一种金属-药物全活性纳米药物及其制备方法与用途
1.本发明涉及一种金属-药物全活性纳米药物及其制备方法与用途,属于生物医药领域。
背景技术:
2.近年来,随着纳米技术的发展,基于纳米载体的药物递送体系(通常称为纳米药物),由于治疗效果提高,毒副作用减少,在疾病诊疗尤其是是癌症治疗中展现了巨大的应用潜力。自1995年美国食品和药物管理局(fda)批准了第一个抗癌纳米药物阿霉素脂质体纳米颗粒用于治疗卵巢癌和艾滋病相关的卡波西肉瘤和多发性骨髓瘤。近年来,越来越多的纳米药物被批准用于肿瘤诊疗的商用或临床使用。例如,2005年首个基于白蛋白的纳米药物白蛋白结合型紫杉醇获得fda批准用于乳腺癌治疗,2012年和2013年又将其适应症扩展到非小细胞肺癌和胰腺癌。负载紫杉醇的聚合物胶束静脉注射液于2007年在韩国市场上推出,用于治疗乳腺癌和非小细胞肺癌。因此,设计合理的纳米药物并优化其制备工艺,成为纳米药物临床转化相关领域的研究热点。
3.基于不同纳米载体如脂质体、聚合物纳米颗粒、树枝状大分子、无机纳米材料的纳米药物,与游离的化疗药物相比,其显著提高了疏水性化疗药物的水分散性及生物利用度,改善了药物的药理特性和药代动力学;纳米药物还可有效延长药物血液循环时间,从而提高药物在肿瘤等病灶部位的有效累积量;同时,基于纳米药物的递送策略可大大提高肿瘤部位靶向性,在进一步增加药物肿瘤部位富集量的同时减少其毒副作用;此外,纳米药物可同时负载多种药物成分或显影剂,易于实现协同治疗或诊疗一体化。
4.然而,目前纳米药物的临床转化仍面临一些亟待解决的问题:1)活性药物成分(active pharmaceutical ingredients,api)含量低,通常api在纳米药物中的质量分数小于10%;2)聚合物、无机纳米载体等难代谢,可能造成潜在的长期毒性、炎症反应等问题;3)制备工艺复杂,难以工业放大;4)成分复杂、成本高,难以实现临床转化等。因此设计安全、高效的纳米药物,并探索操作简单、条件温和、重复性好、利于工业放大的制备方法对实现纳米药物临床转化至关重要。
技术实现要素:
5.为克服现有技术存在的缺陷,本发明的目的之一在于提供一种金属-药物全活性纳米药物(full-api nanodrugs,fand),所述金属-药物全活性纳米药物由路易斯酸金属离子和活性药物成分构成。
6.本发明的目的之二在于提供一种金属-药物全活性纳米药物的制备方法,所述制备方法是将路易斯酸金属离子和活性药物成分通过金属-有机配位作用,同时协同多种分子间弱作用力结合形成金属-药物全活性纳米药物。
7.本发明的目的之三在于提供一种金属-药物全活性纳米药物的应用。
8.为实现本发明的目的,提供以下技术方案。
9.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是由可以形成配位键的路易斯酸金属离子和活性药物成分通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
10.其中,所述活性药物成分为具有配位基团的化疗药物、光敏剂、光热剂、蛋白质和核酸中的至少一种。优选的,所述配位基团为氨基、羧基、羰基、磷酸官能团、磺酸官能团、氮原子或氧原子;
11.优选的,所述活性药物成分为姜黄素、5-氨基酮戊酸(ala)、白藜芦醇、索拉菲尼、吲哚菁绿、阿霉素、亚油酸氢过氧化物、过氧化物酶和可抑制细胞自噬的质粒中的至少一种;
12.优选的,所述金属离子为铁离子、锌离子、锰离子、钙离子、镁离子、铜离子、铈离子或钆离子;进一步优选,所述金属离子为三价铁离子或二价镁离子;更优选的,所述金属离子为三价铁离子。
13.优选的,所述全活性纳米药物的结构为球状纳米结构;更优选的,所述球状纳米结构的平均粒径为20nm~1500nm。
14.一种本发明所述的金属-药物全活性纳米药物的制备方法,所述方法步骤如下:
15.(1)将可以形成配位键的路易斯酸金属离子溶液和活性药物成分溶液滴加混合,或将可以形成配位键的路易斯酸金属离子溶液和活性药物成分溶液依次滴加到去离子水、超纯水或三次重蒸水中,25℃~45℃下连续搅拌10分钟~60分钟,得到待纯化的金属-药物全活性纳米药物溶液。
16.所述金属离子溶液的溶剂为能够完全溶解金属离子的良性溶剂,优选去离子水、超纯水或三次重蒸水;
17.所述活性药物成分溶液的溶剂为能够完全溶解活性药物成分的良性溶剂,优选去离子水、超纯水、三次重蒸水、四氢呋喃、甲醇或乙醇;
18.所述金属离子与活性药物成分的物质的量之比为1:0.7~1:700。优选的,当活性药物成分为化疗药物时,所述金属离子与活性药物成分的物质的量之比为1:0.7~1:200;当活性成分为化疗药物与核酸或活性成分为化疗药物与蛋白质时,所述金属离子与活性药物成分的物质的量之比为1:2.5~1:700。
19.将所述金属离子溶液和活性药物成分溶液依次滴加到去离子水、超纯水或三次重蒸水中时,去离子水、超纯水或三次重蒸水与所述金属离子溶液的体积比为7.5:1~100:1。
20.所述金属离子溶液与活性药物成分溶液的体积比为0.005:1~2:1。优选的,当活性药物成分为化疗药物时,所述金属离子溶液与活性药物溶液成分的体积比为0.005:1~0.5:1;
21.优选的,搅拌转速为300rpm~1500rpm;
22.优选的,所述金属离子溶液中金属离子的物质的量浓度为1mm~50mm;
23.优选的,活性药物成分溶液中活性药物成分的物质的量浓度为1mm~200mm;
24.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液纯化,去除分子态活性药物成分、可以形成配位键的路易斯酸金属离子和有机溶剂,得到金属-药物全活性纳米药物;所述金属-药物全活性纳米药物可以以溶液状态在4℃下储存待用,或经过冷冻干燥,以粉末状态在4℃下储存待用。
25.优选的,所述纯化方式为超滤或透析。
26.一种本发明所述的金属-药物全活性纳米药物在制备抗肿瘤药物或抗炎症药物中的应用。
27.优选的,当所述金属离子为三价铁离子,活性药物成分为5-氨基酮戊酸和姜黄素时,所述全活性纳米药物在制备抗肺癌药物中的应用。
