一种基因测序混合信号的评估方法与流程
1.本发明涉及一种基因测序混合信号的评估方法,属于分子测序的数据处理领域。
背景技术:
2.基因测序是解密基因序列的过程。基因测序的应用范围十分广泛,常见的疾病预测与诊断、病毒识别等都可以应用到基因测序。在高通量dna测序中,原则上,所得到的图像数据中,划定一定区域内的信号为同一dna分子经过扩增反应放大信号得到的结果(即单克隆信号),这也是能通过反应产生的光信号解码dna序列的基础之一。实际操作中,由于工艺限制,如进样反应附着物微球及进样dna模板过程中,不可避免会造成多个dna模板进入同一个观测区域,造成该区域观测的时序信号为多种反应模板叠加产生的混合信号(或简称为多克隆信号),根据该信号进行后续矫正得到dna序列,并不反映被测dna分子的真实组成。所以,需要一种方法来识别这样的信号,进一步能分离出原始信号。
3.ion torrent找到了两种可以反映不同类型信号(混合的或非混合的)特征的指标,ppf(percent positive flows)和ssq(sum of squares)。通过对所找到的两种指标的值进行筛选来鉴别不同种类信号。ppf指的是产生信号为正数的进样流体(flow)次数的百分比,ppf越高,是混合信号的可能性越大。ssq指的是一次flow中信号值与最邻近的正数的平方和,ssq越大,是混合信号的可能性越大。ion torrent在分析程序中允许用户自定义用来估计合理的多克隆筛选的ppf及ssq的阈值的flow的轮数区间。但是ion torrent的方法并不适用于简并测序。主要原因是,在进行简并测序时ppf指标对不同类型的信号并没有很好的区分度。因此,需要开发一种可以有效区分简并测序得到的信号类型的方法。
技术实现要素:
4.ion torrent所采用的使用信号的特征结合分类方法预测信号是否为多种模板产生的混合信号是一种间接的手段。不管是非简并测序或简并测序,多克隆信号都是由两个以上单克隆信号线性组合而成的,其系数由各个dna分子克隆(即信源)占总体的比率确定;故而,高通量测序中多克隆信号的通常特征可以被多个信源信号的线性组合表示,而无法被单个信源信号表示,识别多克隆信号的关键也就在于,判断单信源信号的表示是否存在。
5.本发明公开了一种基因测序混合信号的评估方法,具体采用稀疏编码技术实现这种思想,可以判断某个位点的测序信号是单个片段的信号(单克隆)还是多个片段信号的混合(多克隆)。
6.具体的,本发明公开一种基因测序混合信号的评估方法,其特征在于,包括以下步骤:
7.a.获取数据:测序获得对应于待测序列碱基的测序信号强度结果,将所述强度结果表示为简并多聚物序列或均聚物序列,作为待评估序列;所述简并多聚物序列是由简并多聚物的数目排列组成的序列,所述均聚物序列是由构成均聚物的单体的数目排列组成的序列;
8.b.构建字典:确定参考序列,将待测序列对应的参考序列表示为对应于测序方法的理想简并多聚物序列或理想均聚物序列,从所述理想简并多聚物序列或理想均聚物序列逐位提取长度为k的子信号进行字典构建;
9.c.字典稀疏化:从所述字典中提取子序列,选取与待评估序列距离相近的子序列作为最终字典;
10.d.稀疏表示:运用最优化算法,寻找一个可被所述最终字典矩阵左乘的向量,使得向量的稀疏度与待评估序列的前k位的距离同时被极小化,找到的向量称为稀疏向量;
11.e.过滤结果:根据设定的稀疏度阈值和混合度阈值分析所述的稀疏向量,当稀疏度不高于稀疏度阈值且混合度高于混合度阈值时,所述稀疏向量对应的待评估序列判定为单克隆信号;当稀疏度不高于稀疏度阈值且混合度不高于混合度阈值时,所述稀疏向量对应的待评估序列判定为多克隆信号或混合信号;当稀疏度高于稀疏度阈值时,稀疏表示失败,不能判定信号为单克隆或多克隆。
