用于治疗听力损失的基因疗法系统和相关方法与流程
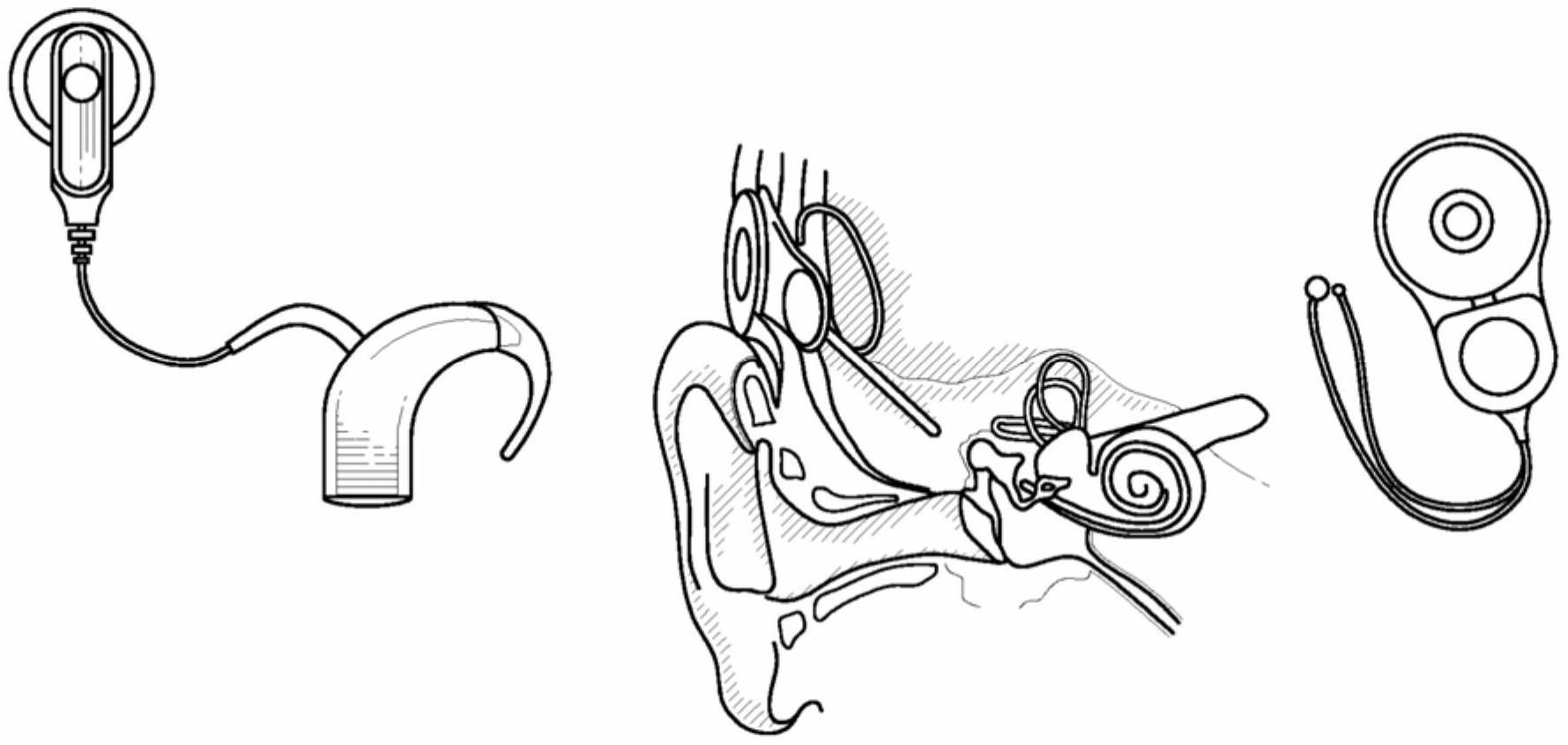
本公开的多个实施方案总体上涉及可用于治疗和/或预防听力损失的基因疗法系统和方法。本文所述的示例性实施方案针对用于预防患者的听力损失的进一步下降的系统和相关方法。更具体说来,本公开中教导的实施方案涉及可用于治疗和/或预防由tmprss3基因或loxhd1基因的基因突变引起的耳聋的基因疗法系统和相关方法。这些系统和方法可以使用针对由基因突变引起的听力损失的基因疗法(例如,分子治疗剂)连同耳蜗植入物的植入的组合。听力损失是人类最常见的感官缺陷。根据2018年世界卫生组织(who)公布的关于致残听力损失程度的估计,全世界有466百万人患有致残听力损失(432百万成人和34百万儿童)。患有致残听力损失的人的数目截至2030年将增长至630百万并且截至2050年将超过900百万(10人中有1人)。全世界超过90%的患有致残听力损失的人(420百万)居住在低收入区(who关于听力损失患病率的全球估计,prevention of deafness who 2018)。目前尚无用于预防或治疗听力损失或耳聋的批准治疗剂。患有致残听力损失的那些人目前的治疗选择是耳蜗植入物。耳蜗植入是一种常见的程序,伴随较大的医疗成本,每名患者的寿命成本超过$1,000,000(mohr pe等人(2000).the societal costs of severeto profound hearing loss in the united states;intj technol assess healthcare;16(4):1120-35)。目前耳蜗植入物的需求超出供应。所制造的耳蜗植入物单元的生产率是每年50,000个单元。基于目前的出生率和新生儿致残听力损失的发生率和患病率,每年需要134,000个耳蜗植入物来为每个受折磨的儿童提供1个耳蜗植入物。如果包括需要双侧(2个)耳蜗植入物的患者,那么这个数字会增加。耳蜗植入物的寿命成本对于大多数人来说是令人望而却步的,特别是对于生活在大多数患有致残听力损失的人所居住的发达国家以外的那些人来说。需要提供耳蜗植入物的具有成本效益的替代品的治疗选择,尤其是对于生活在发达国家以外的那些人来说。超过50%的语前耳聋是遗传的,即遗传性的(centers for disease control andprevention-genetics of hearing loss)。遗传性听力损失和耳聋可能是传导性的、感音神经性的或两者的组合;综合征性的(与外耳或其他器官的畸形或涉及其他器官系统的医疗问题有关)或非综合征性的(无外耳相关的可见异常或任何相关医疗问题);和语前的(在语言发展之前)或语后的(在语言发展之后)(richard jh smith,md,a eliot shearer,michael s hildebrand,phd和guy van camp,phd,deafness and hereditary hearingloss overview,genereviews初始投稿:1999年2月14日;最后修订:2014年1月9日。超过70%的遗传性听力损失是非综合征性的。非综合征性耳聋的不同基因座被指定为dfn(针对耳聋)。基因座是基于遗传模式命名的:dfna(常染色体显性)、df b(常染色体隐性)和dfnx(x连锁)。上述名称后面的数字反映了基因定位和/或发现的顺序。在一般人群中,听力损失的患病率随年龄增长而增加。这种变化反映了遗传和环境的影响,以及环境诱因与个人的遗传倾向之间的相互作用。感音神经性听力损失(snhl)是人类最常见的神经退行性疾病,目前尚无经批准的药物干预。snhl可由遗传病症引起,以及通过诸如声音创伤和耳毒性等损伤获得。