一种基因测序数据智能结构化存储及提取方法与流程
1.本发明涉及基因数据存储或提取领域,具体为一种基因测序数据智能结构化存储及提取方法。
背景技术:
2.近年来,基因数据的分析研究在不断深入,人们也充分意识到了研究基因信息的重要性,由于基因数据庞大、具有不完全性且随机性强,一个样本有几万个基因表达情况,十几万个甲基化和转录因子结合位点,几十万到几百万个突变位点。当样本量上升到数十万个体时,数据量将进一步上升,达到万亿级别。将这些信息快速存入数据库并进行高速查询是一个挑战。传统数据库如mysql数据库等,超过200万-300万条记录就会出现性能问题。为解决上述问题,我们对此做出改进,提出一种基因测序数据智能结构化存储及提取方法。
技术实现要素:
3.为了解决上述技术问题,本发明提供了如下的技术方案:
4.本发明提出了一种基因测序数据智能结构化存储及提取方法,包括以下几个步骤
5.步骤1、建立一个列式数据库,将突变位点、表达谱、chip、甲基化等基因相关数据存在数据库;
6.步骤2、针对生物信息/基因测序信息的特殊性,对数据进行的优化,把基因数据优化到或修改格式到能够方便的存进非关系数据库;
7.步骤3、利用数据库里存储的突变位点、表达谱、chip、甲基化等基因相关数据信息,对照已知信息,结合表型信息进行生物学意义分析;
8.步骤4、设置快速增量统计方法来对数据进行统计,从而将新增加的基因相关数据快速录入到数据库内并进行批量统计。
9.作为本发明的一种优选技术方案,所述的步骤2中突变位点信息同时记录了一个位点的覆盖度和突变信息,且同时记录了一个位点的覆盖度和突变信息的方法是,对于一个样本的基因组位点,如果该位点没有检测到覆盖情况,就不考虑该位点,数据库也不进行记录,如果该基因位点检测到覆盖但是没有突变,则表明该位点无突变,数据库中记录该位点的覆盖情况且标记为无突变,如果该位点有突变且覆盖深度足够,则在数据库中记录该位点的突变信息及对应的覆盖情况,后续进行统计计算。对于表达谱数据,所述步骤2中分样本记录了某个基因的表达。对于该基因表达量的统计,按照表达量从低到高划分出若干区段,并统计每个区域中有多少样本的基因的表达值落在该去榆中。对于chip和甲基化数据而言,所述步骤2中分样本记录了染色体的结合区段和显著性。
10.作为本发明的一种优选技术方案,所述的设置快速增量统计方法来对数据进行统计的方法是,将当需要统计的录取样本数量达到设定的数值m时,则对录取样本内的数据进行录取,首先获得非关系数据库内的现有统计值以及原始样本的数量,并获得待录取样本内的录取数据个数,则数据库内某个统计量指标变为f(现有统计量,现有样本数量,新录入
数据统计量,新录入样本量)。
11.作为本发明的一种优选技术方案,所述的建立一个非关系的数据库的方法是通过列式数据库建立新的数据库。
12.作为本发明的一种优选技术方案,所述的修改格式到能够方便的存进列式数据库里的方法是,通过基因变异文件格式进行存储得到数据格式,把基因变异文件中的位点作为记录进行优化后存入列式数据库中。
13.本发明的有益效果是:1、该种基因测序数据智能结构化存储及提取方法,建立一个非关系数据库,将各个基因测序位点存进数据库里;针对生物信息/基因测序突变位点的特殊性,对数据进行的优化,把基因相关数据优化到或修改格式到能够方便的存进非关系数据库;利用数据库里存储的基因相关数据信息,对照已知信息,结合表型信息进行生物学意义分析;设置快速增量统计方法来对数据进行统计,从而将数据录入到数据库内,该方法采用非关系数据库,能够存储大量的信息且不会发生卡顿现象,利用快速增量统计方法来对数据进行统计,即不对现有样本进行重新整合统计,仅对新样本进行统计并获得统计量,随后与现有统计量进行合并,保证计算资源的消耗处于较小范围内。
附图说明
14.附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一并用于解释本发明,并不构成对本发明的限制。在附图中:
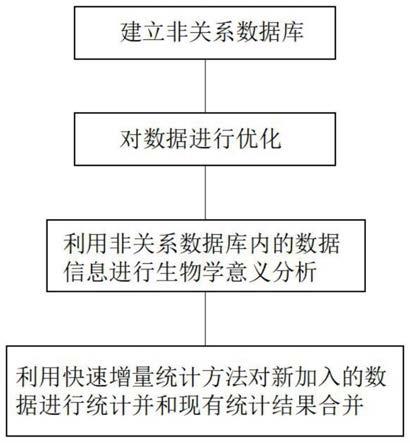
15.图1是本发明一种基因测序数据智能结构化存储及提取方法的流程图。
具体实施方式
16.以下结合附图对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
17.实施例:如图1所示,本发明一种基因测序数据智能结构化存储及提取方法,包括以下几个步骤
18.步骤1、建立一个列式数据库,将各个基因测序位点存进数据库;
19.步骤2、针对生物信息/基因测序突变位点的特殊性,对数据进行的优化,把我们的突变位点的数据优化到或修改格式到能够方便的存进非关系数据库;
20.步骤3、利用数据库里存储的突变位点等数据信息,对照已知的突变位点信息,结合表型信息进行生物学意义分析;
21.步骤4、设置快速增量统计方法来对数据进行统计,并将数据录入到数据库内。
22.所述的步骤2中同时记录了一个位点的覆盖度和突变信息,且同时记录了一个位点的覆盖度和突变信息的方法是,对于一个样本的基因组位点,如果该位点没有检测到覆盖情况,就不考虑该位点,数据库也不进行记录,如果该基因位点检测到覆盖但是没有突变,则表明该位点无突变,数据库中记录该位点的覆盖情况且标记为无突变,如果该位点有突变且覆盖深度足够,则在数据库中记录该位点的突变信息及对应的覆盖情况,后续进行统计计算。对于表达谱数据,所述步骤2中分样本记录了某个基因的表达。对于该基因表达量的统计,按照表达量从低到高划分出若干区段,并统计每个区域中有多少样本的基因的表达值落在该去榆中。对于chip和甲基化数据而言,所述步骤2中分样本记录了染色体的结
合区段和显著性。
23.所述的设置快速增量统计方法来对数据进行统计的方法是,将当需要统计的录取样本数量达到设定的数值m时,则对录取样本内的数据进行录取,首先获得非关系数据库内的现有统计值以及原始样本的数量,并获得待录取样本内的录取数据个数,则数据库内某个统计量指标变为f(现有统计量,现有样本数量,新录入数据统计量,新录入样本量)。
24.在做单细胞测序分析时,经常会对单细胞样本进行标准化,目前的标准化需要将全部样本都引入。如库中有1000个样本,需要新录入1000个样本,此时需要把库中原有1000个样本和新的1000个样本合并,总计2000个样本全部标准化,资源消耗巨大。若后续再录入1000或更多样本,为了标准化这些新样本则需要把原有样本取出全部重新标准化。因此我们设计了增量统计和增量标准化模式,即原有样本不变,仅获取原有样本的统计量,只对新样本进行统计,依赖原有样本参数对新样本进行标准化,并获得新样本统计量,然后将新老样本的统计量进行合并。这样每100个新样本进行统计,内存就会保存在一个比较低的低位。
25.所述的建立一个非关系数据库的方法是基于列式数据库建立。
26.所述的修改格式到能够方便的存进列式数据库的方法是,通过基因变异文件格式进行存储得到数据格式,把基因变异文件中的位点作为记录进行优化后存入列式数据库中。
27.建立一个非关系数据库,将各个基因测序位点存进数据库;针对生物信息/基因测序突变位点的特殊性,对数据进行优化,把突变位点、表达谱、chip、甲基化等基因相关数据优化到或修改格式到能够方便的存进列式数据库;利用数据库里存储的基因相关数据信息,对照已知信息,结合表型信息进行生物学意义分析;设置快速增量统计方法来对数据进行统计,从而将数据录入到数据库内,该方法采用列式数据库,能够存储大量的信息且不会发生卡顿现象,并且利用快速增量统计方法来对数据进行统计,即不对现有样本进行重新整合统计,仅对新样本进行统计并获得统计量,随后与现有统计量进行合并,保证计算资源的消耗处于较小范围内。
28.最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1