12.根据优选的实施方式,所述测序包括多碱基测序。
13.根据优选的实施方式,步骤a中可以选取所述简并多聚物序列或均聚物序列的前k位作为待评估序列。
14.根据优选的实施方式,k的取值优选为8-20,更优选为10-15。
15.根据优选的实施方式,所述长度为k的子信号构成所述字典的每一列。
16.根据优选的实施方式,步骤c确定最终字典包括,从每条待评估序列中提取连续的n位(n《k)作为待评估子序列,将待评估子序列与前述字典中每个子信号的前n位遍历比较,将字典中与实际信号前n位取整相同的项作为备选集合;在备选集合中,逐一计算待评估序列的前k位与字典项的距离,将距离由小到大排列的前m项集合元素取出来,作为最终字典。
17.根据优选的实施方式,所述n的值可以是3或4或5;所述m的取值范围为20-300,优选为50-150。
18.根据优选的实施方式,所述的距离包括但不限于皮尔逊相关系数、斯皮尔曼相关系数、平均互信息、欧几里得距离、汉明距离、车比雪夫距离、马哈兰诺比斯距离、曼哈顿距离、明科斯基距离、对应信号差值的绝对值的最大值或最小值;所述的最优化算法包括但不限于匹配追踪、正交匹配追踪、弱匹配追踪、阈值方法、基追踪、irls算法、lasso算法、加权支持向量机算法。
19.根据优选的实施方式,所述的稀疏度阈值取值范围为2-10,优选为2-5,更优选为2-3;所述的混合度阈值取值范围优选为0.6-1,优选为0.8-1,更优选为0.9-1。
20.本发明还公开一种基因测序混合信号的评估方法,其特征在于,包括以下步骤:
21.1)测序获得对应于待测序列碱基的结果;
22.2)将待测序列对应的参考序列编译为对应于测序方法的理论结果;
23.3)将参考序列的编译的理论结果划分为6-50个碱基的可能结果集群;
24.4)将测序获得的对应于待测序列碱基的结果与步骤3)的可能结果集群进行比对。
25.根据优选的实施方式,利用前面所述任一项中的方法判定多克隆信号或者单克隆信号。
26.根据优选的实施方式,将鉴定出的多克隆信号或混合信号舍弃,将不能判定信号为单克隆或多克隆的信号舍弃,将鉴定出的单克隆信号分离出来用于后续的数据处理和分
析。
27.本发明的有益效果
28.与现有技术相比,本发明的方法具有如下优势:
29.1.本发明的方法能够解决简并测序中多克隆信号的识别问题,现有技术的方法在简并测序上失去效力。
30.2.与现有技术中根据特征进行信号分类的方法相比,本发明的准确度更高。
31.3.本方法适用范围更广,除了适用于简并测序的测序信号的评估,还可以用于非简并测序信号的评估。
32.4.本方法反应信号的本质组成,不依赖寻找、挖掘和重构信号的其他特征这些步骤,而通常这一步通常需要较多专门的领域知识和经验,且耗费相当精力。
附图说明
33.图1.利用本发明的方法得到最终字典的流程示意图。
34.图2.利用稀疏表示鉴定测序信号的流程示意图。
具体实施方式
35.以下讨论旨在使本领域技术人员能够理解和使用所公开的方法,并且这些讨论是针对特定应用及其要求的背景而提供的。对于本领域技术人员来说,对所公开的实施方式的各种修改将是显而易见的,并且在不脱离所公开的技术的精神和范围的情况下,本文中定义的一般原理可以应用于其他实施方式和应用。因此,所公开的方法不限于所示的实施方式,而是包括与本文所公开的原理和特征一致的最大范围。
36.除非另有定义,否则本文使用的所有科技术语具有与本领域普通技术人员通常理解的含义相同的含义。
