用于预测蛋白质翻译后修饰位点的深度学习方法
1.本发明涉及生物信息学领域,尤其是涉及一种用于预测蛋白质翻译后修饰位点的深度学习方法及系统。
背景技术:
2.蛋白质翻译后修饰(ptm)是增加蛋白质组多样性的关键机制,它通过向一个或多个残基添加修饰基团可使蛋白质具有更为复杂的结构和更为完善的功能,实现更为精细的调节(khoury,g.a.;baliban,r.c.;floudas,c.a.,proteome-wide post-translational modification statistics:frequency analysis and curation of the swiss-prot database.scientific reports 2011,1,(90),5.)。磷酸化、糖基化、乙酰赖氨酸化和甲基精氨酸化是其中重要的蛋白质翻译后修饰物,它们与生物的代谢过程有着重要的关联,例如细胞周期、神经活动、肌肉收缩和肿瘤生成等。已有研究表明,异常的蛋白质翻译后修饰与某些疾病的发生有关(eipper,b.betty a.,posttranslational modification of proteins:expanding nature's inventory.the quarterly review of biology 2008,83,(4),403-403)。准确地识别ptm位点,对于更好地理解蛋白质在生命活动中起到的作用以及药物设计具有重要意义。
3.近几年,研究人员已经提出多种ptm位点识别方法,主要分为干实验和湿实验的方法。其中,edman降解法和
32
p标记法是识别ptm位点的两种经典的湿实验方法。最近,lyu等人(lyu,j.w.;wang,y.;mao,j.w.;yao,y.t.;wang,s.j.;zheng,y.;ye,m.l.,pseudotargeted ms method for the sensitive analysis of protein phosphorylation in protein complexes.analytical chemistry 2018,90,(10),6214-6221)提出一种基于平行反应监测技术的伪靶向质谱法识别和定量磷酸肽,该方法产生比过去湿实验方法更多的ptm位点。虽然基于湿实验的方法能够精确的识别ptm位点,但需要消耗大量的成本和时间。而基于干实验的方法假设序列中的残基遵循一种有规律的模式,并利用计算机学习该模式预测真实的ptm位点,具有成本低、速度快的特点。随着技术的发展,越来越多的ptm位点被发现,这为利用干实验的方法挖掘残基上的翻译后修饰模式进而ptm位点预测提供条件。
4.目前,研究人员已经提出一些基于干实验的ptm位点预测方法,主要分为基于机器学习的方法和深度学习的方法,如musite(gao,j.j.;thelen,j.j.;dunker,a.k.;xu,d.,musite,a tool for global prediction of general and kinase-specific phosphorylationsites.molecular&cellular proteomics 2010,9,(12),2586-2600),phospred-rf(wei,l.y.;xing,p.w.;tang,j.j.;zou,q.,phospred-rf:a novel sequence-based predictor for phosphorylation sites using sequential information only.ieee transactions onnanobioscience 2017,16,(4),240-247),musitedeep(wang,d.l.;zeng,s.;xu,c.h.;qiu,w.r.;liang,y.c.;joshi,t.;xu,d.,musitedeep:a deep-learning framework for general and kinase-specific phosphorylation site prediction.bioinformatics 2017,33,(24),3909-3916),capsnet(wang,d.l.;liang,
