主动脉疾病遗传突变自动化解读系统(HTAADVar)
主动脉疾病遗传突变自动化解读系统(htaadvar)
技术领域
1.本技术属于生物信息学分析技术领域,具体涉及一种htaad疾病突变自动化解读系统及其构建方法。
背景技术:
2.随着下一代测序技术的快速发展,基因检测被广泛应用于临床,可辅助遗传性疾病的分子诊断、高危人群疾病筛查和指导生育等。为了准确地评估突变的临床致病性,2015年美国医学遗传学和基因组学学院(acmg)和分子病理学协会(amp)制定了遗传病突变解读标准和指南,极大地推动了临床解读的规范性。但是,由于acmg/amp指南并没有明确规定每一项评判标准的细节和参数,不同的数据解读人员会根据各自的经验和对指南的理解指定具体的解读细则,花费大量人力手工从文献和数据库中搜索各类证据从而进行解读,因此解读过程耗时耗力,且因个人获取信息的能力,具体解读指标和阈值的不同,不同实验室解读一致性往往并不高。为了解决这些瓶颈问题,研究人员开发了解读工具intervar(https://wintervar.wglab.org/)来辅助解读,但其解读过程依赖于目前大型公共数据库,如clinvar和gnomad。此类数据库并非为了突变解读而设计,且大多直接由用户上传,没有经过专家评估,往往存在突变疾病命名不标准,突变致病性分类标准不一致,缺乏解读关键数据,如家系共分离、遗传方式等,限制了解读的准确性。此外,intervar这类通用解读软件目的是能辅助所有遗传性疾病解读,因此在解读过程中缺乏对基因和疾病特异性的考量,在特定疾病的解读中往往表现不佳。开发针对具体疾病的自动解读系统成为了本领域科研人员亟待解决的问题和研究的重点。
3.当前,研究者试图在不同疾病中开发了一些疾病特异性解读工具,譬如听力损伤突变解读工具vip-hl、癌症突变解读工具vic和专门针对brca1的软件nextprot。在心血管疾病领域,也有类似的解读工具发布,例如cardioclassifier和cardiovai。但上述解读工具虽然考虑到了疾病和基因的一些特点,解读结果较通用工具表现有所提高,但它们均为半自动化解读工具,解读过程主要还是从clinvar和gnomad获取公共数据,其解读结果的准确度极大依赖于用户输入的解读证据,在解读效率上并没有很大提高,并未解决解读中的瓶颈问题。因此,如何解决上述难点,实现疾病突变解读的全自动化是亟待解决的问题。
4.主动脉疾病是一种慢性进展性疾病且发病隐匿,如不及时诊断和治疗往往会出现严重的不良事件,导致死亡。有鉴于此,提出本技术。
技术实现要素:
5.针对上述现有技术缺陷,本技术的核心目的是寻求一种能够对突变位点进行自动化解读的系统。
6.为达到上述目的,本技术所采用的技术方案如下:
7.本技术首先提供一种遗传疾病突变数据库的构建方法,其特征在于,包括如下步骤:
8.1)疾病相关基因确定:
9.依据遗传学研究数据、基因与疾病功能学研究数据,和/或结合自产患者及其家属的临床测序数据,确定疾病相关基因;
10.2)文献收集及数据收录:
11.基于相关基因进行文献检索,并进行文献过滤和数据收录;
12.所述文献过滤和数据收录包括:针对每篇文献进行人工审阅,对文献信息、突变信息、家系信息、样本信息及功能学研究结果进行突变解读关键信息的收录;
13.3)突变注释:
14.根据文献提供的突变信息,进行突变注释;
15.所述注释内容包括突变类型、外显子编号、内含子编号、所在蛋白结构域、人群频率、基因组重复区段、突变有害性的多软件预测结果;
16.4)数据库构建
17.根据步骤2)和3)获得的数据,建立底层数据库表,并进行存储;
18.所述底层数据库表包括research表、researchtovar表、sample表、sampletovar表、familytovar表、functional_study表、lit表和variants表。