37.基因测序的时候,一般的,每个微坑(或者称为每个位点)上,有一个dna片段,经过扩增以后,形成该片段的簇。那么在二代测序的时候,经过化学反应,该位点会发生相同的反应,延伸相同的碱基数量。实际在进行测序的时候,一个位点上,可能存在不止一个dna片段的情况,这种情况我们可以称之为多克隆。仅仅从测序信号多少的角度很难判断一个信号是多克隆信号还是单克隆信号。多克隆信号的存在,一般的影响了测序信号的获得,并且使得测序信号难以解读,因此,被判定为多克隆的信号,一般的可以作为被抛弃的数据。本发明提供了一种用于判断多克隆信号的方法,本方法旨在通过将dna测序时校正相位后的信号在特定空间进行表示,以区分出由于实验工艺所限产生的混合信号,以提高测序的正确率。本发明适用于通过检测信号倍率来确定dna序列的测序方法,信号的类型包括但不限于荧光信号,电信号,化学信号。在满足上述条件的情况下,本发明不局限于简并测序,亦可用于非简并测序。
38.具体的,本发明的第一方面公开一种基因测序混合信号的评估方法,其特征在于,包括以下步骤:
39.a.获取数据:测序获得对应于待测序列碱基的测序信号强度结果,将所述强度结果表示为简并多聚物序列或均聚物序列,作为待评估序列;所述简并多聚物序列是由简并多聚物的数目排列组成的序列,所述均聚物序列是由构成均聚物的单体的数目排列组成的
序列;
40.b.构建字典:确定参考序列,将待测序列对应的参考序列表示为对应于测序方法的理想简并多聚物序列或理想均聚物序列,从所述理想简并多聚物序列或理想均聚物序列逐位提取长度为k的子信号进行字典构建;
41.c.字典稀疏化:从所述字典中提取子序列,选取与待评估序列距离相近的子序列作为最终字典;
42.d.稀疏表示:运用最优化算法,寻找一个可被所述最终字典矩阵左乘的向量,使得向量的稀疏度与待评估序列的前k位的距离同时被极小化,找到的向量称为稀疏向量;
43.e.过滤结果:根据设定的稀疏度阈值和混合度阈值分析所述的稀疏向量,当稀疏度不高于稀疏度阈值且混合度高于混合度阈值时,所述稀疏向量对应的待评估序列判定为单克隆信号;当稀疏度不高于稀疏度阈值且混合度不高于混合度阈值时,所述稀疏向量对应的待评估序列判定为多克隆信号或混合信号;当稀疏度高于稀疏度阈值时,稀疏表示失败,不能判定信号为单克隆或多克隆。
44.dna测序方法很多,按照参与每个测序反应的样本分子数,可以分为多分子测序和单分子测序;按照测序反应类型,可以分为合成测序,连接测序,切除测序等;按照每轮反应可以测得的核苷酸数目,又可以分为单碱基测序和多碱基测序,所述多碱基测序指的是每轮测序反应可以测得一至多个核苷酸。常见的illumina测序方法属于单碱基测序,其利用了3端封闭的方法,每次测序延伸一个碱基,因此其信号是0和1。需要说明的是,这里的信号0和1是一个相对比较的数值。可以知道的,实际进行测序的时候,首先进行反应,然后利用ccd等摄像装置获得反应的信号。但是,由于高通量的测序在反应的时候,其每个数据点或者每一簇待测序列的范围都很小,一般的在0.1-3微米之间,这样小的化学反应是难以被准确测量的。因此,调整后的ccd例如获得一个图像,其中的亮点,或者称为反应点的亮度为1000,不反应的点的亮度为500(简单举例,并非实际实验),那么就可以定义亮度接近1000的数据点为1,而亮度更靠近500的数据点为0。
45.本发明中,所述的测序包括多碱基测序。简并测序是一种多碱基测序,是边合成边测序技术的一种新形式,是一种3’端不封闭的基因测序方法,它在一个测序反应中同时进样至少两种碱基底物组成的简并底物,其每次反应延伸的碱基数可能是多个,例如1,2,3,4,5,6等等。多碱基测序方法和每次延伸一个碱基的测序方法进行比较的时候,其后续数据的处理就明显不一样了。测序的过程中,每次延伸多个碱基,那么对应于前面illumina的例子,说明性的,其信号强度有可能是1000,2000,3000,4000,5000,6000等等。可以知道的是,当测序信号需要区分得越精细的时候,其难度也就越大。测序的过程中受到各种因素的影响,例如反应不完全、试剂进入不充分、光源强度不稳定、光源强度不均匀、反应本身的影响等等,都导致了反应可能会超前或者滞后,那么反应的后期,这些积累的因素就会显得越来越严重。那么在定义其信号值1的时候,就更加困难。此外,对于切除测序,其在测序过程中每次测序反应切除不定数目的碱基,也可使用本发明所述方法对测序信号进行评估。