28.优选的,当所述金属离子为三价铁离子,活性药物成分为质粒和索拉菲尼时,所述全活性纳米药物在制备抗肺癌药物中的应用。
29.优选的,当所述金属离子为三价铁离子,活性药物成分为过氧化物酶和亚油酸氢过氧化物时,所述全活性纳米药物在制备抗胰腺癌药物中的应用。
30.优选的,当所述金属离子为三价铁离子,活性药物成分为姜黄素时,所述全活性纳米药物在制备抗肝炎药物中的应用。
31.有益效果
32.1.本发明提供了一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物活性成分理论含量高达100%,无惰性载体成分,稳定性好,生物相容性高,安全性好。其中,参与配位的路易斯金属离子作为活性成分,可以直接用于治疗或作为显影剂用于成像;例如fe
2+
、cu
2+
等通过芬顿(fenton)反应引发化学动力学疗法(chemodynamic therapy,cdt),fe
3+
、fe
2+
引发铁死亡(ferroptosis)以及ca
2+
诱导细胞钙化死亡等;fe
3+
、mn
2+
、gd
3+
等还可以作为显影剂用于成像,协同参与配位作用的其他活性药物成分(化药、蛋白质、核酸、光敏剂、光热剂)通过化疗、光动力治疗、光热治疗等提高疗效。
33.2.本发明提供了一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物具有潜在的多功能性:一方面,金属离子例如mn
2+
或gd
3+
,或具有荧光、或光声成像的光敏剂度和光热剂活性药物可用于成像,实现实时监控;另一方面,金属离子和活性药物成分可以引起不同的治疗方式,例如离子疗法、化疗、光热治疗、光动力治疗、基因治疗等,协同提高疗效。
34.3.本发明提供了一种金属-药物全活性纳米药物的制备方法,所述方法基于金属-有机配位自组装,同时协同其他多种分子间弱作用力制备得到一种金属-药物全活性纳米药物,操作简单、条件温和、重复性好,成本低,易于工业放大。
35.4.本发明提供了一种金属-药物全活性纳米药物的应用,所述金属-药物全活性纳米药物基于活性药物成分不同在制备抗肿瘤药物或抗炎症药物中的应用;当活性药物成分为5-氨基酮戊酸、索拉菲尼、白藜芦醇、姜黄素、吲哚菁绿、阿霉素等抗肿瘤活性药物成分时,所述全活性纳米药物在制备抗肺癌药物或抗胰腺癌药物中的应用;当活性药物成分为白藜芦醇和姜黄素等抗炎症活性药物成分时,所述金属-药物全活性纳米药物在制备抗肝炎药物中的应用。
附图说明
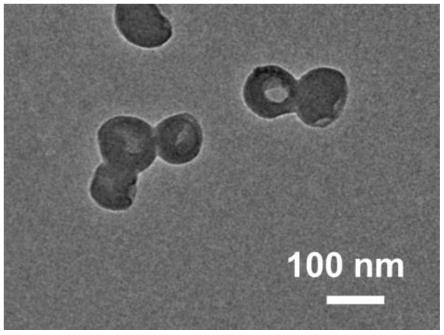
36.图1是实施例1所制备的金属-药物全活性纳米药物的透射电镜图。
37.图2是实施例6所制备的金属-药物全活性纳米药物的透射电镜图。
38.图3是实施例7所制备的金属-药物全活性纳米药物的透射电镜图。
39.图4是实施例8所制备的金属-药物全活性纳米药物的透射电镜图。
40.图5是实施例9所制备的金属-药物全活性纳米药物的透射电镜图。
41.图6是实施例10所制备的金属-药物全活性纳米药物的透射电镜图。
42.图7是实施例11所制备的金属-药物全活性纳米药物的透射电镜图。
43.图8是实施例12所制备的金属-药物全活性纳米药物的透射电镜图。
44.图9是实施例1所制备的金属-药物全活性纳米药物的表面电势图。
45.图10是实施例1所制备的金属-药物全活性纳米药物的稳定性表征图。
46.图11是实施例1所制备的金属-药物全活性纳米药物的作用力表征图。
47.图12是分别由pbs、ala分子、实施例1所制备的金属-药物全活性纳米药物处理后的a549人肺癌细胞(a549细胞)内原卟啉(ppix)的激光共聚焦显微镜荧光成像图。
48.图13是分别由pbs、ala分子、实施例1所制备的金属-药物全活性纳米药物处理后的a549人肺癌细胞(a549细胞)内胆绿素(bv)的激光共聚焦显微镜荧光成像图。
49.图14是分别由pbs、ala分子、实施例1所制备的金属-药物全活性纳米药物处理的a549人肺癌细胞(a549细胞)内一氧化碳(co)的激光共聚焦显微镜荧光成像图。
50.图15是分别由pbs、实施例1所制备的金属-药物全活性纳米药物、实施例1所制备的金属-药物全活性纳米药物和铁离子螯合剂(dfo)处理的a549人肺癌细胞(a549细胞)内脂质活性氧(lipid ros)的激光共聚焦显微镜的荧光成像。
51.图16是分别由pbs、纯激光、ala分子和激光、实施例1所制备的金属-药物全活性纳米药物和激光处理的a549人肺癌细胞(a549细胞),胞内活性氧(ros)的激光共聚焦显微镜荧光成像图。
52.图17是a549人肺癌细胞(a549细胞)经不同浓度实施例1所制备的金属-药物全活性纳米药物处理后的细胞生存率。
53.图18是经不同处理的荷瘤小鼠在14天内的肿瘤体积变化表征图。
54.图19是l02正常人类肝脏细胞(l02细胞)经不同处理后胞内ros的激光共聚焦显微镜荧光成像图。
55.图20是panc-1人胰腺癌细胞(panc-1细胞)分别经磷酸缓冲盐溶液(1
×
pbs)和实施例10所制备的金属-药物全活性纳米药物处理后的细胞生存率。
56.图21是a549人肺癌细胞(a549细胞)分别经pbs和实施例11所制备的金属-药物全活性纳米药物处理后的细胞生存率。
具体实施方式
57.下面结合附图和具体实施例来详述本发明,但不作为对本发明专利的限定。
58.以下实施例中:
59.a549人肺癌细胞(a549细胞)、l02正常人类肝脏细胞(l02细胞)和panc-1人胰腺癌细胞(panc-1细胞)购自北京协和医院;
60.小鼠免疫缺陷模型balb/c-nu小鼠(4~5周,雌性)购自北京大学医学部(实验动物科学部);
61.5-氨基酮戊酸盐酸盐购自阿拉丁试剂(上海)有限公司;
62.过氧化物酶(型号:c0278-6.25ku)和姜黄素购自美国sigma-aldrich公司;
63.氯化铁、氯化镁、氯化钯、氯化钠、尿素、十二烷基硫酸钠、乙二胺四乙酸、四氢呋喃