19.在一些方式中,步骤2)中,所述文献过滤和数据收录具体为:对文献中每个突变携带人数进行核查,排除已在先前发表文献中报道过的样本;记录突变携带者的具体信息:突变遗传方式(优选的,所述遗传方式为新发突变或遗传自父/母),是否是嵌合突变,携带者是否在先前文献中报道过,携带者是否具有疾病家族史,突变携带者的表型或家族史是否高度符合某种单基因遗传疾病(比如表型是否对马凡/血管型ehlers-danlos综合征/loeys-dietz综合征高度特异);记录突变在家系中的携带情况,计算共分离值;对于功能学研究,按照实验类型(优选的,所述实验类型为动物实验/细胞实验)和模拟表型的特异性(优选的,所述特异性为否直接模拟出患者表型或导致蛋白功能异常)把功能学证据分成不同类型。
20.在一些方式中,所述步骤3)的突变注释后还包括人工校对步骤:注释后对突变进行统一清洗,过滤因文献提供信息不足导致无法注释的突变、将突变信息统一为hgvs命名规则,将参考基因组坐标统一注释到grch37/hg19参考基因组。
21.在一些方式中,所述步骤3)中,所述注释采用vep软件和内部程序;
22.所述突变有害性预测结果根据预测软件原理不同分为四类:1)进化保守性预测算法:sift、mutation assessor和fathmm;2)基于蛋白质结构和功能与进化保守性预测算法:polyphen2 hdiv、polyphen2 hvar和mutation taster;3)综合预测算法:cadd;4)可变剪接预测算法:dbscsnv、spliceai和spidex。
23.在一些方式中,步骤4)中,所述存储采用mongodb数据库存储上述数据;所述research表是对文献中样本信息的描述,包括家系个数、患者个数、对照个数、种族、人群、性别比例、年龄比例、实验方法和研究类型;所述researchtovar表给出每篇文章收录的突变以及携带该突变的先证者个数;所述sample表记录突变携带者信息,包括样本编号、家系关系、性别、年龄、诊断结果、具体临床表型、是否有家族史、表型特异性;所述sampletovar表记录突变和携带者的对应关系及突变的遗传方式、杂合度、样本突变是否在之前文献报道;所述familytovar表包含突变和家系的对应关系及家系共分离值;所述functional_
study表包含针对突变的体内外功能实验信息及对蛋白功能和剪接影响的证据分级;所述lit表包含所有收录文献的基本信息;所述variants表包含所有收录突变的注释和解读信息。
24.在一些方式中,上述所述疾病可以为遗传性主动脉病htaad;
25.在一些方式中,所述步骤1)中的相关基因包括:acta2、col3a1、fbn1、lox、myh11、mylk、prkg1、smad3、tgfb2、tgfbr1、tgfbr2、bgn、foxe3、hcn4、mat2a、mfap5、smad2和tgfb3。
26.本技术还提供一种疾病突变数据库,所述数据库通过上述方法进行构建。
27.本技术还提供一种遗传突变自动化解读程序的构建方法,其特征在于,包括如下步骤:
28.1)制定解读规范
29.依据acmg/amp遗传病突变解读标准和指南,及clingen序列突变解释(svi)专家组对其规则的细化和更新,制定解读规范;
30.优选的所述规范:1)纳入pvs1最新解读指南并提出具体实现算法;2)阈值的界定和解读依据基本来自于构建的突变数据库;3)对照人群也使用内部明确排除相关疾病的人群数据;4)新发突变增加考虑父/母为嵌合,但无症状或症状轻、孩子为患者的情况;5)按照模型和模拟表型的差异,对功能学研究结果进行了不同证据水平的分类;6)在公共数据中查找突变时同时考虑此位点的测序深度,在满足一定深度的情况下确定突变是否存在;7)对家系共分离进行定量并在不同家系中做累计,根据不同证据水平进行打分;8)多种软件预测工具先按照算法原理不同分类,以大多数类型的预测结果作为突变的最终预测结果。
31.2)获取标准数据:读取用户输入数据及上述疾病突变数据库中存储的标准化数据;
32.优选的,所述用户输入包括待解读突变和解读辅助证据;所述标准化数据包括:突变携带者数量、突变遗传方式、突变杂合度、携带者表型特异性、家系共分离值和突变的功能学研究结果;
33.3)引入突变注释:根据用户输入的突变信息,采用vep软件和内部程序进行自动化注释;
34.优选的,所述注释信息包括人群频率、蛋白结构域、基因组重复区域、突变位置保守型、突变有害性预测和可变剪接预测;
35.4)建立突变解读:基于1)的解读规范,提出每条规则具体的阈值设定、升降级方法和解读方法,自动进行突变解读;
