一种基于k-mer的序列联配方法与流程
1.本发明属于生物信息领域,具体地,本发明涉及一种基于k-mer的序列比对方法。
背景技术:
2.随着新一代测序技术454 (roche 公司)、solexa (illumina 公司)和 solid (abi公司)的诞生,测序通量得到迅速提升,而测序成本急剧下降,这种突破极大地推动了基因组科学的发展。通过一代测序技术进行菌种鉴定是比传统生化鉴定更加快速、准确的鉴定方法。一代测序菌种鉴定的一般步骤就是通过检测荧光信号得到整条序列信息,然后将序列与数据库比对从而获得菌种鉴定信息。通过转录物组学和蛋白质组学等相关技术对基因表达谱、基因突变等进行匹配分析,可获得与疾病相关基因的信息。通过序列信息与基因组序列或特定基因序列(参考序列)进行联配、分析,并揭秘患病的根源。如何准确快速的从浩瀚的测序结果数据中得到基因信息的关键是序列比对。
3.序列比对是指通过一定算法对两条dna或蛋白质序列进行比较,找出两者之间的最大相似性匹配。它已经成为序列比对问题和数据库搜索的基础。在现有技术中,最具有代表性的比对算法有点阵图法和动态规划算法,但是这些算法在面对大量数据时存在着处理速度慢、占用内存大等缺点。因此,对于核苷酸序列的联配方法仍需要进一步地开放和改进。
技术实现要素:
4.为了克服现有技术的不足,本发明的目的是提供一种基于k-mer的序列联配方法,该方法应用于核苷酸序列联配中,能明显提高序列联配效率并减少程序运算量,快速获得两条序列的联配信息。
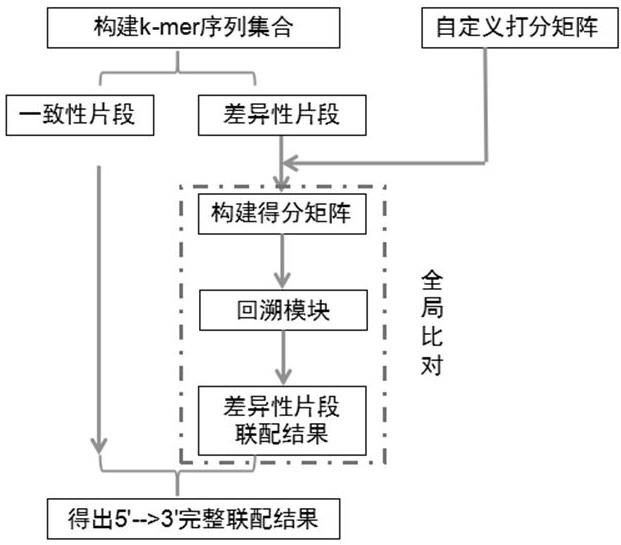
5.一种基于k-mer的序列联配方法,包括以下步骤:步骤一,分别对输入的seq1序列和seq2序列根据第一预定长度进行k-mer分析,获得k-mer序列集合,所述k-mer第一预定长度大于联配所允许的错配数;步骤二,比较步骤一所述的k-mer序列集合,获得两条序列公共的k-mer,以此来寻找一致性片段;如果同一k-mer在同一条序列中有多个,则取第一个位置作为一致性片段;如果两条序列有多个连续k-mer共有,则将多个连续k-mer合并以作为一致性片段;步骤三,利用步骤二所述的一致性片段将序列划分成若干段差异性片段,进行差异性片段序列全局联配获得最优联配结果;如果差异性片段序列长度小于第一预定长度,则向前或向后截取一个第一预定长度的碱基并入一起联配分析;步骤四,根据步骤三所述的最优联配结果,从5’端到3’端输出最终完整序列联配结果。
6.所述第一预定长度为奇数。
7.所述差异性片段序列全局联配包括全局比对模块和回溯模块。
8.所述全局比对模块实行步骤如下:
1)初始化阶段:获取待联配的subseq1序列和subseq2序列各位置上的单元信息;构建(m+1)
×
(n+1)的得分矩阵m,其中,m为subseq1的单元数目,n为subseq2的单元数目,subseq1序列沿顶部展开,subseq2序列沿左侧展开,得分矩阵初始化值全填充为 0;2)计算单元得分值:用于计算得分矩阵中的单元值通过以下三个途径到达每个单元:a.来自上面的单元,代表将左侧的字符与空格比对;b.来自左侧的单元,代表将上面的字符与空格比对;c.来自左上侧的单元,代表与左侧和上面的字符比对,可能匹配也可能不匹配;即当矩阵m(i-1,j-1)、m(i,j-1)和m(i-1,j)值计算结束后,m(i,j)值才能计算;该单元的值来自于以下4个中的最大值:a. 上面单元的值-空格罚分预定值;b. 左侧单元的值-空格罚分预定值;c. 左上侧单元值+相应单元打分;d. 0;其中,所述单元值计算公式如下:其中,m
(i-1,j-1)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i ,j-1)
表示所述subseq1序列中第i个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i-1 ,j)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j个单元的比对得分;g表示空格罚分预定值;e(qi,sj)是基于所述subseq1序列中第i个单元qi与所述subseq2序列中第j个单元sj确定的数值,该数值根据自定义打分矩阵获得;其中,当qi与sj相同时,e(qi,sj)为第一预定打分值,当qi与sj不相同时,e(ri,sj)为第二预定打分值,所述第二预定打分值小于所述第一预定打分值。
9.所述空格罚分预定值为-50。
10.所述第一预定打分值和第二预定打分值依据自定义打分矩阵。
11.所述单元为碱基。
12.所述回溯模块是根据下列步骤确定的:(a) 确定回溯起始位置模块,所述确定回溯起始位置模块用于确定矩阵m