遗传诊断已经证明,至少有100个基因导致非综合征性snhl。遗传学和基因疗法技术的最新进展已经示出,通过基因疗法有可能挽救许多隐性类型的耳聋(akil等人,2012;askew等人,2015)。已经使用腺相关病毒载体(aav)实现了向内耳的长期基因递送(shu,tao,wang等人,2016)。首个使用基因疗法来解决耳聋和听力损失的人类临床试验(cgf166)在2014年6月启动并且在2019年12月完成。cgf166的主要研究者是hinrich staecker博士并且所述试验由novaris赞助。(https://clinicaltrials.gov/ct2/show/nct02132130)。这一领域的转化研究的理想疾病目标是隐性遗传性听力损失,它影响内耳内的一组确定细胞并在出生后在言语发育后发生。突变的流行是一个额外的考虑因素。如本文所述,通过仔细评估听力损失的常见隐性原因的发生率并且考虑基因的大小,有可能开发出一种基因疗法计划,所述基因疗法计划具有一个容易接近并且相当常见的患者群体。例如,尽管tmprss3较其他突变不太常见,但它是听力损失的一个相当常见的原因,其严重程度足以保证耳蜗植入。另外,具有tmprss3突变的患者可能与具有其他突变的患者一样对耳蜗植入没有反应(shearer等人,2017)。这提供了机会来靶向tmprss3或诸如loxhd1等其他基因,作为独立治疗剂或与其他治疗剂和/或耳蜗植入组合以改进这一病症的植入效果。表1(由(miyagawa,nishio和usami,2016)改编)证实tmprss3突变可能是语后隐性听力损失的最常见原因,其在耳蜗内的分布相当有限,并且由于基因的大小,可能被构建到现有的aav载体中。表1:176名成人耳蜗植入患者中不同突变的发生率。人跨膜蛋白酶丝氨酸3(tmprss3;还称作dfnb10、dfnb8、echos1、tadg12;acc:hgnc:11877)通过其与先天性(出生时存在)和童年发病常染色体隐性耳聋的相关性进行鉴定。tmprss3基因突变与dfnb8和10型常染色体隐性非综合征性听力损伤相关。tmprss3是编码丝氨酸蛋白酶的1646碱基对基因并且与dfna 8/10相关,并且可能占经历耳蜗植入的听力损失患者的1-5%(weegerink等人,2011)。这一基因的功能缺失似乎会导致广泛的听力表型,视突变位点而定。先天性和成年发病的进行性听力损失都与这一基因的缺失有关。dfnb8听力损失的发病是语后的(10-12岁),而dfnb10听力损失的发病是语前的(先天性)。这种表型差异反映了基因型差异。引起dfnb8的变体是一种剪接位点变体,表明低效剪接与减少的正常蛋白质的量有关,所述减少的正常蛋白质的量足以预防语前耳聋,但不足以预防最终的听力损失。(参见richard jh smith,md等人(2014).genes known tocause autosomal recessive nonsyndromic hearing impairment:deafness andhereditary hearing loss overview;genereviews)。表2描述了染色体21上已知引起听力损失的tmprss3突变。脂氧合酶同源结构域1基因(loxhd1;还称作lh2d1、dfnb77、flj32670;omim:613072;acc:hgnc:26521)编码完全由plat(多囊蛋白/脂氧合酶/α-毒素)结构域组成的高度保守的蛋白质,被认为参与将蛋白质靶向质膜。对小鼠的研究示出,这一基因在内耳中的机械感觉毛细胞中表达,并且这一基因的突变导致听觉缺陷,表明这一基因对正常毛细胞功能至关重要。对分离性耳聋的人类家族进行筛查,鉴定出这一基因的突变,所述突变引起dfnb77,这是一种常染色体隐性非综合征性听力损失(arnshl)的进行性形式。