46.实际测序的时候,随着测序反应的进行,同一簇的dna分子,可能会出现不同长度的延伸的情况,并且这种情况随着测序长度的增加,是不断积累的。这会对测序信号的解读造成一定的困难,破坏测序信号的整倍性。例如的,当一个dna片段进行测序的时候,开始得到的测序信号通过比较,是比较接近整数的,这个时候,测序反应进行得比较充分,并且没
有太多杂反应的发生;当测序进行到50个(简单举例,并非实际实验)碱基的时候,测序信号结果可能变得不接近整数。不接近整数的信号,会影响对于多克隆信号判断的准确性。
47.对于本方法的步骤a,在测序的过程中,首先利用常见的多碱基测序方法,将一段基因序列进行测序,通过测序仪读取数据的方式获得对应于基因序列的测序信号。这个测序信号一般是由图像的强度值或者电流值等转换而来的。测序信号处理的过程不属于本发明的具体关注范围。申请人之前的专利cn201510944878.5,或者cn202010061629.2,或者cn201610899880.x等详细介绍了获得更加精确的测序信号的方法或者测序信号的处理方法。在此只是简单描述测序信号处理过程:测序得到原始测序数据后,首先判断此数据是否合规,对于合规的数据,提取测序信号强度结果,减去背景噪声并归一化,一般的,结果为依照采集时间排列的整型或浮点型向量,例如某单个碱基的测序信号为x,那么当得到的测序信号为2x的时候,可以知道的是,本次测序可能延伸了2个碱基。多碱基测序以2+2简并测序为例,每一轮测序反应通入两种核苷酸底物,对于所有的奇数轮反应,通入的是两种核苷酸底物,而偶数轮通入的是另外两种核苷酸底物,例如奇数轮通入a/c,偶数轮通入g/t,这种方法可以保证每一轮反应都会发生碱基的延伸,不会有空反应,因此反应速度较快,由简并测序反应得到的序列不是精确的碱基序列,而是简并碱基序列。例如mmkmkkkmmmkkkm
…
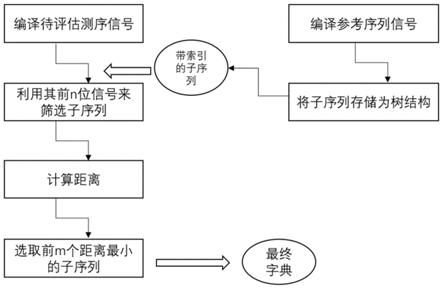
,其中m表示a/c,k表示g/t。对于简并测序,将测序信号强度结果表示为简并多聚物序列,简并多聚物序列是指由简并多聚物的数目排列组成的序列,例如,对于序列mmkmkkkmmmkkkm,其简并多聚物序列为(2,1,1,3,3,3,1)。此外,多碱基测序方法还包括例如ion torrent的测序方法,每轮测序反应只通入一种核苷酸底物,发生延伸的核苷酸的个数可以是一个或多个(不考虑不发生延伸的情况),此即为均聚物,即只由一种单体聚合而成的聚合物。均聚物序列指的是由构成均聚物的单体的数目排列组成的序列。例如延伸了4个a,则均聚物为aaaa,此均聚物序列为4。特殊的,当延伸的核苷酸的个数为1时,此时从严格意义上来讲,并不能称之为均聚物,但本发明将其表示为1处理。碱基序列、简并碱基序列、简并多聚物序列(以mk测序为例)及均聚物序列的对应关系可参见表1所示的例子,即简并多聚物序列为(4,5,3,3,5),均聚物序列为(3,1,3,2,2,1,3,3,2)。
48.表1.多种序列表示方式
49.碱基序列aaactttggccatttaaacc简并碱基序列mmmmkkkkkmmmkkkmmmmm简并多聚物序列4
ꢀꢀꢀ5ꢀꢀꢀꢀ3ꢀꢀ3ꢀꢀ5ꢀꢀꢀꢀ
均聚物序列3
ꢀꢀ
13
ꢀꢀ
2 2 13
ꢀꢀ3ꢀꢀ