购自北京伊诺凯科技有限公司;
64.去铁胺(dfo)购自上海麦克林生化科技有限公司;
65.ros探针(2',7'-二氯荧光黄双乙酸盐,dcfh-da)和mtt细胞增殖及细胞毒性检测试剂盒购自北京索莱宝科技有限公司;
66.c11 bodipy 581/591脂质过氧化荧光探针购自美国cayman chemical公司;
67.磷酸缓冲盐溶液(1
×
pbs)、dmem(dulbecco's modified eagle medium)培养基、rmi 1640(roswell park memorial institute 1640)培养基、胰蛋白酶和青霉素/链霉素混合液(100
×
)购自中科迈晨(北京)科技有限公司;
68.胎牛血清(fetal bovine serum,fbs)购自德国pan-biotech公司;
69.bd基质胶/基质膜(bd:356234)购自美国corning公司;
70.rmi 1640完全培养基:以含体积分数为10%的胎牛血清和体积分数为1%的青霉素/链霉素混合液(100
×
)的rmi 1640培养基。
71.dmem完全培养基:以含体积分数为10%的胎牛血清和体积分数为1%的青霉素/链霉素混合液(100
×
)的dmem培养基。
72.细胞培养条件:在37℃含体积分数为5%的co2的培养箱中培养。
73.对以下实施例进行如下测试:
74.1.对实施例1进行如下测试:
75.(1)形貌测试:采用透射电子显微镜,型号:ht-7700,日立,日本;将所制得的样品滴在铜网上,干燥后进行形貌观察。
76.(2)表面电势测试:zeta电位分析仪,型号:en 3600,malvern,英国。
77.(3)稳定性测试:将实施例1中制备得到的金属-药物全活性纳米药物溶液在室温下放置14天,观察其稳定性。
78.(4)作用力分析:向实施例1中制备得到的金属-药物全活性纳米药物溶液中分别加入等体积去离子水、四氢呋喃、氯化钠(50mm)、尿素(50mm)、十二烷基硫酸钠(50mm)和乙二胺四二钠(50mm),37℃孵育2小时后,通过荧光分光光度计测量其在525nm激发下,470~640nm处的荧光发射图谱。
79.(5)肿瘤细胞内生成原卟啉(ppix)性能分析:以a549人肺癌细胞作为肿瘤细胞模型。
80.实验步骤:a549人肺癌细胞经2ml胰蛋白酶消化2分钟后接种在3个共聚焦小皿中,在37℃含体积分数为5%的co2的培养箱中培养过夜。去除旧培养基,分别用含1
×
pbs、5-氨基酮戊酸(ala)、实施例1中制备得到的金属-药物全活性纳米药物(nps)(等量ala含量:150μg/ml,250μl)的rmi 1640完全培养基1ml孵育,继续在37℃含体积分数为5%的co2的培养箱中培养24小时。培养结束后,1
×
pbs清洗,用激光共聚焦显微镜(型号:a1,nikon,日本)观察不同组别代谢产生ppix的自身红色荧光信号。
81.(6)肿瘤细胞内产生胆绿素(bv)性能分析:肿瘤细胞模型及其培养条件同测试(5)。
82.实验步骤:a549人肺癌细胞经2ml胰蛋白酶消化2分钟后接种在3个共聚焦小皿中,在37℃含体积分数为5%的co2的培养箱中培养过夜。去除旧培养基,分别用含1
×
pbs、5-氨基酮戊酸(ala)、实施例1中制备得到的金属-药物全活性纳米药物(等量ala含量:150μg/
ml,250μl)的rmi 1640完全培养基1ml孵育,继续在37℃含体积分数为5%的co2的培养箱中培养24小时。培养结束后,1
×
pbs清洗,用激光共聚焦显微镜观察(型号:a1,nikon,日本)不同组别代谢产生bv的自身绿色荧光信号。
83.(7)肿瘤细胞内产生一氧化碳(co)性能分析:肿瘤细胞模型及其培养条件同测试(5)。
84.实验步骤:a549人肺癌细胞经2ml胰蛋白酶消化2分钟后接种在3个共聚焦小皿中,在37℃含体积分数为5%的co2的培养箱中培养过夜。去除旧培养基,分别用含1
×
pbs、5-氨基酮戊酸(ala)、实施例1中制备得到的金属-药物全活性纳米药物(等量ala含量:150μg/ml,250μl)的rmi 1640完全培养基1ml孵育,继续在37℃含体积分数为5%的co2的培养箱中培养24小时。随后,加入含1μm co探针(氯甲酸丙烯酯功能化荧光素,fl-co-1)和1μm氯化钯的rmi 1640培养基1ml,在37℃下含体积分数为5%的co2的培养箱中培养30分钟。培养结束后,1
×
pbs清洗,用激光共聚焦显微镜(型号:a1,nikon,日本)观察不同组别代谢产生co的荧光信号。
85.(8)肿瘤细胞内引起铁死亡性能考察:肿瘤细胞模型及其培养条件同测试(5)。
86.实验步骤:a549人肺癌细胞经2ml胰蛋白酶消化2分钟后接种在3个共聚焦小皿中,在37℃含体积分数为5%的co2的培养箱中培养过夜。去除旧培养基,分别用含1
×
pbs、实施例1中制备得到的金属-药物全活性纳米药物(nps)(ala含量:150μg/ml,250μl)、nps+铁离子螯合剂(去铁胺,dfo)(ala含量150μg/ml,250μl,dfo含量10mm,10μl)的rmi 1640完全培养基1ml孵育,继续在37℃含体积分数为5%的co2的培养箱中培养24小时。随后,加入含2μm c11 bodipy 581/591脂质过氧化荧光探针的rmi 1640培养基1ml,在37℃含体积分数为5%的co2的培养箱中培养30分钟。培养结束后,1
×
pbs清洗,用激光共聚焦显微镜(型号:a1,nikon,日本)观察不同组别诱发铁死亡的能力。
87.(9)光照条件下,在肿瘤细胞内产生活性氧(ros)的性质验证:肿瘤细胞模型及其培养条件同测试(5)。
88.实验步骤:a549人肺癌细胞经2ml胰蛋白酶消化2分钟后接种在4个共聚焦小皿中,在37℃含体积分数为5%的co2的培养箱中培养过夜。去除旧培养基,4个皿分别设定为实验组:pbs组、激光组(laser组)、ala+laser组、nps+laser,依次分别用含1
×
pbs、1
×
