、人类表型本体数据以及基因表达数据;将多种类型的数据转换成基因关联网络,包括基因共表达网络和基于疾病基因关联的基因语义相似性网络;
基因关联网络的计算方法如下:为将基因表达谱数据应用到基因特征挖掘中,计算人类基因表达谱基因之间的皮尔逊系数
其中d1i表示与基因g1关联的疾病,d2j表示与基因g2关联的疾病;n表示与基因g1关联的疾病数量;m表示与基因g2关联的疾病数量;sim(d1i,d2j)表示疾病d1i和疾病d2j之间的相似性或者关联。
3.根据权利要求2所述的基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法,其特征在于,所述步骤1.1后还包括步骤1.2:获取多类型的蛋白质物理相互作用网络,所蛋白质物理相互作用网络包括调控网络、代谢网络、信号传导网络、蛋白质复合物网络、蛋白质激酶网络、高通量二元相互作用网络、文献验证的蛋白质相互作用网络的一种或者多种。
进一步的,步骤2中具体包括:
步骤2.1:为实现多网络融合,将m个基因网络中对应相同基因的节点相互连接起来,构建多层基因网络的邻接矩阵,其邻接矩阵表示为a=aintra+ainter∈rnm×nm,aintra表示独立网络层的超矩阵,ainter表示层间网络超矩阵,n表示各网络层中的基因数目;独立网络层的超矩阵定义为,
其中a(α)表示α网络层的邻接矩阵;层间网络超矩阵定义为,
其中al∈rm×m表示网络层连接矩阵,i∈rn×n为单位矩阵,
步骤2.2:通过对多层基因网络的邻接矩阵的列归一化,计算多层基因网络的扩散矩阵,扩散矩阵的每个元素值的计算方法为:wij=aij/∑iaij;其中,aij为矩阵a的元素值。
进一步的,步骤3中具体包括:
步骤3.1:抑郁障碍基因驱动的多层基因网络扩散动力学方程定义为,
其中抑郁障碍基因设为扩散源,yi(t)表示在t时刻多层网络节点i的状态,其中i=1~nm;wij作为网络节点之间的扩散系数;其向量化表示为,
其中l=d-w为多层基因网络的拉普拉斯矩阵,d为对角矩阵,对角元素dii=∑jwij;
步骤3.2:依据定义的多层网络扩散动力学方程,将抑郁障碍的已知致病基因作为扩散源,激发抑郁障碍基因驱动的多层基因网络扩散动力学过程,计算多层基因网络中每个节点的状态随时间的演化过程。
进一步的,步骤4中具体包括:
步骤4.1:依据抑郁障碍基因驱动的多层基因网络扩散动力学过程中网络节点的状态随时间的演化行为,提取扩散过程中的峰值特征
步骤4.2:基于抑郁障碍基因驱动的多层网络节点扩散特征的量值,计算多层基因网络中对应相同基因的网络节点的扩散特征的几何均值,获得综合的抑郁障碍基因特征,计算公式为
在另外一方面,本发明还公开了一种基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘系统,包括:
至少一个处理器;以及与所述处理器通信连接的至少一个存储器,其中:
所述存储器存储有可被所述处理器执行的程序指令,所述处理器调用所述程序指令能够执行如上述任一项所述的基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法。
在另外一方面,本发明还公开了一种非暂态计算机可读存储介质,所述非暂态计算机可读存储介质存储计算机指令,所述计算机指令使所述计算机执行如上述任一项所述的基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法。
(三)有益效果
本发明的上述技术方案具有如下优点,该方法将多种类型的数据转换成基因关联网络,通过多层网络融合多个类型的基因关联网络,通过列归一化计算多层基因网络的扩散矩阵,将抑郁障碍基因作为扩散源,激发抑郁障碍基因驱动的多层基因网络扩散动力学过程,挖掘抑郁障碍基因特征,通过多层网络节点扩散特征的几何均值构建综合的基因特征,从而更有效的挖掘抑郁症相关基因。在真实的数据集上的实验结果表明,与多个现有方法相比,本方法挖掘的抑郁障碍基因特征具有更强的鉴别能力,其网络建模和动力学激发过程能够很好地应用于各个基因之间相关性强的抑郁障碍基因的特征挖掘工作。