36.5)结果展示:基于突变解读结果及解读依据自动生成突变解读结果报告;
37.优选的,所述突变解读结果包括突变致病性分类:“致病”、“可能致病”、“临床意义不明”、“可能良性”或“良性”及详细的解读依据。
38.本技术还提供一种遗传突变自动化解读程序,特征在于,是利用上述遗传突变自动化解读程序的构建方法构建而成。
39.本技术还提供一种htaad突变自动化解读系统,包括如下模块:
40.1)突变预解读模块:用于上述自动化解读程序和上述疾病突变数据库中存储的标准化数据,对突变数据库中的所有突变预先进行致病性解读,最终致病性分类和解读依据一并存入数据库variants表中;
41.2)输入突变解读模块:用于读取用户输入,判读输入的待解读突变是否在于权利要求1-6任一所述构建方法构建的突变数据库;如在且用户未提供额外证据,系统将直接获取数据库中保存的致病性分类和解读依据;如在且用户提供了额外证据,系统将根据突变数据库数据和用户提供的额外证据重新对突变进行解读;如未在数据库中,系统将对突变进行自动化解读,以获取最终分类。
42.3)系统交互模块:用于提供可视化交互页面。
43.本技术还提供一种计算机可读介质,其存储有计算机程序,所述计算机程序被处理器执行时,实现上述任一所述构建方法。
44.本技术还提供一种电子设备,包括处理器以及存储器,所述存储器上存储一条或多条可读指令,所述一条或多条可读指令被所述处理器执行时,实现上述任一所述方法。
45.本技术还提供上述自动化解读程序或上述疾病突变数据库在htaad突变自动化解读中的应用。
46.与现有技术相比,本技术至少具有如下优势:
47.1)现有疾病特异的解读工具在很大程度上仍然依赖于公共疾病数据库来解释一些重要证据,这些数据库中大多数突变是直接从文献或用户上传数据中获取的,数据质量存在很多问题,比如录入标准不一、致病性分类不一致以及对同一突变未经考察的重复报道等。本技术构建的解读系统在解读工具中引入自建突变数据库,同时对每个突变的基因组区域、分子功能和表型信息进行注释,以支持深入的突变解读,具有灵敏准确等优势。
48.2)相比于现有技术,比如intervar或cardioclassifier等,本技术的htaadvar在检测灵敏度和特异性方面优于这些工具至少2-5倍。此外,针对临床患者检测,采用本技术的htaadvar工具对患者的突变位点进行解读,其“可能致病/致病”突变的检出率为43.04%,“可能致病/致病/临床意义不明”突变的检出率为70.89%,这与以往报道的人工解读对应的检出率一致性高,表明其在临床应用中表现良好。
49.3)本技术构建的htaad疾病突变自动化解读系统,能够帮助解读人员快速地依照acmg/amp遗传病突变解读标准和指南,及clingen序列突变解释(svi)专家组对其规则的细化和更新进行解读,同时允许用户输入自己的证据,极大减少解读过程的人力投入,提高解读效率。本系统解读后还能够提供每个突变解读标准的详细证据,供用户审核。
附图说明
50.为了更清楚地说明本技术具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本技术的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
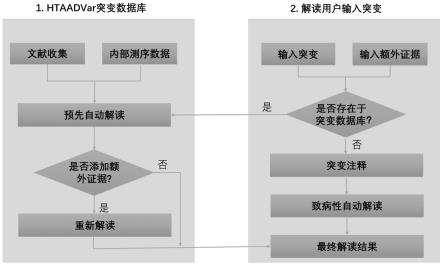
51.图1、本技术遗传疾病突变自动化解读系统流程图;
52.图2、本技术网站页面图;
53.图3、htaadvar与intervar、cardioclassifier的解读结果比较图。
具体实施方式
54.下面将结合附图对本技术的技术方案进行清楚、完整地描述,显然,所描述的实施