pbs、5-氨基酮戊酸(ala)、实施例1中制备得到的金属-药物全活性纳米药物(nps)(等量ala含量:150μg/ml,250μl)的rmi 1640完全培养基1ml孵育,继续在37℃含体积分数为5%的co2的培养箱中培养24小时。随后,加入含10μm ros探针(2',7'-二氯荧光黄双乙酸盐,dcfh-da)的rmi 1640培养基1ml,在37℃含体积分数为5%的co2的培养箱中培养30分钟。培养结束后,1
×
pbs清洗,laser组、ala+laser组、nps+laser组分别用激光(635nm,300mw/cm2)照射小皿中的细胞5分钟。最后,用激光共聚焦显微镜(型号:a1,nikon,日本)评估不同组别的光动力性质。
89.(10)体外抗肿瘤能力考查:肿瘤细胞模型及其培养条件同测试(5)。
90.实验通过标准mtt比色法测定实施例1中制备得到的金属-药物全活性纳米药物(nps)的体外细胞毒性。将a549细胞接种于96孔板中(1万个细胞/孔),在37℃含体积分数为5%的co2的培养箱中培养过夜;用含不同浓度(0、25、50、100μg/ml)nps的rmi 1640完全培养基孵育(每个浓度设置5个复孔),继续在37℃含体积分数为5%的co2的培养箱中培养24
小时;激光(635nm,300mw/cm2)照射细胞5分钟,在37℃含体积分数为5%的co2的培养箱中继续培养12小时;随后,在避光的条件下,加入mtt(含0.5mg/ml的rmi1640完全培养基,100μl/孔);在37℃含体积分数为5%的co2的培养箱中培养4小时后,加入dmso(110μl/孔),震荡15分钟,用酶标仪(a51119600dpc,thermofisher,美国)测定各孔在490nm处的吸光度值。
91.(11)体内抗肿瘤性质评估:选用4~6周的balb/c-nu小鼠,通过皮下注射混合含体积分数为10%基质胶的含有107个细胞的细胞悬液到小鼠腹股沟的方法,构建荷瘤小鼠模型。至肿瘤体积至90mm3时,将所述小鼠随机分成4组,每组3只,分别命名为:pbs组、cur组、ala+laser组、nps+laser组,4组的小鼠分别通过尾静脉注射:1
×
pbs、0.2mg/ml姜黄素、0.77mg/ml ala、1mg/ml实施例1中制备得到的金属-药物全活性纳米药物(nps),200μl/只。其中ala+laser组和nps+laser组在注射药物后24小时用激光(635nm,300mw/cm2)照射肿瘤部位10分钟。对上述小鼠继续培养14天,每两天记录肿瘤体积,绘制变化曲线,评估nps的体内抗肿瘤性质。
92.2.对实施例2~12制备得到的金属-药物全活性纳米药物进行形貌测试:透射电子显微镜,型号:ht-7700,日立,日本;将所制得的样品滴在铜网上,干燥后进行形貌观察。
93.3.对实施例9制备得到的金属-药物全活性纳米药物进行体外抗炎性能考察。
94.实验步骤:l02细胞经2ml胰蛋白酶消化1.5分钟后接种在3个共聚焦小皿中,分别命名为pbs组、h2o2组、h2o2+nps组,在37℃含体积分数为5%的co2的培养箱中用dmem完全培养基培养过夜。去除旧培养基,pbs组、h2o2组h2o2+nps组用1
×
pbs孵育,h2o2+nps组细胞用含有实施例9中制备得到的金属-药物全活性纳米药物(nps)(50μg/ml,250μl)的1ml dmem完全培养基孵育,继续培养6小时;随后,pbs组无需进一步处理,h2o2组、h2o2+nps组中分别吸弃旧培养基并加入含30μm过氧化氢的dmem完全培养基1ml,继续培养3小时。然后,所有实验组,用含10μm ros探针(2',7'-二氯荧光黄双乙酸盐,dcfh-da)的dmem培养基1ml培养30分钟。培养结束后,1
×
pbs清洗,用激光共聚焦显微镜观察不同组别的ros含量。
95.4.对实施例10制备得到的金属-药物全活性纳米药物进行体外抗肿瘤能力考查。实验通过标准mtt比色法测定实施例中制备得到的金属-药物全活性纳米药物(nps)的体外细胞毒性。
96.实验步骤:将panc-1细胞接种于96孔板中(1万个细胞/孔),在37℃含体积分数为5%的co2的培养箱中培养过夜;用含nps(0、100μg/ml)的dmem完全培养基孵育(每个浓度设置5个复孔),继续在37℃含体积分数为5%的co2的培养箱中培养24小时;随后,在避光的条件下,加入mtt(含0.5mg/ml的dmem完全培养基,100μl/孔);在37℃含体积分数为5%的co2的培养箱中培养4小时后,加入dmso(110μl/孔),震荡15分钟,用酶标仪(a51119600dpc,thermofisher,美国)测定各孔在490nm处的吸光度值。
97.5.对实施例11制备得到的金属-药物全活性纳米药物进行体外抗肿瘤能力考查。实验通过标准mtt比色法测定实施例11中制备得到的金属-药物全活性纳米药物(nps)的体外细胞毒性。
98.实验步骤:将a549细胞接种于96孔板中(1万个细胞/孔),在37℃含体积分数为5%的co2的培养箱中培养过夜;用含nps(0、100μg/ml)的rmi 1640完全培养基孵育(每个浓度设置5个复孔),继续在37℃含体积分数为5%的co2的培养箱中培养24小时;随后,在避光的条件下,加入mtt(含0.5mg/ml的rmi 1640完全培养基,100μl/孔);在37℃含体积分数为5%
的co2的培养箱中培养4小时后,加入dmso(110μl/孔),震荡15分钟,用酶标仪(a51119600dpc,thermofisher,美国)测定各孔在490nm处的吸光度值。
99.实施例1
100.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
101.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
102.(1)分别配制1mm的氯化铁的水溶液、1mm的姜黄素的四氢呋喃溶液和1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