附图说明
通过参考附图会更加清楚的理解本发明的特征和优点,附图是示意性的而不应理解为对本发明进行任何限制,在附图中:
图1为本发明挖掘方法ddsm的流程示意图;
图2为在采用不同控制集策略时本发明挖掘方法ddsm和rwrh、chn、rwrmp、rwrmg和rwrdrs的性能指标比较图。
具体实施方式
下面将结合附图和实施例对本发明进行清楚、完整地描述,同时也叙述了本发明技术方案解决的技术问题及有益效果,需要指出的是,所描述的实施例仅旨在便于对本发明的理解,而对其不起任何限定作用。
如图1所示,本发明提供了一种基于多网络融合和多层网络扩散的抑郁障碍基因特征挖掘方法,包括以下步骤:
步骤1:构建多类型基因关联网络
将多种类型的生物数据建模转换成基因关联网络:计算人类基因表达谱的皮尔逊系数,获取每个基因最相似的k个邻居,构建稀疏化的k近邻基因共表达网络;通过疾病相似性网络和疾病基因关联,计算基因语义相似性网络;
步骤1中具体包括:
步骤1.1:获取抑郁障碍及各种相关疾病已知的致病基因数据、疾病表型关联注释数据、人类表型本体数据以及基因表达数据;将多种类型的数据转换成基因关联网络,包括基因共表达网络和基于疾病基因关联的基因语义相似性网络;
基因关联网络的计算方法如下:为将基因表达谱数据应用到基因特征挖掘中,计算人类基因表达谱基因之间的皮尔逊系数
其中d1i表示与基因g1关联的疾病,d2j表示与基因g2关联的疾病;n表示与基因g1关联的疾病数量;m表示与基因g2关联的疾病数量;sim(d1i,d2j)表示疾病d1i和疾病d2j之间的相似性或者关联;
步骤1.2:获取多类型的蛋白质物理相互作用网络(例如:调控网络、代谢网络、信号传导网络、蛋白质复合物网络、蛋白质激酶网络、高通量二元相互作用网络和/或文献验证的蛋白质相互作用网络);
步骤2:构建标准化的多层基因网络
将所述基因关联网络中的k近邻基因共表达网络、基因语义相似性网络和多类型的蛋白质物理相互作用网络中对应相同基因的节点相互连接,构建多层基因网络的邻接矩阵,实现多网络融合;通过多层基因网络的邻接矩阵的列归一化,计算多层基因网络的扩散矩阵;
步骤2中具体包括:
步骤2.1:为实现多网络融合,将m个基因网络中对应相同基因的节点相互连接起来,构建多层基因网络的邻接矩阵,其邻接矩阵表示为a=aintra+ainter∈rnm×nm,aintra表示独立网络层的超矩阵,ainter表示层间网络超矩阵,n表示各网络层中的基因数目;独立网络层的超矩阵定义为,
其中a(α)表示α网络层的邻接矩阵;层间网络超矩阵定义为,
其中al∈rm×m表示网络层连接矩阵,i∈rn×n为单位矩阵,
步骤2.2:通过对多层基因网络的邻接矩阵的列归一化,计算多层基因网络的扩散矩阵,扩散矩阵的每个元素值的计算方法为:wij=aij/∑iaij;其中,aij为矩阵a的元素值。
步骤3:激发抑郁障碍基因驱动的多层基因网络散动力学过程
依据抑郁障碍基因驱动的多层基因网络扩散动力学方程,将抑郁障碍的已知致病基因作为扩散源,激发抑郁障碍基因驱动的多层基因网络扩散动力学过程,计算多层基因网络中每个节点的状态在扩散过程中的扩散特征;
步骤3中具体包括:
步骤3.1:抑郁障碍基因驱动的多层基因网络扩散动力学方程定义为,
其中抑郁障碍基因设为扩散源,yi(t)表示在t时刻多层网络节点i的状态,其中i=1~nm;wij作为网络节点之间的扩散系数;其向量化表示为,
其中l=d-w为多层基因网络的拉普拉斯矩阵,d为对角矩阵,对角元素dii=∑jwij;