针对这一基因已经注意到编码不同同种型的替代剪接转录变体。loxhd1的临床特征:·常染色体隐性;·听力损失,感音神经性,双侧(在低频率时听力损失较轻);·在德系犹太人家族中,先天性发病导致7-10岁耳蜗植入;·在一个有血缘关系的伊朗家族中,7-8岁发病,在成年期逐渐发展为中度至重度中高频缺失。有证据表明,常染色体隐性非综合征性听力损失-77(dfnb77)是由染色体18q21上loxhd1基因(613072)的纯合突变引起的。在胚胎13.5天和16天,原位杂交检测到发育中的小鼠内耳中的loxhd1表达,但在任何其他组织中均未检测到。在出生后第4天,在耳蜗和前庭毛细胞中检测到表达,其中细胞核中的浓度最高。loxhd1逐渐定位于细胞质,并且在成体中,loxhd1沿静纤毛长度在毛细胞中表达。使用n-乙基-n-亚硝基脲(enu)诱变筛选,grillet等人(2009)发展'samba'小鼠系,所述小鼠系在3周龄时听力受损并且在8周龄时耳聋。纯合的samba小鼠未显示其他神经或前庭异常,并且杂合samba小鼠显得完全正常。纯合samba小鼠的静纤毛发育不受影响,但毛细胞功能紊乱并且毛细胞最终退化。grillet等人(2009)发现samba是小鼠loxhd1基因的突变,它使plat结构域10的β-三明治结构不稳定。所述突变并未改变loxhd1蛋白的mrna或蛋白质稳定性或沿静纤毛长度的定位。然而,截至出生后第21天,一些毛细胞显示形态缺陷,具有融合的静纤毛和在顶端细胞表面的膜皱褶。截至出生后第90天,深度退行性变化很明显,包括毛细胞缺失和螺旋神经节神经元减少。grillet等人(2009)假设螺旋神经节神经元的退化可能是继发于毛细胞功能和维持的紊乱。表3描述了染色体18上已知引起听力损失的loxhd1突变。美国申请公布号2013/0095071(以引用的方式整体并入本文中)描述了使用突变的酪氨酸腺相关病毒载体将x-连锁凋亡抑制蛋白(xiap)递送至内耳的圆窗膜来恢复年龄相关性听力损失的基因疗法方法。然而,所述公布未考虑递送编码功能性tmprss3或loxhd1的核酸序列来预防或延迟由tmprss3或loxhd1基因的基因突变引起的听力损失或耳聋的发病或恢复听力损失或耳聋,如本文所公开。另外,开发用于听力障碍的临床基因疗法的现有技术中的一个重要陷阱是缺乏反映人类听力损失的动物模型。针对成年发病的人类遗传性听力损失的许多可用的小鼠模型都存在先天性听力损失,这使得递送研究变得复杂。很少有模型在听力发育后出现遗传性听力损失的发病。载体在新生小鼠体内的递送导致与在成年小鼠体内的递送不同的转染模式(shu,tao,li等人,2016)。需要新颖动物模型,它们可用于使用不同的载体系统和基因靶来评估听力的挽救。综上所述,耳蜗植入是一种用于解决介于严重到深度范围内的听力损失的常见的治疗方法选择。耳蜗植入物是一种小型、复杂的电子器件,它可以帮助向深度耳聋或严重听力障碍的人提供听觉。所述植入物由位于耳朵后面的外部部分和通过手术放置在皮肤下方的第二部分组成。虽然耳蜗植入物的设计和性能近年来取得了巨大的进步,但仍有患者使用植入物后在语言效果方面表现不佳。最近的研究已证实,引起耳聋的两种基因tmprss3和loxhd1的突变在耳蜗植入结果方面也具有不良效果1。具体说来,tmprss3突变患者具有螺旋神经节功能障碍2。在小鼠tmprss3突变模型的评估期间,已证实毛细胞最初退化,随后不久是螺旋神经节细胞退化3。内耳毛细胞的永久性损伤会导致感音神经性听力损失,从而导致在很大比例的人口中存在沟通困难。毛细胞是转导声刺激的受体细胞。受损毛细胞的再生将为治疗目前除了假体器件外没有其他疗法的疾患提供一条途径。在具有tmprss3突变的人类患者的评估期间,已证实耳蜗植入功能随年龄增长而下降,这表明在人类群体中还发生螺旋神经节细胞的延迟退化4。上述情况表明,单独耳蜗植入物可能不足以对抗听力损失。因此,有机会与耳蜗植入组合来提供用于听力损失的分子治疗剂(例如基因疗法)的组合。