2 50.本发明中,所应用的测序信号是经过归一化的测序信号(简称为归一化信号),也就是对应于单碱基测序信号的比值。
51.根据优选的实施方式,当待测序列的归一化信号长度比较长的时候,并不需要将所有的归一化信号都用作单克隆与多克隆的计算,只需要选取其中的一部分作为该方法的输入即可。经验性的,可以选取更加接近整数值的前k位连续归一化信号用作输入,即选取所述简并多聚物序列或均聚物序列的前k位作为待评估序列;k的值与参考序列的大小以及测序循环数有关,k的取值优选为8-20,更优选为10-15。不同的选取方式,对单克隆和多克隆的最终区分影响不大。多克隆信号有更加明确的特征,无论从哪个位置开始选取,都是多个测序信号的混合。选取的位置不同,但是得到的结果是相同的。
52.对于与待测序列对应的参考序列,确定参考序列是较为容易的过程,例如当待测序列为小鼠的某组织rna时,参考序列为小鼠的转录组序列;当待测序列为人类的某组织dna时,则参考序列为人类基因组序列。确定参考序列后,需要将待测序列对应的参考序列表示为对应于测序方法的理想简并多聚物序列或理想均聚物序列,也就是说当待测序列进行的是简并测序时,则将参考序列表示为对应于该简并测序方法的简并多聚物序列;当待测序列进行的是普通多碱基测序时(即每轮反应只加入一种核苷酸底物),则将参考序列表示为对应的均聚物序列。
53.稀疏字典学习
54.字典学习(dictionary learning)和稀疏表示(sparse representation)在学术界的正式称谓应该是稀疏字典学习(sparse dictionary learning)。该算法理论包含两个阶段:字典构建阶段(dictionary generate)和利用字典(稀疏的)表示样本阶段(sparse coding with a precomputed dictionary)。稀疏表示本质上是一种信号表示方法,它从原始信号中尽可能少地选取基本信号,并将这些基本信号通过线性组合来表达大部分或全部的原始信号。通过稀疏表示可以获得信号更为简洁的表示方式,从而更容易地获取信号中所蕴含的信息,更方便进一步对信号进行加工处理。而识别混合信号的本质是通过计算信号能否被字典中的某一列向量近似表示。本发明的方法具体采用sparse coding稀疏编码技术实现这种思想,从而完成对多克隆信号的识别。
55.字典构建
56.用于将待评估序列进行展示的线性空间被称为“字典”。其中空间为一个线性代数概念,用一个整数矩阵表示,可理解为所有潜在的信号种类的集合。对待评估序列构建出相应的合理的字典,是该方法的关键。图1所示为构建最终字典的流程示意图,所述最终字典是稀疏化的字典,下面详述其具体构建过程。
57.本方法中,字典的集合为将待测序列对应的参考序列表示为对应于测序方法的理想简并多聚物序列或理想均聚物序列,从所述理想简并多聚物序列或理想均聚物序列逐位提取长度为k的子信号的集合,所述长度为k的子信号构成所述字典的每一列。k的取值与所述参考序列的大小以及测序循环数有关,k的取值优选为8-20,更优选为10-15。为了方便后续的查询,这些长度为k的字符串,更通用的术语是k-mer,用一定的数据结构进行存储方便后续查询。数据结构包括但不限于,二叉树,红黑树,inverted index等等。考虑到随着基因组增大该集合的增大而导致的后续矩阵计算所增长的计算量,“字典”构建过程中采取一定筛选方式不断挑选出子集合作为潜在的真正用来进行信号表示的空间。下面详细描述这个筛选过程,即字典稀疏化过程。
58.对于每条待评估序列,从中提取连续的n位(n《k)作为待评估子序列,将待评估子序列与前述字典中每个子信号的前n位遍历比较,将字典中与实际信号前n位取整相同的项作为备选集合;若字典中没有前n位完全相同的,则直接进入下一步骤。n可以取值为3,或4,或5。