((m+1)
×
(n+1))
中最右侧或最下方得分最大值所对应的回溯起始位置;如果所述回溯起始位置在最右侧而不是最右下方,subseq1序列前端引入gap ("-"),gap 个数由subseq2序列起始位置决定;如果所述回溯起始位置在最下方而不是最右下方,subseq2序列前端引入gap ("-"),gap 个数由subseq1序列起始位置决定;(b) 确定下一回溯位置模块,所述确定下一回溯位置模块用于确定基于所述回溯位置上游相邻三个位置的数值,确定下一回溯位置,其中,所述上游相邻三个位置包括行相邻位置、对角线相邻位置和列相邻位置,其中,选择数值最大的位置作为所述下一回溯位置,并且优先选择所述对角线相邻位置;如果所述最大值出现在该单元上方,则subseq1序列引入一个gap ("-"),subseq2序列取该处碱基;如果所述最大值出现在左侧,则subseq2
序列引入一个gap ("-"),subseq1序列取该处碱基; 如果所述最大值出现在左上方,则不引入gap,subseq1和subseq2均取相应的碱基;(c) 重复步骤(b),直到步骤(b)中所确定的所述下一回溯位置的行号和列号的至少之一为0;(d) 比对结果输出模块,所述比对结果输出模块用于基于步骤(a)-(c)中所确定的回溯路线,确定所述subseq1序列与所述subseq2序列的比对结果。
13.所述回溯模块与所述得分矩阵单元相关联,用于基于所述得分矩阵m
((m+1)
×
(n+1))
的数值,进行回溯处理,以便获得所述的subseq1序列和subseq2序列的比对结果,其中,所述单元m
(i ,j)
计算公式如下:其中,m
(i-1,j-1)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i ,j-1)
表示所述subseq1序列中第i个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i-1 ,j)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j个单元的比对得分;g表示空格罚分预定值;e(qi,sj)是基于所述subseq1序列中第i个单元qi与所述subseq2序列中第j个单元sj确定的数值,该数值根据自定义打分矩阵获得。其中,当qi与sj相同时,e(qi,sj)为第一预定打分值,当qi与sj不相同时,e(ri,sj)为第二预定打分值,所述第二预定打分值小于所述第一预定打分值。
14.所述联配结果采用三行结构输出,第一行为seq1序列,包含引入的gap;第二行为联配结果,其中“*”表示碱基匹配,“n”表示碱基不匹配,
“‑”
表示引入gap;第一行为seq2序列,包含引入的gap。
15.本发明的有益效果在于:1)通过采取k-mer算法模式,大大的减少了比对运算量,节约了计算资源和时间成本。
16.2)通过全局比对,极大地满足了两条分段序列的最佳比对结果。
17.3)提升了序列联配的效率和准确性。
附图说明
18.图1为本发明实施例中的序列联配示意图。
19.图2 为本发明实施例中的序列分段示意图。
20.图3为本发明实施例中的打分矩阵和回溯模块示意图。
21.图4为本发明实施例中的比对结果图。
具体实施方式
22.为了更好的说明本发明,下面结合实施例做进一步说明,所述实施例的示例在附图中展示出。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
23.本发明提供的序列联配方法基于全局比对的核心思想,在此基础上进一步引入k-mer思想,依照自定义打分和特定的回溯规则,实现序列的联配。如图1所示,具体的核心步骤如下:第一步,分别对输入的seq1序列和seq2序列根据第一预定长度进行k-mer分析,获得k-mer序列集合,所述k-mer预定长度大于联配所允许的错配数;第二步,比较步骤一所述的k-mer序列集合,获得两条序列公共的k-mer,以此来寻找一致性片段。如果同一k-mer在同一条序列中有多个,则取第一个位置作为一致性片段;如果两条序列有多个连续k-mer共有,则会将多个连续k-mer合并以作为一致性片段;第三步,利用步骤二所述的一致性片段将序列划分成若干段差异性片段,进行分段全局联配获得最优联配结果。如果差异性片段序列长度小于第一预定长度,则会向前或向后截取一个k-mer长度的碱基并入一起联配分析;第四步,根据步骤三所述的最优联配结果,从5’端到3’端输出最终完整序列联配结果。根据本发明实施例的上述联配方法通过k-mer分割模式,减少了比对的运算量,极大提高了比对速度。