背景技术:
技术实现思路
1、本公开的实施方案尤其涉及可用于治疗和/或预防听力损失的基因疗法系统和方法。本文所述的系统和方法涉及组合基因疗法和耳蜗植入,以根据干预时间修复和/或挽救退化的毛细胞和/或退化的螺旋神经节细胞。
2、本文所公开的每个实施方案均可以包括结合任何其他所公开的实施方案描述的一个或多个特征。
3、本文公开一种包括核酸序列seq id no:1或seq id no:2或与核酸seq id no:1或seq id no:2具有至少90%序列同一性的核酸序列的表达载体,其中所述核酸序列可操作地连接至启动子。本文还公开一种用于治疗或预防听力损失的方法中的药物组合物,所述药物组合物包括具有核酸序列seq id no:1或seq id no:2或与核酸seq id no:1或seq idno:2具有至少90%序列同一性的核酸序列的表达载体,其中所述核酸序列可操作地连接至启动子。在一些实施方案中,所述核酸序列与核酸序列seq id no:1或seq id no:2具有至少75%、至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%或至少99%序列同一性。在一些实施方案中,所述表达载体选自腺相关病毒载体、腺病毒载体、单纯疱疹病毒载体、牛痘病毒载体、辅助病毒依赖型腺病毒载体或慢病毒载体。在一些实施方案中,所述载体选自aav2、aav2/anc80、aav5、aav6、aav6.2、aav7、aav8、aav9、aavrh8、aavrh10、aavrh39、aavrh43aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、anc80或腺相关病毒载体血清型的合成形式的腺相关病毒载体。在一些实施方案中,所述腺相关病毒载体为aav2、anc80或腺相关病毒载体血清型的合成形式。在一些实施方案中,所述启动子选自在早期发育时驱动可操作地连接的核酸的表达并在整个生命中维持表达的任何毛细胞启动子,例如tmprss3启动子、人巨细胞病毒(hcmv)启动子、巨细胞病毒/鸡β-肌动蛋白(cba)启动子、myo7a启动子或pou4f3启动子。
4、本文公开一种细胞,所述细胞具有包括核酸序列seq id no:1或seq id no:2或与核酸seq id no:1或seq id no:2具有至少90%序列同一性的核酸序列的表达载体,其中所述核酸序列可操作地连接至启动子。在一些实施方案中,所述核酸序列与核酸序列seq idno:1或seq id no:2具有至少75%、至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%或至少99%序列同一性。在一些实施方案中,所述细胞是干细胞。在一些实施方案中,所述干细胞是诱导性多能干细胞。
5、本文公开一种用于治疗或预防听力损失的方法,所述方法包括向有需要的受试者施用有效量的包括核酸序列seq id no:1或seq id no:2或与核酸seq id no:1或seq idno:2具有至少90%序列同一性的核酸序列的表达载体,其中所述核酸序列可操作地连接至启动子。在一些实施方案中,所述核酸序列与核酸序列seq id no:1或seq id no:2具有至少75%、至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%或至少99%序列同一性。在一些实施方案中,所述表达载体选自腺相关病毒载体、腺病毒载体、单纯疱疹病毒载体、牛痘病毒载体、辅助病毒依赖型腺病毒载体或慢病毒载体。