59.在备选集合中,逐一计算待评估序列的前k位与字典项的距离,所述距离包括但不限于皮尔逊相关系数、斯皮尔曼相关系数、平均互信息、欧几里得距离、汉明距离、车比雪夫距离、马哈兰诺比斯距离、曼哈顿距离、明科斯基距离、对应信号差值的绝对值的最大值或最小值,将距离由小到大排列的前m项集合元素取出来,作为最终字典。所述m的选取要考虑
包含可能信号的完备性和计算量的平衡,视参考序列的情况,m的取值范围为20-300,优选为50-150。
60.稀疏表示(sparse representation)
61.稀疏表示也被称为稀疏编码。一个含有大多数零元素的向量可以被称为稀疏向量。对于一个线性方程,x=d*α,当d是一个欠定的矩阵的时候,d被称为一个字典,x是我们感兴趣的信号。稀疏表示指的是,用一个尽可能稀疏的向量α表示x与d之间的关系。对于一个维度固定的向量α,当向量α里面的零元素越多的时候,其越稀疏。从数学的角度说,稀疏表示算法就是解一个同时极小化稀疏度以及从d*α到x的距离的双目标优化问题,如图2所示,本发明需要运用适当的最优化算法,寻找一个尽可能稀疏的向量,使得前述最终字典矩阵左乘该向量的结果与待评估序列的前k位尽可能接近,找到的向量称为稀疏向量。所述的最优化算法包括但不限于匹配追踪、正交匹配追踪、弱匹配追踪、阈值方法、基追踪、irls算法、lasso算法、加权支持向量机算法等。
62.举例性的,当一个待测序列的简并多聚物序列是(2,1,2,1,1,2,1,1,1,2,2)的时候,可以选取前面的k个信号作为待评估序列(当k取值为8时),待评估序列为(2,1,2,1,1,2,1,1)。实际测序的时候,归一化信号可能不完全是整数组成的,可以选取前面比较接近整数的部分,或者其中比较接近整数的部分。简单的,选取靠前的测序归一化信号作为输入。可以选取前面的5-50个连续归一化信号用作输入;更优选的,选取前面的6-45个信号,更优选的选取7-40个信号,或者8,9,10,11,12,13,14,15个信号。其中所述的前面的并不需要精确的定义,可以是从第一个测序信号开始选取,也可以从第2,3,4个测序信号开始选取。
63.举例性的,某个待测基因序列的归一化信号为:2.3,2.2,2.5,3,1,1,2,1,3,3.1,3.5,3.6,1.3,1.2
…
;当选取的时候,可以选取前面的例如前9个信号,或者从第三个开始的8个信号。理论上,选取的位置不影响最终结果;单克隆信号与多克隆信号有着明显的区别,多克隆是两个甚至更多个单克隆信号的混合体。但由于实际测得的信号会受到失相、衰减等因素的影响,总的来说,采集顺序越靠后的信号质量就越差,也越有可能偏离整数;故而,作为优选的实施方式,应当尽可能选用靠前的子信号作为待评估信号。本发明中,利用了数学的方式描述了这种规律,并且没有引入额外的意义。这种方式对于多克隆和单克隆的选择正确率是比较高的。当然,当信号极差,测序准确度例如低于90%的时候,准确率可能会下降,但是可以理解的,这并不归咎于本发明方法的准确性问题。
64.过滤结果
65.稀疏向量α是一个由非零元素与零元素组成的向量,其含有很多零或者接近零的元素。如图2所示,对稀疏表示得到的优化后的α,第一步是检查稀疏度(sparsity),即非零元素的个数。只有稀疏度不高于稀疏度阈值的α向量,才是本方法所关注的,稀疏度阈值的取值范围为2-10,优选为2-5,更优选为2-3;其他的情况统一称为异常,异常情况说明稀疏表示算法没能按照设计产生稀疏度较低的向量,提示原始信号与字典中基本信号的差异较大,可能是低质量序列,或者序列不在参考序列中,无法用本方法判断单克隆或多克隆信号。在目前所观察到的情况下,稀疏度高于稀疏度阈值是非常罕见的。