103.然后,在搅拌的条件下,依次将1ml的1mm 5-氨基酮戊酸溶液、0.05ml的1mm氯化铁溶液、0.2ml的1mm的姜黄素溶液滴加到2ml的去离子水中,25℃,750rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;
104.(2)将步骤(1)得到的金属-药物全活性纳米药物溶液通过50kda超滤管超滤纯化,得到本实施例所述一种金属-药物全活性纳米药物;所述全活性纳米药物为溶液状态,4℃储存待用。
105.实施例2
106.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
107.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
108.(1)分别配制1mm的氯化铁的水溶液、1mm的姜黄素的四氢呋喃溶液、1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加三乙胺溶液并测试ph值至ph=7.0,利用三乙胺溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
109.然后,在搅拌的条件下,依次将1ml的1mm 5-氨基酮戊酸溶液、0.05ml的1mm氯化铁溶液、0.2ml的1mm的姜黄素溶液滴加到5ml的去离子水中,45℃,600rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;
110.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
111.实施例3
112.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
113.一种本实施例所述全活性纳米药物的制备方法,所述方法具体步骤如下:
114.(1)分别配制1mm的氯化铁的水溶液、1mm的姜黄素的甲醇溶液、1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph
=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
115.然后,在搅拌的条件下,依次将1ml的1mm 5-氨基酮戊酸溶液、0.05ml的1mm氯化铁溶液、0.2ml的1mm的姜黄素溶液滴加到2ml的去离子水中,25℃,1500rpm连续搅拌反应20分钟,得到待纯化的金属-药物全活性纳米药物溶液;
116.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
117.实施例4
118.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
119.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
120.(1)分别配制1mm的氯化铁的水溶液、1mm的姜黄素的四氢呋喃溶液、1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
121.在搅拌的条件下,依次将0.8ml的1mm 5-氨基酮戊酸溶液、0.05ml的1mm氯化铁溶液、0.2ml的1mm的姜黄素溶液滴加到2.2ml的去离子水中,25℃,300rpm连续搅拌反应60min分钟,得到待纯化的金属-药物全活性纳米药物溶液。
122.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
123.实施例5
124.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
125.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
126.(1)配置1mm的氯化铁的水溶液、1mm的姜黄素的四氢呋喃溶液、1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
127.在搅拌的条件下,依次将1ml的1mm 5-氨基酮戊酸溶液、0.05ml的1mm氯化铁溶液、0.2ml的1mm的姜黄素溶液滴加到3ml的去离子水中,25℃,900rpm连续搅拌反应10分钟,得到待纯化的金属-药物全活性纳米药物溶液。
128.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
129.实施例6
130.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、
5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
131.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
132.(1)分别配制1mm的氯化铁的水溶液、1mm的姜黄素的四氢呋喃溶液、1mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
133.然后,在搅拌的条件下,依次将1ml的1mm 5-氨基酮戊酸溶液、0.01ml的1mm氯化铁溶液、1ml的1mm的姜黄素溶液滴加到不含去离子水的圆底烧瓶中,25℃,500rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液。
134.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
135.实施例7
136.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
137.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