tgfbr2 or mfs2 or tgfbr1 or msse or ess1 or prkg1 or prkgr1b or prkg1b or tgfb2 or acta2 or myh11 or mylk or lox or bgn or hcn4 or mat2a or mfap5)not review[publication type]and("1000/01/01"[date-publication]:"2020/10/20"[date-publication]),共检索到1564篇研究文章。为确保遗传突变收录的完整性,本技术同时从clinvar和人类基因突变数据库(hgmd)中检索相关基因和疾病文献,另补充了617篇文章。针对此2181篇文献,通过文献摘要阅读进行人工审查以删除不相关的文章,最终保留了697篇相关文献。此外,本技术还整合了实验室内部790名htaad患者及其家属的遗传突变和表型数据。基于上述两组数据,本技术构建的htaadvar数据库共收集上述18个基因中共计4379个高质量突变。
[0069]
2)针对每篇文献,由研究人员人工审阅,对文献信息、突变信息、家系信息、样本信息及功能学研究结果按照一定规范和格式进行收录,包括突变解读的关键信息。对于文献中每个突变携带的人数进行核查,排除已在之前发表文献中报道过的样本,避免因样本复用导致过多计算突变携带人数;记录突变携带者的具体信息:突变遗传方式(优选的,所述遗传方式为新发突变或遗传自父/母),是否是嵌合突变,携带者是否在先前文献中报道过,是否具有疾病家族史,突变携带者的表型或家族史是否高度符合某种单基因遗传疾病(比如其表型是否对马凡/血管型ehlers-danlos综合征/loeys-dietz综合征高度特异);记录突变在家系中的携带情况,采用jarvik方法[19]计算共分离值;对于功能学研究,按照实验类型(动物实验/细胞实验)和模拟表型的特异性(是否直接模拟出了患者表型还是导致蛋白功能异常)把功能学证据分成不同的类型。对于收录的所有元数据(meta-data)进行双人核查校对,对疾病命名进行统一。
[0070]
3)突变的注释和标准化。根据文章提供的突变信息,采用vep软件和内部程序进行注释和人工校对,注释内容包括突变类型、外显子编号、内含子编号、所在蛋白结构域、人群频率,基因组的重复区段,突变有害性的多软件预测结果等,其中突变有害性预测结果根据预测软件原理不同分为四类:1)进化保守性预测算法:sift、mutationassessor和fathmm;2)基于蛋白质结构和功能与进化保守性预测算法:polyphen2 hdiv、polyphen2 hvar和mutationtaster;3)综合预测算法:cadd;4)可变剪接预测算法:dbscsnv、spliceai和spidex。另外,采用maxentsan工具对pvs1证据中是否存在新的或者隐藏的可变剪接位点进行预测。注释后对突变进行统一清洗,过滤掉因文献提供信息不足导致无法注释的突变、将不标准的突变信息统一为目前学术界公认的hgvs命名规则、将参考基因组坐标统一注释到grch37/hg19参考基因组。此突变信息的清洗和标准化解决了因作者发表时期不同、基因组版本不同、书写错误等原因导致的突变命名有差异,无法直接合并比较。
[0071]
4)建立底层数据库表、构建突变数据库。
[0072]
针对收录的元数据,设计research、researchtovar、sample、sampletovar、familytovar、functional_study、lit和variants八张底层数据库表,详见表1-8。research表是对文献中样本信息的详细描述,包括家系的个数、患者个数、正常对照样本个数、种族、人群、性别比例、年龄比例、实验方法、研究类型等;researchtovar表具体给出每篇文章收录的突变以及携带该突变的先证者个数;sample表记录突变携带者信息,包括样本编号、家系关系、性别、年龄、诊断结果、具体临床表型、是否有家族史、表型特异性;等;sampletovar表记录了突变和携带者的对应关系及突变的遗传方式、杂合度、此样本此突变是否在之前
文献中被报道过;familytovar表包含突变和家系的对应关系及家系共分离值;functional_study表包含针对突变的体内外功能实验信息及对蛋白功能和剪接影响的证据分级;lit表包含所有收录文献的基本信息;variants表包含所有收录突变的注释和解读信息。因mongodb数据库具有高性能、易部署、易使用和存储方便等优点,用于存储上述数据,从而建立交互网站,供用户浏览查询收录数据。其中researchtovar、sample、sampletovar、familytovar、functional_study和variant六个表用于突变解读。