66.优选的,对稀疏向量的所有元素进行归一化,其中,稀疏向量中的最大元素是我们关心的部分,其表示了x与d中的某个列向量接近的程度,将其称作混合度。当稀疏向量的混合度接近数字1的时候,其表示的意义是,该测序信号与参考序列的某个理想归一化信号相
近,则该测序信号为单克隆信号(mono-clonal)。相反的,当稀疏向量的混合度不接近数字1,或者有多个相差不多的元素的时候,则该测序信号判断为多克隆信号(poly-clonal)或混合信号。据此可对稀疏向量中混合度的值设定一个阈值,大于此阈值,判定为单克隆信号;小于此阈值,判定为多克隆信号,或混合信号;该阈值被称为混合度阈值。经验性的,混合度阈值在0.6-1范围内的时候,认为该测序信号为单克隆信号;优选的,混合度阈值在0.8-1范围内的时候,认为该测序信号为单克隆信号;更优选的,混合度阈值在0.9-1范围内的时候,认为该测序信号为单克隆信号;更优选的,混合度阈值在0.95-1范围内的时候,认为该测序信号为单克隆信号;反之则为多克隆信号或混合信号。
67.正常的,利用本发明的方法区分出多克隆信号后,由于多克隆信号中的混杂信息通常是不可用的,所以将鉴定出的多克隆信号或混合信号舍弃,将不能判定信号为单克隆或多克隆的信号舍弃,将鉴定出的单克隆信号分离出来用于后续的数据处理和分析,例如用于碱基识别等。
68.优选的,可以用数学符号、正负号表示多碱基测序中的不同的化学反应。例如的,2+2测序中以km进样顺序测序,得到简并多聚物序列为2,3,1,1,3,3,2,1,则k信号可以表示为正值,m信号可以表示为负值,即:2,-3,1,-1,3,-3,2,-1。
69.本发明中,选用了数学判定中字典与稀疏表示的判定方式。这种方式能够表示一个向量或者一组数据与参考值的接近标准。本发明中,所使用的方法并没有额外增加限制或者判定,更进一步的说,数学算法中,每个数字或者计算的实际意义都是实际测序信号与参考序列接近程度的计算,通过给定的阈值,可以判断测序信号属于单克隆信号或者多克隆信号。因此,所用的数学方式完全是对于本发明的要求保护技术方案的具体实现方式之一。
70.d为由参考序列的可能片段的理想归一化信号组成的矩阵。参考序列的基因序列是已知的。其表示的意义是,每一列是一种可能的理想归一化信号。并且的,选取的时候,其信号是连续的,并且和x的维数是相同的。x的维数也是其测序信号的个数。例如的,某个已知序列的参考基因组,其包含500bp的碱基长度。当x的维数是10的时候;那么d是参考基因组的理想归一化信号中连续选取的,每10个信号组成的列向量,构成的矩阵。其意义是将所有可能的10个信号的情况,组成了一个矩阵。当然实际计算的时候,由于知道的信息更多或者更少,会有更加简单的组成方式简化矩阵,但是其原理是相同的。
71.本专利中,虽然使用了矩阵的计算方式,其实际意义还是测序结果同参考序列的理想测序结果的比较。简单的数学方式的选取,并不会引入额外的物理意义或者限定。
72.α为稀疏向量,稀疏表示的计算方式是数学中的常见知识。适当的距离选择可以将求解α的过程转变为一个凸二次规划(convex quadratic optimization)的问题。目前有很多针对这类问题的解法器,如内点法(interior point methods),定点连续法(fixed-point continuation),in-crowd algorithm等。本发明中不再详述稀疏表示的计算方式,此部分内容并不属于本发明的关注重点。
73.本发明的第二方面公开了一种基因测序混合信号的评估方法,其特征在于,包括以下步骤:
74.1)测序获得对应于待测序列碱基的结果;