138.(1)分别配置5mm的氯化铁的水溶液、5mm的姜黄素的四氢呋喃溶液、5mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
139.然后,在搅拌的条件下,依次将0.5ml的5mm 5-氨基酮戊酸溶液、0.5ml的5mm氯化铁溶液、0.5ml的5mm的姜黄素溶液滴加到不含去离子水的圆底烧瓶中,25℃,800rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液。
140.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
141.实施例8
142.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、5-氨基酮戊酸和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
143.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
144.(1)分别配制10mm的氯化铁的水溶液、50mm的姜黄素的四氢呋喃溶液、5mm的盐酸化5-氨基酮戊酸的水溶液,向盐酸化5-氨基酮戊酸溶液中滴加5m氢氧化钠水溶液并测试ph值至ph=7.0,利用氢氧化钠水溶液对盐酸化5-氨基酮戊酸溶液进行脱盐酸处理;
145.然后,在搅拌的条件下,依次将0.1ml的10mm 5-氨基酮戊酸溶液、0.1ml的50mm氯化铁溶液、0.5ml的5mm的姜黄素溶液滴加到0.75ml的去离子水中,25℃,800rpm连续搅拌反
应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;
146.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
147.实施例9
148.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子和姜黄素通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
149.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
150.(1)分别配制1mm的氯化铁的水溶液和5mm的姜黄素的四氢呋喃溶液;
151.然后,在搅拌的条件下,依次将0.05ml的1mm氯化铁溶液、0.2ml的5mm的姜黄素溶液滴加到2ml的去离子水中,25℃,800rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;
152.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
153.实施例10
154.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、过氧化物酶(c0278-6.25ku)和由亚油酸氧化制备得到的亚油酸氢过氧化物通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
155.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
156.(1)分别配制1mm的氯化铁的水溶液、21.818units/ml的过氧化物酶的水溶液和200mm的亚油酸氢过氧化物的乙醇溶液;
157.然后,在搅拌的条件下,依次将0.8ml的21.818units/ml过氧化物酶溶液、0.1ml的1mm氯化铁溶液、0.35ml的200mm的亚油酸氢过氧化物溶液滴加到4ml的去离子水中,37℃,800rpm连续搅拌反应40分钟,得到待纯化的金属-药物全活性纳米药物溶液;所述药物溶液中金属离子和活性药物成分的物质的量之比为1:700;
158.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
159.实施例11
160.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是三价铁离子、可抑制细胞自噬的质粒和索拉菲尼通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
161.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
162.(1)分别配制1mm的氯化铁的水溶液、10μg/ml的质粒的水溶液和10mm的索拉菲尼的四氢呋喃溶液;
163.然后,在搅拌的条件下,依次将5μl的10μg/ml的质粒溶液、0.02ml的1mm氯化铁溶液、5μl的10mm的索拉菲尼溶液滴加到2ml的去离子水中,37℃,800rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;所述药物溶液中金属离子和活性药物成分的物质的量之比为1:2.5;
164.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
165.实施例12
166.一种金属-药物全活性纳米药物,所述金属-药物全活性纳米药物是二价镁离子、神经营养因子和白藜芦醇通过金属-配位作用和分子间弱作用力结合在一起形成的纳米颗粒。
167.一种本实施例所述金属-药物全活性纳米药物的制备方法,所述方法具体步骤如下:
168.(1)分别配制1mm的氯化镁的水溶液、10ng/ml的神经营养因子的水溶液和1mm的白藜芦醇的四氢呋喃溶液;
169.然后,在搅拌的条件下,依次将0.8ml的10ng/ml神经营养因子溶液、0.04ml的1mm氯化镁溶液、0.2ml的1mm的白藜芦醇溶液滴加到3ml的去离子水中,37℃,800rpm连续搅拌反应30分钟,得到待纯化的金属-药物全活性纳米药物溶液;所述药物溶液中金属离子和活性药物成分的物质的量之比为1:5;