24.根据本发明的实施例,上述方法进一步包括如下技术特征:根据本发明的实施例,所述全局比对是通过如下方式进行的:获取待联配的subseq1序列和subseq2序列各位置上的单元信息;基于所述单元信息,构建(m+1)
×
(n+1)的得分矩阵m,其中,m为subseq1的单元数目,n为subseq2的单元数目。subseq1序列沿顶部展开,subseq2序列沿左侧展开,得分矩阵第一行和第一列数值全填充为 0;其中所述得分矩阵中的单元m
(i,j)
表示所述subseq1序列中第i个单元与所述subseq2序列中第j个单元的比对得分;基于所述得分矩阵的数值,进行回溯处理,以便获得所述的subseq1序列和subseq2序列的比对结果,其中单元m
(i,j)
是基于以下计算公式确定:其中,m
(i-1,j-1)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i ,j-1)
表示所述subseq1序列中第i个单元与所述subseq2序列中第j-1个单元的比对得分;m
(i-1 ,j)
表示所述subseq1序列中第i-1个单元与所述subseq2序列中第j个单元的比对得分;g表示空格罚分预定值;e(qi,sj)是基于所述subseq1序列中第i个单元qi与所述subseq2序列中第j个单元sj确定的数值,该数值根据自定义打分矩阵获得。其中,当qi与sj相同时,e(qi,sj)为第一预定打分值,当qi与sj不相同时,e(ri,sj)为第二预定打分值,所述第二预定打分值小于所述第一预定打分值。
25.需要说明的是,本技术所述的“比对所允许的错配数”是指在具体比对时,所允许的容错碱基数。
26.根据本发明的实施例,所述第一预定长度为奇数。通过模拟数据测试,发明人发现,所述第一预定长度数值并不是所有序列联配设置为统一值是最佳的,可以根据序列长
度和实际情况进行选择;所述第一预定长度过长,可能会因为碱基错配导致获取不到共有k-mer,从而增加比对运算时长,比对速度较慢,也可能会影响比对结果准确性。
27.根据本发明的实施例,所述空格罚分预定值为-50。
28.根据本发明的实施例,所述第一预定打分值和第二预定打分值依据自定义 打分矩阵,如图3所示。
29.根据本发明的实施例,所述单元为碱基。
30.根据本发明的实施例,所述回溯模块(如图3所示)是根据下列步骤确定的:确定回溯起始位置模块,所述确定回溯起始位置模块用于确定矩阵m
((m+1)
×
(n+1))
中最右侧或最下方得分最大值所对应的回溯起始位置;如果所述回溯起始位置在最右侧而不是最右下方,subseq1序列前端引入gap ("-"),gap 个数由subseq2序列起始位置决定。如果所述回溯起始位置在最下方而不是最右下方,subseq2序列前端引入gap ("-"),gap 个数由subseq1序列起始位置决定;确定下一回溯位置模块,所述确定下一回溯位置模块用于确定基于所述回溯位置上游相邻三个位置的数值,确定下一回溯位置,其中,所述上游相邻三个位置包括行相邻位置、对角线相邻位置和列相邻位置,其中,选择数值最大的位置作为所述下一回溯位置,并且优先选择所述对角线相邻位置;如果所述最大值出现在该单元上方,则subseq1序列引入一个gap ("-"),subseq2序列取该处碱基;如果所述最大值出现在左侧,则subseq2序列引入一个gap ("-"),subseq1序列取该处碱基; 如果所述最大值出现在左上方,则不引入gap,subseq1和subseq2均取相应的碱基。重复确定下一回溯位置,直到所确定的所述下一回溯位置的行号和列号的至少之一为0;比对结果输出模块,所述比对结果输出模块用于基于所确定的回溯路线,确定所述subseq1序列与所述subseq2序列的比对结果。
31.根据本发明的实施例,所述方法可描述为:1) 输入序列seq1和seq2进行k-mer切分,构建k-mer序列集合;2) 根据k-mer序列集合获取共有的k-mer序列并记录位置,如果同一k-mer在同一条序列中有多个,则取第一个位置作为一致性片段;如果两条序列有多个连续k-mer共有,则会将多个连续k-mer合并以作为一致性片段,如图2所示;3) 根据一致性片段将序列划分成若干段差异性片段,并对分段序列进行全局比对以便获得最优联配结果。如果差异性片段序列长度小于第一预定长度,则会向前或向后截取一个k-mer长度的碱基并入一起联配分析,如图2所示;4) 根据分段最优联配结果,从5’端到3’端输出最终完整序列联配结果,如图4所示。
32.以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本领域技术人员而言,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1