一种基于多通道神经网络的抗癌药物筛选方法
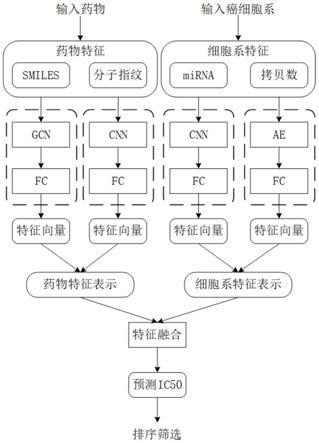
1.本发明属于计算机科学领域,涉及人工智能技术在生物医药问题中的应用,具体涉及一种基于多通道神经网络的抗癌药物筛选方法。
背景技术:
2.由于癌症治疗的特殊性,精准医学已经成为全世界科学家想要攻克的难题。而预测癌细胞系对特定药物的反应,进而筛选出具有研究意义的抗癌药物,对精准医疗具有重大意义。一些经典的机器学习算法得益于它们在数据和模型集成方面的强大能力,对药物反应预测进行了初步的探索。然而,由于抗癌药物反应数据维度高和标签少的特点,这些方法显得力不能支。深度学习是机器学习的一个更先进的分支,因其能够从复杂数据中提取特征而被广泛应用于各个领域的数据挖掘。
3.然而,有效融合多个数据源的信息仍是抗癌药物响应预测中一个具有挑战性的问题。原因是很难找到有效的方法克服数据集成的一系列问题,例如数据结构的差异和数据的复杂性。此外,模型的实用性也是一个值得思考的问题。实际应用场景中的癌细胞系数据通常并不理想,从而可能导致模型无法发挥实际作用。
技术实现要素:
4.本发明所要解决的技术问题是有效融合药物和细胞系的多个特征,从而进行更高效的筛选。针对现有技术不足,提供了一种基于多通道神经网络的抗癌药物筛选方法。本发明方法结合一维卷积神经网络、栈式自动编码器和图卷积网络,利用不同的输入通道提取不同数据结构的输入信息。通过有效融合药物的全局和局部结构信息,以及来自不同组学数据的细胞系特征,提高药物敏感性预测的精度,进而提高抗癌药物筛选的效率。同时,多信息源的融合提高了模型的鲁棒性,使其在训练数据规模减小时,仍能保持稳定的性能。
5.本发明所采用的技术方案是:
6.一种基于多通道神经网络的抗癌药物筛选方法,包括以下步骤:
7.1)将表示药物全局结构信息的smiles序列转化为graph形式,并利用gcn提取其特征;
8.2)利用一维卷积神经网络分别提取表示药物局部结构信息的分子指纹特征,以及癌细胞系的转录组学特征mirna;
9.3)将超高维的基因组学特征基因拷贝数通过栈式自编码器降维,得到低维度的特征表示;
10.4)利用全局最大池化和全连接网络,将各个通道提取出的不同格式和维度的特征规范化至指定维度向量;
11.5)拼接融合规范化后的药物的全局和局部结构特征,以及癌细胞系的基因组学和转录组学特征;
12.6)融合后的特征通过全连接的神经网络,预测药物-细胞系反应的ic50值,筛选出
具有研究价值的抗癌药物。
13.作为本发明的进一步改进,
14.所述步骤1)中,将给定药物的smiles序列通过rdkit转化为分子图(graph),并以特征矩阵x和邻接矩阵a的形式存储,x是一个n
×
f大小的矩阵,其中n是化合物的原子节点个数,每个节点由一个f维向量表示,a是一个n
×
n大小的矩阵,表示节点之间的边;使用三个图卷积层对药物分子图进行特征提取,其中σ表示非线性激活函数,h表示层,上标l表示层数,当l=0时,h
(0)
即为特征矩阵x;w是可训练参数矩阵,l=0、1、2时的w
(0)
、w
(1)
、w
(2)
的维度分别为f
×
f、f
×
2f、f
×
4f,对应的h
(1)
、h
(2)
、h
(3)
的维度分别为n
×
f、n
×
2f、n
×
4f;是添加了自环的图邻接矩阵,是图的对角度矩阵。
15.作为本发明的进一步改进,
16.所述步骤2)中,将药物的分子指纹信息和细胞系的mirna信息通过三个一维卷积层层其中z
l+1
(i)表示第l+1个卷积层输出的特征向量的第i个元素,z
l
和w
l+1
分别表示第l+1个卷积层的输入特征和卷积核,b表示偏移量,k
l
为输入通道数,m表示卷积核大小,是第k个通道的输入特征,s0i+x表示特征向量索引,s0是卷积步长,是第k个通道的卷积核,x表示卷积核元素索引,l
l
和l
l+1
分别为第l+1个卷积层的输入和输出特征向量长度,p是填充层数量;在每个卷积层提取特征后,将输出的特征数据z
l+1
传递给池化层进行特征选择和信息过滤,其中s1i+x表示特征向量的索引,s1表示池化步长,q是一个预定义参数,被设置为q
→
∞,即最大池化。
17.作为本发明的进一步改进,
18.所述步骤3)中,超高维度的基因拷贝数信息通过栈式自编码器降维,即使用比输入更少的隐藏节点来预测输入:h(t)≈t,其中t为输入,h为学习函数;并在各隐层之间添加非线性激活函数relu,实现非线性降维;训练时的目标函数采用mse:其中y为真实值,为预测值,num表示训练样本数量。