170.(2)将步骤(1)得到的待纯化的金属-药物全活性纳米药物溶液利用透析袋(mw3500)透析纯化,得到本实施例所述一种金属-药物全活性纳米药物,所述全活性纳米药物为溶液状态,4℃储存待用。
171.对以上实施例制备得到的金属-药物全活性纳米药物进行如下分析:
172.(1)形貌分析,实施例1制备得到的金属-药物全活性纳米药物形貌如图1所示,所述全活性纳米药物为具有核-壳球形结构的纳米颗粒,平均粒径约为65nm;实施例2~8与实施例1相比,虽然所制备的金属-药物全活性纳米药物都是基于三价铁、ala和姜黄素,但实施例2~5改变水溶液体积、转速、反应温度、反应时间参数,制备得到的金属-药物全活性纳米药形貌变化不明显,因此实施例2~5制备得到的金属-药物全活性纳米药物进行形貌分析测试结果与实施例1相似。
173.而实施例6~8改变不同活性成分浓度、体积,对所形成的金属-药物全活性纳米药物的形貌影响较大,因此实施例6制备得到的金属-药物全活性纳米药物进行形貌如图2所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约在200nm;实施例7制备得到的金属-药物全活性纳米药物进行形貌如图3所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约在40nm;实施例8制备得到的金属-药物全活性纳米药物进行形貌如图4所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约在20nm。
174.实施例9制备得到的全活性纳米药物进行形貌如图5所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约为60nm。
175.实施例10制备得到的全活性纳米药物进行形貌如图6所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约为300nm。
176.实施例11制备得到的全活性纳米药物进行形貌如图7所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约为500nm。
177.实施例12制备得到的全活性纳米药物进行形貌如图8所示,所述全活性纳米药物为具有球形结构的纳米颗粒,粒径约为1300nm。
178.(2)表面电势测试:
179.对实施例1制备得到的金属-药物全活性纳米药物进行表面电势分析,结果如图9所示,测得该样品表面平均电荷为-16.1mv,可以得知本实施例所制备的金属-药物全活性纳米药物具有较好的生物相容性,同时由于粒子间排斥力大于吸引力,还具有良好的稳定性。
180.实施例2~8的测试结果与实施例1相类似,制备得到的金属-药物全活性纳米药物具有较好的生物相容性,同时由于粒子间排斥力大于吸引力,还具有良好的稳定性。
181.实施例9~12的制备得到的金属-药物全活性纳米药物溶液呈负电,具有较好的生物相容性和良好的稳定性。
182.(3)稳定性测试:将实施例1中制备得到的金属-药物全活性纳米药物溶液在室温下放置14天,观察其稳定性。结果如图10所示,在14天中,纳米药物溶液始终未见明显聚集沉淀产生,证明其具有良好的颗粒稳定性。
183.实施例2~8制备得到的金属-药物全活性纳米药物溶液测试结果与实施例1相同,在室温下放置14天,纳米药物溶液始终未见明显聚集沉淀产生,证明其具有良好的颗粒稳定性。
184.将实施例9~12制备得到的金属-药物全活性纳米药物溶液在室温下放置14天,纳米药物溶液始终未见明显聚集沉淀产生,证明其具有良好的颗粒稳定性。
185.(4)作用力分析:
186.对实施例1所制备的金属-药物全活性纳米药物的形成的主要驱动力进行分析,结果如图11所示,加入去离子水(water)的样品,结构未发生变化,聚集态的姜黄素分析发生聚集荧光淬灭,无明显荧光信号。加入四氢呋喃(thf)的样品,由于thf为姜黄素的良性溶剂,使该金属-药物全活性纳米药物完全瓦解,姜黄素恢复分子态,具有明显的荧光信号。加入氯化钠(nacl)、尿素(urea)、十二烷基硫酸钠(sds)、乙二胺四乙酸(edta)分别可以破坏纳米结构中的静电作用、氢键、疏水作用、配位作用,使纳米结构瓦解,姜黄素由聚集态变为游离态,恢复荧光信号。由图11可知,加入sds的样品荧光信号明显,加入edta的样品,也有一定程度的荧光信号,由此可见实施例1所制备的金属-药物全活性纳米药物的形成的驱动力有疏水作用和金属-有机配位作用。此外,由于fe
3+
与ala部分带不同电荷,姜黄素中含有共轭基团,该全活性纳米药物的驱动力中还受静电作用以及π-π堆叠的影响。
187.实施例2~8的作用力分析结果与实施例1相类似。
188.对实施例9所制备的金属-药物全活性纳米药物的形成的驱动力进行分析可知,所述驱动力为姜黄素之间的疏水作用、π-π堆叠以及fe
3+
与姜黄素之间的金属-有机配位作用。
189.对实施例10所制备的金属-药物全活性纳米药物的形成的驱动力进行分析可知,所述驱动力为fe
3+
与过氧化物酶和亚油酸氢过氧化物之间的金属-有机配位作用,同时协同疏水作用、π-π堆叠、氢键和静电作用。
190.对实施例11所制备的金属-药物全活性纳米药物的形成的驱动力进行分析可知,
fe
3+
与抑制细胞自噬的质粒和索拉菲尼之间的金属-有机配位作用,同时协同疏水作用、π-π堆叠、氢键和静电作用。
191.对实施例12所制备的金属-药物全活性纳米药物的形成的驱动力进行分析可知,mg
2+
与神经营养因子和白藜芦醇之间的金属-有机配位作用,同时协同疏水作用、π-π堆叠、氢键和静电作用。
192.(5)肿瘤细胞内生成原卟啉(ppix)性能分析
193.对实施例1中制备得到的金属-药物全活性纳米药物在肿瘤细胞内生成ppix性能分析,ala是美国食品和药物管理局(fda)批准的光敏剂前药,其在细胞内代谢产生的ppix,在外源性光源激发的条件下,可以产生ros杀伤肿瘤细胞,实现光动力治疗。通过共聚焦激光扫描显微镜,分析实施例1中制备得到金属-药物全活性纳米药物(nps)中ala在细胞内代谢产生ppix的荧光信号。由图12可知,pbs组作为阴性对照,无ppix荧光信号;nps组显示比ala组显示明显的ppix红色荧光信号,表明nps中包含更大量的ala,可代谢产生了ppix。