步骤3.2:依据定义的多层网络扩散动力学方程,将抑郁障碍的已知致病基因作为扩散源,激发抑郁障碍基因驱动的多层基因网络扩散动力学过程,计算多层基因网络中每个节点的状态随时间的演化过程;
步骤4:挖掘抑郁障碍基因驱动的多层基因网络扩散动力学特征
通过计算多层基因网络中对应相同基因的网络节点的扩散特征的几何均值,获得综合的抑郁障碍基因特征;依据综合的抑郁障碍基因特征优选抑郁障碍相关基因。
步骤4中具体包括:
步骤4.1:依据抑郁障碍基因驱动的多层基因网络扩散动力学过程中网络节点的状态随时间的演化行为,提取扩散过程中的峰值特征
步骤4.2:基于抑郁障碍基因驱动的多层网络节点扩散特征的量值,计算多层基因网络中对应相同基因的网络节点的扩散特征的几何均值,获得综合的抑郁障碍基因特征,计算公式为
为了体现本发明的优点,在另外一个实施例中,还进一步通过实验验证本发明方法的有效性,本发明通过收集抑郁障碍及相关疾病基因数据、疾病表型数据、表型本体数据、基因表达数据以及多类型的蛋白质物理相互作用数据,对本方法的性能进行综合评估:
(1)数据准备:通过omim数据库(https://omim.org/)获取抑郁症等疾病基因数据;通过hpo数据库获取疾病表型注释数据和表型本体数据;通过gtex获取基因表达数据;然后,构建基因语义相似性网络和基因共表达网络;通过文献获取多类型的蛋白质物理相互作用数据(https://science.sciencemag.org/content/suppl/2015/02/18/347.6224.1257601.dc1);
(2)评价策略:采用留一验证,每次将一个已知的疾病基因关联作为阳性测试集,其它作用训练集;采用ali控制集和wg控制集;ali对于阳性测试集的每个基因,选择与其在相同染色体上距离最近并且不属于训练集的99个基因作为控制集;wg将训练集和阳性测试集之外的所有基因作为控制集;
(3)评价指标:将auroc和auprc指标作为预测性能的评估指标;auroc,又称为auc,是接收着工作特性曲线(roc)下的面积,已被广泛用于全面衡量预测算法的全局性能,其中roc曲线是以真阳性率tpr(又称召回率、敏感性)为纵坐标,以假阳性率fpr为横坐标的性能曲线;auprc是精度-召回率曲线(prc)下的面积,其中prc曲线以精度为纵坐标,以召回率为横坐标;
(4)评价结果
从图2可以看出,无论在ali控制集下还是在wg控制集下,本方法ddsm的auroc值和auprc值都优于rwrh、chn、rwrmp、rwrmg和rwrdrs等其它方法的结果;这表明本方法通过多类型的基因关联网络融合和抑郁障碍基因驱动的多层基因网络扩散动力学过程,能够更有效的挖掘抑郁障碍基因特征,从而获得更强的识别抑郁障碍基因的能力。
另外需要说明的是,上述本发明的基因特征挖掘方法可以作为软件程序或者计算机指令在非暂态计算机可读存储介质中执行或者在带有存储器和处理器的控制系统中对大量数据进行批量执行,且其计算程序简单快速。在本发明各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用硬件加软件功能单元的形式实现。上述以软件功能单元的形式实现的集成的单元,可以存储在一个计算机可读取存储介质中。上述软件功能单元存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)或处理器(processor)执行本发明各个实施例所述方法的部分步骤。而前述的存储介质包括:u盘、移动硬盘、只读存储器(read-onlymemory,rom)、随机存取存储器(randomaccessmemory,ram)、磁碟或者光盘等各种可以存储程序代码的介质。
最后说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!