19.作为本发明的进一步改进,
20.所述步骤4)中,将gcn提取的特征矩阵h
(3)
,通过全局最大池化,转换为一个维度为4f的向量,并通过全连接层,将维度规范至128,得到特征向量v1,表示提取后的药物分子图信息;将1d cnn提取的药物分子指纹和细胞系mirna,分别通过全连接层将维度规范至128,得到特征向量v2和v3,分别表示提取后的分子指纹和mirna信息;将ae降维的基因拷贝数信息通过全连接层,将维度规范至128,得到特征向量v4,表示提取后的拷贝数信息。
21.作为本发明的进一步改进,
22.所述步骤5)中,将包含药物分子图信息的特征向量v1和分子指纹信息的特征向量v2进行拼接,得到药物特征的最终表示v
drug
;将包含细胞系mirna信息的特征向量v3和基因
拷贝数信息的特征向量v4进行拼接,得到细胞系特征的最终表示v
cell-line
;融合v
drug
和v
cell-line
,得到药物-细胞系对的最终特征v
fusion
。
23.作为本发明的进一步改进,
24.所述步骤6)中,融合后的药物-细胞系对特征v
fusion
,通过全连接的神经网络,输出节点数定为1,再通过sigmoid函数,将输出映射到0~1之间;对已有ic50标签的数据进行训练,并预测尚未进行实验验证的药物-细胞系对的ic50值,根据预测的ic50值大小,筛选出具有研究价值的抗癌药物。
25.与现有技术相比,本发明所具有的有益效果为:
26.本发明提供了一种基于多通道神经网络的抗癌药物筛选方法,针对不同输入特征数据结构不同和数据复杂度高的问题,通过图卷积网络提取图结构的药物分子图特征,一维卷积提取常规向量格式的药物分子指纹和细胞系mirna特征,自编码器提取超高维的基因拷贝数特征,从而有效融合多源信息。本发明方法可以显著提高药物敏感性的预测效果,精确筛选具有研究价值的抗癌药物。与现有的方法相比,本方法不仅可以在训练数据充足时,更精确地进行抗癌药物筛选,还可以在训练数据不足时,保持稳定的预测性能,具备更强大的实际应用能力。
附图说明
27.图1是本发明实施例的总体流程图;
28.图2是预测-真实值散点图。
具体实施方式
29.下面结合附图和具体实施例对本发明进一步详细说明。
30.参照附图1,本发明提出的一种基于多通道神经网络的抗癌药物筛选方法,具体通过以下步骤实现:
31.步骤1,将表示药物全局结构信息的smiles序列转化为graph形式,并利用gcn提取其特征。
32.本实施例中,将给定药物的smiles序列通过rdkit转化为分子图(graph),并以特征矩阵x和邻接矩阵a的形式存储,x是一个n
×
f大小的矩阵,其中n是化合物的原子节点个数,每个节点由一个f维向量表示,a是一个n
×
n大小的矩阵,表示节点之间的边;使用三个图卷积层对药物分子图进行特征提取,其中σ表示非线性激活函数,h表示层,上标l表示层数,当l=0时,h
(0)
即为特征矩阵x;w是可训练参数矩阵,l=0、1、2时的w
(0)
、w
(1)
、w
(2)
的维度分别为f
×
f、f
×
2f、f
×
4f,对应的h
(1)
、h
(2)
、h
(3)
的维度分别为n
×
f、n
×
2f、n
×
4f;是添加了自环的图邻接矩阵,是图的对角度矩阵。
33.本实施例中,特征向量维度f为78,药物分子图的节点个数n因药物的不同而变化;w
(0)
、w
(1)
、w
(2)
的维度分别设置为78
×
78、78
×
156、78
×
312,故h
(1)
、h
(2)
、h
(3)
的维度分别为n
×
78,n
×
156,n
×
312;非线性激活函数σ使用的是relu函数。
34.步骤2,利用一维卷积神经网络分别提取表示药物局部结构信息的分子指纹特征,以及癌细胞系的转录组学特征mirna。
35.本实施例中,将药物的分子指纹信息和细胞系的mirna信息通过三个一维卷积层本实施例中,将药物的分子指纹信息和细胞系的mirna信息通过三个一维卷积层其中z
l+1
(i)表示第l+1个卷积层输出的特征向量的第i个元素,z
l
和w
l+1
分别表示第l+1个卷积层的输入特征和卷积核,b表示偏移量,k
l
为输入通道数,m表示卷积核大小,是第k个通道的输入特征,s0i+x表示特征向量索引,s0是卷积步长,是第k个通道的卷积核,x表示卷积核元素索引,l
l
和l
l+1
分别为第l+1个卷积层的输入和输出特征向量长度,p是填充层数量;在每个卷积层提取特征后,将输出的特征数据z
l+1