75.2)将待测序列对应的参考序列编译为对应于测序方法的理论结果;
76.3)将参考序列的编译的理论结果划分为6-50个碱基的可能结果集群;4)将测序获得的对应于待测序列碱基的结果与步骤3)的可能结果集群进行比对。
[0077][0078]
根据优选的实施方式,利用前面所述任一项中的方法判定多克隆信号或者单克隆信号。
[0079]
在本发明的第一方面具体实施部分中所讨论的特征中的每个特征同样适用于本发明的第二方面的具体实施。如上所示,部分其他特征在此处不再重复,并且应被视为以引用方式重复。本领域普通技术人员将理解在这些具体实施中识别的特征可如何容易地与在其他具体实施中识别的基本特征组组合。
[0080]
实施例1
[0081]
对λ噬菌体进行ws 2+2简并测序。选择k=10,m=256,尝试用稀疏表示的方法区分单克隆和多克隆信号。
[0082]
一个被判定为单克隆的信号为:
[0083]2ꢀ‑
2 2
ꢀ‑
2 2
ꢀ‑
2 2
ꢀ‑
2 2
ꢀ‑2[0084]
该mono-clonal信号进行表示的字典(展示与该信号距离最近的前10列)为
[0085][0086]
该单克隆信号的稀疏表示如下,其实际是一个列向量,为了更加清楚地表示,现表示α的转置如下:
[0087][0088]
一个被判定为多克隆的信号为
[0089]2ꢀ‑
1 3
ꢀ‑
2 2
ꢀ‑
1 2
ꢀ‑
3 2
ꢀ‑2[0090]
一个poly-clonal信号的字典(展示与该信号距离最近的前10列)
[0091][0092]
一个多克隆信号的稀疏表示结果
[0093][0094]
实施例2
[0095]
识别烟草花叶病毒(tmv)链特异性逆转录文库测序中的多克隆信号
[0096]
烟草花叶病毒的基因组为单链rna,长度为6395,以km进样顺序对cdna进行简并测序时,合并重复的0测序信号,可直接得到一个1509列,12行的矩阵:
[0097][0098]
如图,每一列为参考基因组中一种可能的测序信号,正数为k信号,负数为m信号。
[0099]
将该矩阵按列归一化,就得到字典d:
[0100][0101]
对于以下四组测序信号:
[0102]
1.无噪声的理想单克隆信号sm:
[0103]4ꢀ‑
2 1
ꢀ‑
1 3
ꢀ‑
2 1
ꢀ‑
1 6
ꢀ‑
1 3
ꢀ‑2[0104]
2.带噪声的实测单克隆信号s
′m:
[0105]
3.653
ꢀ‑
2.095 1.221
ꢀ‑
1.178 3.190
ꢀ‑
1.637 1.429
ꢀ‑
0.879 5.978
ꢀ‑
1.264 2.479
ꢀ‑
2.024
[0106]
3.无噪声的理想多克隆信号s
p
:
[0107]
2.5
ꢀ‑
2.5 2.5
ꢀ‑
1.5 2
ꢀ‑
1.5 2
ꢀ‑
1.5 3.5
ꢀ‑
1 4.5
ꢀ‑
1.5
[0108]
4.带噪声的实测多克隆信号s
′
p
:
[0109]
2.444
ꢀ‑
2.523 2.529
ꢀ‑
1.435 1.957
ꢀ‑
1.477 1.991
ꢀ‑
1.422 3.472
ꢀ‑
0.999 4.675
ꢀ‑
1.546
[0110]
运用lasso方法优化如下函数:
[0111][0112]
并设混合度阈值为0.6,
[0113]
对于x=sm,混合度为0.889,判定为单克隆。
[0114]
对于x=s
′m,混合度为0.651,判定为单克隆。
[0115]
对于x=s
p
,
[0116][0117]
,混合度为0.367,判定为多克隆。
[0118]
对于x=s
′
p
,
[0119][0120]
混合度为0.366,判定为多克隆。
[0121]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1