194.实施例2~8的肿瘤细胞内生成原卟啉(ppix)性能测试结果与实施例1相类似。
195.(6)肿瘤细胞内产生胆绿素(bv)性能分析
196.对实施例1中制备得到的金属-药物全活性纳米药物在肿瘤细胞内产生bv性能分析:ala在细胞内代谢产生的ppix之后,在其代谢的下游通路中会进一步产生bv,而bv是一种已报道的光热剂,在外源性光源激发的条件下,产生热量杀伤肿瘤细胞,实现热治疗。通过共聚焦激光扫描显微镜,分析实施例1中制备得到的金属-药物全活性纳米药物(nps)中ala在细胞内代谢产生bv的荧光信号。由图13可知,pbs组作为阴性对照,无bv荧光信号;nps组显示比ala组显示明显的bv绿色荧光信号,表明nps中包含更大量的ala,可代谢产生了bv。
197.实施例2~8的肿瘤细胞内产生胆绿素(bv)性能测试结果与实施例1相类似。
198.(7)肿瘤细胞内产生一氧化碳(co)性能分析:
199.对实施例1中制备得到的金属-药物全活性纳米药物在肿瘤细胞内产生co性能分析:ala在细胞内代谢产生的ppix之后,在其代谢的下游通路产生bv的同时,也会进一步产生co。当细胞内co浓度高时,癌细胞中的ros水平将大大增加,从而杀死肿瘤细胞,实现气体治疗。通过共聚焦激光扫描显微镜,利用fl-co-1探针,分析实施例1中制备得到的金属-药物全活性纳米药物(nps)中ala在细胞内代谢产生co的荧光信号。由图14可知,pbs组作为阴性对照,无co荧光信号;nps组显示比ala组显示明显的co荧光信号,表明nps中包含更大量的ala,可代谢产生了co。
200.实施例2~8的肿瘤细胞内产生一氧化碳(co)性能测试结果与实施例1相类似。
201.(8)肿瘤细胞内引起铁死亡性能考察:对实施例1中制备得到的全活性纳米药物在肿瘤细胞内引起铁死亡的性能考察:实施例1中制备得到的全活性纳米药物(nps)中的三价铁离子被肿瘤细胞中高表达的谷胱甘肽还原为二价铁离子,随后通过芬顿反应产生羟基自由基(
·
oh),
·
oh在膜脂上通过脂质过氧化形成lipid ros,引发铁死亡。使用共聚焦激光扫描显微镜,通过c11 bodipy581/591探针表征铁死亡信号。如图15所示,相比于pbs对照组,nps组显示明显的lipid ros信号,并且在加入铁离子螯合剂dfo后,lipid ros的信号明显减弱,证明nps在细胞中确实引起了铁死亡。
202.实施例2~8的肿瘤细胞内肿瘤细胞内引起铁死亡性能结果与实施例1相类似。
203.(9)光照条件下,在肿瘤细胞内产生活性氧(ros)的性质验证:
204.实施例1中制备得到的金属-药物全活性纳米药物在光照条件下,在肿瘤细胞内产生ros的性质验证:光照条件下,在肿瘤细胞内产生ros的性质验证:ala代谢产生的ppix是一种光敏剂,在外源性光源激发的条件下,可以产生ros杀伤肿瘤细胞,实现光动力治疗。利用ros探针dcfh-da,通过共聚焦显微镜观察实施例1中制备得到的全活性纳米药物(nps)中ala在细胞内实现光动力治疗的能力。如图16所示,相比于对照的pbs组和laser组未观察到ros绿色荧光信号,ala组能观察到较弱的ros信号,nps组的ros绿色荧光信号明显强于ala组,表明其具有更好的光动力性质。
205.实施例2~8的肿瘤细胞内肿瘤细胞内产生活性氧(ros)的性质验证结果与实施例1相类似。
206.(10)体外抗肿瘤能力考查:
207.实施例1中制备得到的金属-药物全活性纳米药物在体外抗肿瘤能力考查结果如图17所示,用实施例1制得的全活性纳米药物(nps)培养的a549细胞的存活率随其浓度增加而降低,表明所述全活性纳米药物对肿瘤细胞具有很强的杀伤作用。实施例2~8的体外抗肿瘤能力考查结果与实施例1相类似,实施例2~8制得的全活性纳米药物(nps)培养的a549细胞的存活率随其浓度增加而降低,表明所述全活性纳米药物对肿瘤细胞具有很强的杀伤作用。
208.对实施例10中制备得到的金属-药物全活性纳米药物在体外抗肿瘤能力考查结果如图20所示,与对照组(pbs组)相比,用实施例10制得的金属-药物全活性纳米药物(nps)培养的panc-1人胰腺癌细胞的存活率显著降低,表明所述全活性纳米药物有理想的抗肿瘤作用。
209.对实施例11中制备得到的金属-药物全活性纳米药物在体外抗肿瘤能力考查结果如图21所示,与对照组(pbs组)相比,用实施例11制得的金属-药物全活性纳米药物(nps)培养的a549细胞的存活率显著降低,表明所述全活性纳米药物对肿瘤细胞具有明显的杀伤作用。
210.(11)体内抗肿瘤性质评估:
211.实施例1中制备得到的金属-药物全活性纳米药物在体内抗肿瘤性质评估结果如图18所示,与不同对照组相比,实施例1中制备得到的金属-药物全活性纳米药物治疗组的小鼠肿瘤被有效抑制,证明所述全活性纳米药物具有良好的体内抗肿瘤活性。
212.实施例2~8的体内抗肿瘤能力考查结果与实施例1相类似,与不同对照组相比,实施例2~8中制备得到的金属-药物全活性纳米药物治疗组的小鼠肿瘤被有效抑制,证明所述全活性纳米药物具有良好的体内抗肿瘤活性。
213.(12)体外抗炎性能考察:
214.实施例9中制备得到的金属-药物全活性纳米药物在体外抗炎能力考查结果如图19所示,用实施例9制得的全活性纳米药物(nps)中姜黄素可有效清除由过氧化氢培养后的l02正常人类肝脏细胞中高表达的ros,起到抗氧化作用,缓解炎症。使用共聚焦激光扫描显微镜,通过ros探针评估细胞内ros含量。如图18所示,相比于pbs对照组,过氧化氢处理的细胞显示明显的ros信号,并且在nps后,ros的信号明显减弱,证明nps可起到抗氧化作用,缓解炎症。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1