传递给池化层进行特征选择和信息过滤,其中s1i+x表示特征向量的索引,s1表示池化步长,q是一个预定义参数,被设置为q
→
∞,即最大池化。
36.本实施例中,l=0、1、2时三个卷积层的通道数k0、k1、k2分别设置为4、8和16,卷积核长度m设置为8,卷积步长s0=1,填充层数量p=0,w和b为可学习参数。池化层中,预定义参数q
→
∞,即使用最大池化,池化步长s1设置为3。
37.步骤3,将超高维的基因组学特征基因拷贝数通过栈式自编码器降维,得到低维度的特征表示。
38.本实施例中,超高维度的基因拷贝数信息通过栈式自编码器降维,即使用比输入更少的隐藏节点来预测输入:h(t)≈t,其中t为输入,h为学习函数;并在各隐层之间添加非线性激活函数relu,实现非线性降维;训练时的目标函数采用mse:其中y为真实值,为预测值,num表示训练样本数量。
39.本实施例中,栈式自编码器包含输入输出层和6个隐层,其中3个属于编码器,另外3个属于解码器。输入输出层的节点个数均为23316,即基因拷贝数的特征向量长度。隐层的节点个数分别为1024、512、256、256、512、1024,其中,编码器的输出,一个256维的特征向量,被用作非线性降维后的特征向量。
40.步骤4,利用全局最大池化和全连接网络,将各个通道提取出的不同格式和维度的特征规范化至指定维度向量。
41.本实施例中,将gcn提取的特征矩阵h
(3)
,通过全局最大池化,转换为一个维度为4f的向量,并通过全连接层,将维度规范至128,得到特征向量v1,表示提取后的药物分子图信息;将1d cnn提取的药物分子指纹和细胞系mirna,分别通过全连接层将维度规范至128,得到特征向量v2和v3,分别表示提取后的分子指纹和mirna信息;将ae降维的基因拷贝数信息通过全连接层,将维度规范至128,得到特征向量v4,表示提取后的拷贝数信息。
42.本实施例中,gcn提取的特征矩阵h
(3)
维度为n
×
312,通过全局最大池化转化为一个312维的特征向量,并通过全连接层转化为128维特征向量v1。药物分子指纹提取通道的输出为464维的特征向量,mirna提取通道的输出为368维特征向量,同样经过全连接层转化为128维特征向量v2和v3。ae编码的基因拷贝数特征是一个256维向量,经全连接层转化为
128维特征向量v4。
43.步骤5,拼接融合规范化后的药物的全局和局部结构特征,以及癌细胞系的基因组学和转录组学特征。
44.本实施例中,将包含药物分子图信息的特征向量v1和分子指纹信息的特征向量v2进行拼接,得到药物特征的最终表示v
drug
;将包含细胞系mirna信息的特征向量v3和基因拷贝数信息的特征向量v4进行拼接,得到细胞系特征的最终表示v
cell-line
;融合v
drug
和v
cell-line
,得到药物-细胞系对的最终特征v
fusion
。
45.步骤6,融合后的特征通过全连接的神经网络,预测药物-细胞系反应的ic50值,筛选出具有研究价值的抗癌药物。
46.本实施例中,融合后的药物-细胞系对特征v
fusion
,通过全连接的神经网络,输出节点数定为1,再通过sigmoid函数,将输出映射到0~1之间;对已有ic50标签的数据进行训练,并预测尚未进行实验验证的药物-细胞系对的ic50值,根据预测的ic50值大小,筛选出具有研究价值的抗癌药物。
47.本实施例中,训练损失函数使用均方误差其中为预测值,y为真实值,num表示训练样本数量。训练次数设置为300轮,学习率lr=1
×
10-4
。由于ic50值越小,敏感性越高,故对药物细胞系对按照ic50值升序排列,并选择前1%供进一步研究使用。
48.为了验证本发明的有效性,图2给出了预测结果散点图。由图2结果可知,本发明对抗癌药物敏感性做出了高精度的预测,预测值与真实值呈现高相关性分布,其皮尔森相关系数达到了0.866,均方误差仅为0.087。该结果说明,本发明通过不同的特征提取通道有效融合了不同结构和维度的特征。准确的预测结果使得更具研究价值的药物被筛选出来,从而提高大规模药物筛选的效率,为临床研究节约时间成本和费用成本。
49.上述说明示出并描述了发明应用的实施例,但如前所述,应当理解本发明并非局限于本文所披露的形式,不应看作是其他实施例的排除,而可用于各种其他组合、修改和环境,并能在本文所述发明构想范围内,通过上述教导或相关领域的技术进行改动。而本领域人员所进行的改动和变化不脱离发明的精神和范围,则都应在发明所附权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1