[0073]
表1.research表
[0074][0075]
表2.researchtovar表
[0076][0077]
表3.sampletovar表
[0078][0079]
表4.familytovar表
[0080][0081][0082]
表5.sample表
[0083][0084]
表6.functional_study表
[0085][0086]
表7.lit表
[0087]
[0088][0089]
表8.variants表
[0090]
[0091][0092]
本实施例进一步对数据库性能进行评估,与目前普遍用于突变致病性解读的公共突变数据库clinvar、hgmd和umd-lsdb相比,效果优势如表9所示,htaadvar从已发表文献中收录了更多的主动脉相关突变。有别于用户提交不同,htaadvar采用专家设计、从文献中人工收集最全面的信息,并通过双人校验以确保数据的高质量。值得注意的是,只有htaadvar包含对于突变致病性解读至关重要的结构化信息。此外,htaadvar还提供了最为全面的突变注释,如突变在gnomad数据库中的最大人群频率及测序深度,突变有害性的多软件预测,
蛋白质结构域等等。htaadvar同时记录了由解读工具统一解读的突变致病性分类,构成了标准化、可参考的高质量突变集合。总之,htaadvar突变数据库可提供更可靠的资源以支持主动脉病相关突变详细解读。
[0093]
表9.数据库比较
[0094][0095]
实施例2自动化解读系统建立
[0096]
本实施例在数据库基础上,进一步构建自动化解读系统
[0097]
步骤一、确定突变解读的基础规范
[0098]
2015年acmg/amp制定了遗传病突变解读标准和指南,随后clingen svi专家组对指南中一些标准的应用提出了更具体更细化的指导和建议。在上述指南和框架下,本技术基于两者制定了htaad疾病和基因特异的基础解读规范。
[0099]
示例性的,具体细则内容和大体实现方法如表10所示,该解读规范的特点包含:1)纳入pvs1最新解读指南并提出具体实现算法,弥补了现有解读工具的缺陷;2)阈值的界定和解读依据基本来自于构建的突变数据库,而非公共疾病数据库;3)对照人群也使用内部明确排除相关疾病的人群数据,而非普通人群的公共数据库;4)新发突变增加考虑父/母为嵌合,但无症状或症状轻,孩子为患者的情况;5)按照模型和模拟表型的差异,对功能学研究结果进行了不同证据水平的分类;6)在公共数据中查找突变时同时考虑此位点的测序深度,在满足一定深度的情况下确定突变是否存在;7)对家系共分离进行定量并在不同家系中做累计,根据不同证据水平进行打分;8)多种软件预测工具先按照算法原理不同分类,以大多数类型的预测结果作为突变的最终预测结果。
[0100]
表10.解读细则及确定方法
[0101]
[0102]
[0103][0104]apm4不适用于同时应用pvs1(任何强度水平)证据的突变。
[0105]bpp3不适用于同时应用pvs1(任何强度水平)/ps3/pm4证据的突变。
[0106]cbp4不适用于同时应用pvs1(任何强度水平)/ps3/pm4证据的突变。
[0107]dpp1必须与pm2结合使用。
[0108]
步骤二、构建遗传突变全自动解读系统
[0109]
在上述步骤基础上,进一步构建解读系统,此系统可以全自动地对用户输入突变进行解读,给出“良性”、“可能良性”、“意义不确定”、“可能致病性”或“致病性”类别,并展示详细的解读证据。
[0110]
1)建立自动化解读流程。为了实现突变的自动化解读,程序会解析步骤2所述的htaadvar数据库中的6个表,同时还会读取其他公共数据库来源的信息,比如重复区域的染色体位置、人群频率数据库突变位点的覆盖度和最大人群频率、18个基因的外显子区域等等。根据制定解读细则和阈值,本技术采用perl编程语言实现解读算法。
[0111]
2)预判读突变数据库突变。采用上述自动化解读程序对构建的突变数据库中所有突变,即variants表记录的所有突变进行解读,将解读结果和所有证据填入variants表相应字段。