y.c.;xu,d.,capsule network for protein post-translational modification site prediction.bioinformatics 2019,35,(14),2386-2394),hybridsucc(薛宇;宁万山;许浩东;邓万锟;郭亚萍,蛋白质编码方法及蛋白质翻译后修饰位点预测方法及系统,cn110033822,2019.03.29)等方法。其中,musite、phospred-rf是典型的机器学习方法,这些方法提供有效的特征提取,并优化模型的关键参数以拟合实验数据。musite使用k近邻评分、蛋白质紊乱特征和氨基酸频率特征来表示潜在ptm位点周围的局部序列信息。phospred-rf是一种基于随机森林的预测模型,它应用信息论特征、重叠属性特征、二十位特征和skip-n-gram特征来捕获ptm位点和非ptm位点之间的差异。然而,基于机器学习的方法性能依赖特征工程,对方法的使用与改进有着很大的限制。与其相比,基于深度学习的方法可以自动对蛋白质序列的特征进行提取,通过端到端的策略直接预测ptm位点,摆脱特征工程和专家领域的依赖。其中musitedeep和capsnet是两种常用的基于一级序列信息的深度学习的ptm位点预测方法,hybridsucc则是基于多级结构与进化信息的ptm位点预测方法,它们表现出比基于机器学习方法更好的预测效果。musitedeep发布基于web的在线预测服务,研究人员可以直接输入faste文件获得蛋白质序列中潜在的ptm位点位置。
5.虽然已有的ptm位点预测方法能够获得良好的预测性能,但仍存在以下不足:一是对序列信息的利用率不高。现有的预测ptm位点的方法主要侧重于利用潜在ptm位点与周围位点的短程耦合信息进行预测,而没有考虑长程耦合信息;二是一些方法采用蛋白质的多级结构信息,难以进行获取;三是模型的特征表征能力不足。研究发现,相对于非ptm位点周围的残基,ptm位点周围的残基通常有更高度的相关性;并且潜在ptm位点附近的残基对该位点是否为ptm位点更为重要,而目前大多数的模型设计时没有关注到这方面的特性。三是目前的在线预测服务仍存在着一些问题,如当用户有大量数据需要预测时,由于服务器吞吐量和性能的限制需要等待较长时间。当出现浏览器崩溃或不慎误关网页的情况,很容易导致预测失败。这不仅会造成服务器资源的浪费,也消耗用户的时间。
技术实现要素:
6.本发明的目的在于针对现有技术存在的信息利用率不足、模型可解释性差、web服务等待时间过长等问题,提供可提升预测ptm位点的预测准确度,缓解深度学习模型可解释性差,节省用户等待时间的一种用于预测蛋白质翻译后修饰位点的深度学习方法。
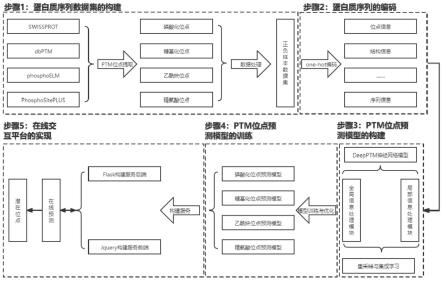
7.本发明包括以下步骤:
8.1)蛋白质序列数据集的构建:从公开的数据库中收集带有翻译后修饰位点(ptm)的蛋白质序列,删除冗余的蛋白质序列,得到不同ptm位点类型的蛋白质序列数据集;
9.2)蛋白质序列的编码:通过补零或剪切的方式归一化蛋白质序列的长度,并对蛋白质序列进行one-hot编码;
10.3)ptm位点预测模型的构建:利用基于深度神经网络构建ptm位点的预测模型,设计并行的特征提取模块分别提取蛋白质序列的短程和长程的耦合信息;
11.4)ptm位点预测模型的训练:根据ptm位点的类型,利用蛋白质序列数据集分别训练预测模型;
12.5)在线交互系统的实现:将训练后的神经网络模型部署于服务器上,实现在线预测和结果可视化,并将预测结果输出。
13.步骤1)中,所述蛋白质序列数据集的构建,具体包括以下步骤:
14.(1)从公开的数据库中收集并整理有ptm位点的蛋白质序列,获取ptm位点的位置,整理成为初始数据集;所述公开的数据库包括swissprot、dbptm、phosphoelm、phosphositeplus等ptm位点数据库;
15.(2)使用蛋白质序列聚类工具cd-hit从初始数据集中去除冗余的蛋白质序列;
16.(3)对去冗余的蛋白质序列进行数据清洗,删除可信度低的位点注释信息,得到干净的蛋白质序列数据集。
17.在步骤2)中,所述蛋白质序列的编码,具体包括以下步骤:
18.(1)将长序列进行截断操作,短序列进行零填充,使蛋白质序列具有相同的长度;
19.(2)对统一长度的蛋白质序列进行one-hot编码,转化为计算机可识别的向量。
20.在步骤3)中,所述ptm位点预测模型的构建,具体步骤为:构建一个基于深度神经网络的模型,模型包含短程耦合特征提取模块和长程耦合信息处理模块,分别用于提取蛋白质序列的短程和全局耦合信息;短程耦合特征提取模块,依次包含有1个卷积神经网络cnn,2个挤压与激励网络senet,1个双向长短期记忆网络bi-lstm和2个全连接层fc。