在一些实施方案中,所述载体选自aav2、aav2/anc80、aav5、aav6、aav6.2、aav7、aav8、aav9、aavrh8、aavrh10、aavrh39、aavrh43、aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8或anc80或腺相关病毒载体血清型的合成形式的腺相关病毒载体。在一些实施方案中,所述腺相关病毒载体为aav2、anc80或腺相关病毒载体血清型的合成形式。在一些实施方案中,所述启动子选自在早期发育时驱动可操作地连接的核酸序列的表达并在整个生命中维持表达的任何毛细胞启动子,例如tmprss3启动子、人巨细胞病毒(hcmv)启动子、巨细胞病毒/鸡β-肌动蛋白(cba)启动子、myo7a启动子或pou4f3启动子。在一些实施方案中,所述表达载体例如通过注射施用至所述受试者的内耳中。在一些实施方案中,所述递送方法选自耳蜗底转、圆窗膜、管造口术或其任何组合(参见erin e.leary swan等人(2008)inner ear drug deliveryfor auditory applications.adv drug deliv rev.60(15):1583-1599)。在一些实施方案中,所述表达载体通过内淋巴囊递送至蜗管中(colletti v等人(2010)evidence ofgadolinium distribution from the endolymphatic sac to the endolymphaticcompartments of the human inner ear.audiol neurootol.15(6):353-63;marcomandalà,md等人(2010)induced endolymphatic flow from the endolymphatic sac tothe cochlea in ménière’s disease.otolaryngology-head and neck surgery.143,673-679;yamasoba t等人(1999)inner ear transgene expression after adenoviralvector inoculation in the endolymphatic sac.hum gene ther.10(5):769-74)。在一些实施方案中,所述受试者具有一种或多种与听力损失相关的遗传风险因素。在一些实施方案中,所述遗传风险因素之一是tmprss3基因的突变。在一些实施方案中,所述tmprss3基因的突变选自已知引起听力损失的任何一种或多种tmprss3突变(参见例如表2)。在一些实施方案中,所述遗传风险因素之一是loxhd1基因的突变。在一些实施方案中,所述loxhd1基因的突变选自已知引起听力损失的任何一种或多种loxhd1突变(参见例如表3)。在一些实施方案中,所述受试者未表现出听力损失的任何临床指标。
6、在一些实施方案中,本文所述的表达载体与一种或多种包含其他核酸序列的表达载体一起和/或与一种或多种用于治疗听力损失的其他活性药剂一起作为组合疗法施用。例如,组合疗法可以包括具有核酸序列seq id no:1的第一表达载体和具有核酸序列seqid no:2的第二表达载体,其中两种表达载体作为组合疗法的一部分施用于受试者以治疗听力损失。
7、本文公开一种具有人tmprss3基因的转基因小鼠,所述人tmprss3基因具有选自已知引起听力损失的任何一种或多种tmprss3突变的突变(参见例如表2)。本文公开一种具有人loxhd1基因的转基因小鼠,所述人loxhd1基因具有选自已知引起听力损失的任何一种或多种loxhd1突变的突变(参见例如表3)。
8、可以理解,前述一般描述和以下详细描述均仅为例示性和解释性的,并且对要求保护的本发明不具限制性。
- 还没有人留言评论。精彩留言会获得点赞!