[0112]
3)解读用户输入突变。首先判断用户输入突变是否在突变数据库中,如果输入突变在htaadvar数据库中存在,可以直接获取其致病性的分类,同时用户可以根据提供的额外证据重新对判读结果进行解读。如果输入突变在htaadvar数据库中不存在,解读工具将调用vep软件和内部程序生成相关注释,然后采用自动化判读流程对突变进行解读,以获取最终分类,具体可参见图1。
[0113]
实施例3基于自动解读系统的交互网站开发和本地程序运行方式
[0114]
为了帮助用户更好的浏览突变数据库信息及交互式解读突变,本技术构建了具有强大浏览、查询和解读功能的交互式网站(http://htaadvar.fwgenetics.org)。网站页面主要包括首页、基因详情页、突变页、文献页、解读页和帮助页等(参见图2)。其中,首页包含了系统的简单介绍和基本搜索功能,用户可以在搜索框中输入基因、突变和文献的相关信息进行查询;基因页包含基因概述页和基因注释页,前者提供了18个基因的基本信息以及
按基因外显子和结构域分组的突变统计,后者给出了当前基因的相关注释信息,包括基本信息、表型数据库、表达数据库、转录后修饰、go数据库、通路数据库、蛋白互作数据库、药物相关数据库等;突变页展示了突变数据库收录的所有突变,里面包含突变位点的基本信息、致病性解读结果和详细解读依据等;文献页展示了突变数据库收录的所有文章,根据研究类型分为测序研究和功能研究。解读工具页包含单位点解读页和多位点解读页。对于单位点解读,可以按照提示输入染色体位置信息和其他相关证据,其中染色体位置是必填项,其他证据可以根据用户数据情况选择性填入,点击提交后可查看并下载结果;对于多位点解读,默认输入格式为制表符分隔的vcf文件,其中前八列是必须填入的,如果用户有额外证据可以选择性填入后面九列内容。用户可以根据页面上的提示填入正确格式的数据,同时也可以上传相同格式的文件,递交后可以下载多位点解读结果和详细解读依据。帮助页是本系统的说明文档,包括htaadvar数据库概述、如何进行数据库浏览、检索、解读的输入输出数据格式、网页版和本地版解读工具的使用等。
[0115]
为了方便用户进行大规模突变解读和重新定制阈值进行解读,本技术同时提供了本地版程序。本地版的使用方法主要包含三步:首先,安装perl、ensembl variant effect predictor(vep)和samtools软件,下载grch37版本的参考基因组文件和vep cache文件(homo_sapiens_refseq_vep_103_grch37.tar.gz);第二步,下载htaadvar本地版的安装包,包括两个perl脚本(interpretation.pl和splicing.pl),配置文件(config.ini),maxentscan软件,突变数据库(htaadvar1.0_data)和其他数据资源(source)。第三步,在本地运行脚本,如使用默认参数进行解读,命令如下:interpretation.pl[-c config file]-i infile[-o outfile]。
[0116]
本地版程序允许用户根据自定义解读阈值进行突变解读。首先,需在配置文件(config.ini)中修改解读阈值。以下阈值是可修改:pvs1_4,ps2_verystrong,ps2,ps4,ps4_moderate,ps4_supporting,pm2,pm5_strong,pm5,pm5_supporting,bs1,ba1;其次,当配置文件准备好后,就可以根据自定义的阈值重新解读htaadvar数据库中的所有突变,解读结果将更新到“htadvar1.0_data/variants.txt”文件中。用法:interpretation.pl-c config file-u。更新“htadvar1.0_data/variants.txt”文件后,可以使用新的配置文件解读感兴趣的突变,用法:interpretation.pl[-c config file]-i infile[-o outfile]。
[0117]
实施例4 htaad疾病突变的自动化解读相比传统工具的灵敏度和特异性优势