21.在步骤4)中,所述ptm位点预测模型的训练,具体步骤包括:
22.(1)由于ptm位点的正负样本通常是不平衡的,故将训练数据集随机分为n个训练子集,样本量不足的训练子集采用重采样方法补齐,得到n个平衡的训练子集;
23.(2)用n个训练子集分别对模型进行训练,再对训练结果进行集成学习得到预测模型。
24.在步骤5)中,所述在线交互系统的实现,具体步骤包括:
25.(1)利用python和javascript语言编程实现在线的ptm位点预测模型;
26.(2)设计在线模型的输入输出模块;
27.(3)用户通过系统提交蛋白质序列文件,选择待预测的ptm类型,系统将预测结果通过http和邮件的方式返回给用户;所述预测结果包括蛋白质序列的信息、位点位置、ptm位点种类等信息。
28.与现有技术相比,本发明包括以下优点和技术效果:
29.1、本发明针对信息利用率不足的问题,通过引入长程耦合信息估计ptm位点的先验分布,进而提升预测ptm位点的预测准确度;
30.2、本发明针对模型的特征表征能力问题,在模型设计时引入bi-lstm和senet网络来分别捕获以上两种信号,在提升性能的同时缓解深度学习模型可解释性差的问题。
31.3、本发明针对已有的web服务等待时间过长的问题,提供的在线预测技术在处理大型fasta文件任务时,利用stmp邮件功能库预设需要发送邮件的账户。用户只需留下邮箱地址,本发明提供的服务便可以在模型运行结束后将结果以邮件的形式发送给用户,节省用户的等待时间。
32.4、本发明所述deepptm模型在构建数据集时使用多个数据库中的数据,相比于其它同类型的预测模型有着更大的训练集;
33.5、本发明所述deepptm模型有着更高的信息利用率和更有效的模型设计,具有更高的预测性能;且编码时不需要获取蛋白质的多级结构,只需要蛋白质的一级结构,预测过程更加便捷;
34.6、本发明所述基于deepptm模型的ptm位点预测的在线预测系统,采用更简洁的可视化界面,使用户可以更加直观了解到预测的结果。采用通过邮件后台发送预测结果的设计,解决用户需要长时间保存网站页面等待预测结果的问题。
35.7、本发明可以同时预测多种ptm位点,为生物医学研究和药物设计提供相关的蛋白质信息。
附图说明
36.图1是本发明总体流程图。
37.图2是本发明中deepptm神经网络模型结构示意图。
38.图3是本发明中长程耦合信息处理模块和短程耦合信息处理模块结构示意图。
39.图4是deepptm神经网络模型在磷酸化位点的预测任务上,基于pre、re、f1-score、mcc等指标和其他方法的对比。
40.图5是本发明中的deepptm神经网络的本地信息处理模块中第二个senet块的输出。
具体实施方式
41.以下实施例将结合附图对本发明作进一步的说明。
42.如图1,本发明实施例包括以下步骤:
43.1)蛋白质序列数据集的构建
44.从网上公开的蛋白质翻译后修饰(ptm)位点数据库中收集并整理有ptm位点的蛋白质序列,获取ptm位点的位置,整理成为数据集。对整理好的初始数据集进行去冗余的处理后,为每一类型的ptm位点构建一个独立的蛋白质序列数据集。将每个数据集的蛋白质序列按10︰1的比例随机分为训练集和测试集,将标注的ptm位点定义为正样本,其余的潜在ptm位点定义为负样本。具体地,ptm位点数据集从swissprot,dbptm,phosphoelm,phosphositeplus等ptm位点数据库中收集蛋白质序列和ptm位点的数据,并使用cd-hit web服务器从数据集中去除相似度超过0.5的冗余序列。
45.2)蛋白质序列的编码
46.得到数据集后,根据蛋白质序列上的氨基酸相互作用分为长程耦合和短程耦合两类。长程耦合定义为蛋白质序列上的所有氨基酸对ptm位点的可能作用,而短程耦合则定义为ptm位点周围少数几个氨基酸的作用;为了统一模型输入数据的维数,将长程耦合的最大距离限制为n,而将长度大于n蛋白质序列进行截断,长度小于n的蛋白质序列进行零填充。同时将短程耦合的距离限制为m,若潜在ptm位点的上下游的氨基酸数目不足m,则使用“*”填充,并将其视为一个氨基酸。
47.使用ont-hot编码方式,对两类蛋白质序列进行编码,将处理好的蛋白质序列转化为计算机可识别的向量。具体的,每个残基可以编码为一个向量,序列中相应的索引氨基酸表示为“1”,其余的向量均为“0”,具体步骤可如下:
48.采用2000
×
20矩阵和51
×