[0118]
为进一步证实本技术的数据库和解读系统优势,本实施例比较了本技术的htaadvar与现有技术中其他解读工具(intervar和cardioclassifier),比较三者解读出来的分类的灵敏度和特异性。
[0119]
因为htaadvar和cardioclassifier解读的目标基因存在较大差异,只有fbn1重叠,故在三软件比较中,本技术选取clinvar数据库中多人提交且致病性分类一致的fbn1突变作为“金标准”,包括299个一致认为“可能致病/致病”突变和312个一致认为“可能良性/良性”突变。
[0120]
其中,在299个“可能致病/致病”突变中,279个与本技术的结果一致,而intervar和cardioclassifier与其一致的个数分别是109和113个;类似,在312个“可能良性/良性”的突变中,238个与本技术的结果一致,而intervar和cardioclassifier与其一致的个数分别是52和17个,详见表11,可见,本技术的htaadvar灵敏度(htaadvar:93.31%
vs.intervar:36.45%,cardioclassifier:37.79%)和特异性(htaadvar:76.28%vs.intervar:16.67%,cardioclassifier:5.45%)是其他两个工具的2-5倍(具体参见图3),具有显著优势。
[0121]
为了证明htaadvar的优势不仅仅局限在fbn1基因上,本技术进一步拓展了“金标准”突变集合,选取18个目标基因上所有clinvar数据库中多人提交且致病性分类一致1764个突变,包括416个一致认为“可能致病/致病”突变和1348个一致认为“可能良性/良性”突变。如表12所示,与intervar相比,htaadvar在灵敏度(htaadvar:85.58%vs.intervar:35.10%)和特异性(htaadvar:80.27%vs.intervar:28.12%)上仍具有极大优势。
[0122]
表11.三款软件比较
[0123][0124][0125]
表12.htaadvar与intervar比较
[0126][0127]
实施例5 htaad疾病突变的自动化解读的临床样本检测
[0128]
本实施例进一步采用临床样本进行系统准确性评估,在本技术收录的790名临床住院患者中,采用htaadvar工具对临床患者的突变位点进行解读,其“可能致病/致病”突变的检出率为43.04%,“可能致病/致病/临床意义不明”突变的检出率为70.89%(表13),这与以往报道的人工解读对应的检出率的一致性高,充分表明本技术在临床应用中的可信度。
[0129]
此外,本技术也比较了htaadvar判读速度与人工判读效率的差异。在16gb内存,intel(r)xeon(r)gold 6151cpu(3.00ghz)中央处理器的机器上测试,如待解读突变收录于突变数据库中,用户可瞬间(《1秒)获得结果,如待解读突变未收录,htaadvar解读一个突变大约需10秒。因此,在批量解读中,htaadvar返回速度会随着待解读突变中数据库收录比例有所变化。假定解读100个突变,其中36%是突变数据库未收录的新突变,htaadvar大约需13秒解读完毕;假定解读100个未收录的新突变,htaadvar大约需35秒。而一个经验丰富的解读人员手工解读一个突变约需5-10分钟,100个突变就要8-17个小时。由此可见htaadvar在保证临床检出率的同时大大提高了解读效率。
[0130]
表13.htaadvar与人工解读的比较
[0131][0132][0133]
前述对本技术的具体示例性实施方案的描述是为了说明和例证的目的。这些描述并非想将本技术限定为所公开的精确形式,并且很显然,根据上述教导,可以进行很多改变和变化。对示例性实施例进行选择和描述的目的在于解释本技术的特定原理及其实际应用,从而使得本领域的技术人员能够实现并利用本技术的各种不同的示例性实施方案以及各种不同的选择和改变。本技术的范围意在由权利要求书及其等同形式所限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1