21矩阵分别表示残基的长程耦合信息和短程耦合信息。将长程耦合信息中的蛋白质序列统一长度为2000,长度大于2000的蛋白质序列进行截断操作,长度小于2000的蛋白质序列进行零填充,以适应模型的输入。使用一个51
×
1的窗口来
获取短程耦合信息,每个窗口的中心为潜在的ptm位点,窗口两侧为位点左右的各25个残基,若潜在ptm位点的上下游的氨基酸数目不足25,则空缺的部分使用“*”进行填充,将它视为一种额外的氨基酸。然后使用one-hot对蛋白质进行编码,每个残基可以编码为一个向量,每个序列里仅有一个向量为“1”,其余的为“0”,其中“1”代表蛋白质序列中相应的索引氨基酸。
49.3)ptm位点预测模型的构建
50.构建基于深度神经网络的预测模型,记为deepptm,结构如图2所示。该模型包括短程耦合信息处理模块(short-rangecouplingmodule)和长程耦合信息处理模块(long-rangecouplingmodule),两个信息处理模块分别用于处理长程耦合信息和短程耦合信息,分别提取蛋白质序列上来自邻近氨基酸的作用和来自远距离氨基酸的作用,结构如图3所示。其中,长程耦合信息处理模块和短程耦合信息处理模块的特征提取模块使用网络架构,包含有1个卷积神经网络(convolutionalneuralnetwork,cnn),2个挤压与激励网络(sequeeseandexcitationnetworks,senet),1个双向长短期记忆网络(bi-directionallongshort-termmemory,bi-lstm)和2个全连接层(fullyconnectedlayers,fc。长程和短程耦合特征提取模块通过一系列非线性变换提取蛋白质序列中的高级特征,并将其输入全连接层使用softmax激活函数生成位点预测结果。
51.ptm位点预测模型构建的具体步骤可为:
52.1)首先使用卷积层来提取蛋白质序列的特征,本发明中使用的是1
×
1的卷积核,它可以实现跨通道交互和信息集成。卷积块的输出可以写成公式(1):
[0053][0054]
其中,m
jk
为在k
th
特征图中的j
th
位置,x为输入矩阵,f为relu的激活函数,w
d,i
为滤波器的d
th
行的i
th
列,bk为k
th
滤波器的偏置。
[0055]
2)在卷积层提取好特征后,使用senet层来压缩和激励特征,以此对特征图进行优化。压缩操作是对特征进行压缩,然后将特征图转化为一个能反映特征的数字。压缩操作可以表示为公式(2):
[0056][0057]
其中m
ic
为c
th
特征图的i
th
位置,h为特征图的个数,zc为c
th
特征图的分布。同时,利用激励运算可以构造特征图之间的相关性。这一操作由两个全连接层完成,第一个全连接层用于减少图的数量并降低计算复杂度,第二个全连接层将维数恢复为输入的维数。激励操作可以表示为公式(3):
[0058]
wc=f1(f2(zc,w1),w2)3其中z为压缩操作后的c
th
特征图,w1和w2为两个全连接层的权重。f1和f2分别为sigmoid和relu激活函数。wc可以作为c
th
特征图的重要性度量,接着将对所有的特征图进行加权求和得到优化后的特征图,可以表示为公式(4):
[0059]m′
=w*m4其中m为senet层的输入矩阵,w为压缩操作的输出,m
′
为senet层的输出矩阵。
[0060]
3)使用一个bi-lstm层来进一步获取senet层的特征。在每个bi-lstm层中有三个
门,分别是遗忘门,输入门,输出门。遗忘门用来丢弃不重要的信息,输入门决定需要添加多少新的信息进入状态信息中,而输出门则提供每个单元的输出结果,可以表示为公式(5):
[0061][0062]
其中f
t
,i
t
,o
t
分别为遗忘门,输入门,输出门的输出,c
t
,h
t
分别为候选单元状态,未标注单元状态,隐藏单元状态。wf,wi,wc,wo和bf,bi,bc,bo分别为相应门和单元状态的权重和偏置。
[0063]
4)使用另一个senet层估计潜在ptm位点附近的每个位置的残基贡献。上一个senet层的输出会被调换并输入到另一个senet层。短程耦合信息处理模块会产生一个具有短程耦合信息的输出,接着将所有特性提供给全连接层。对于长程耦合信息处理模块,也可以使用相似的结构获取长程耦合信息。然后使用其它的全连接层和softmax输出层获取最后的预测结果。
[0064]
深度学习模型由于其高度非线性的特征,常常引起黑箱效应并导致数据解释难以进行。deepptm模型中引入了注意力机制,从而一定程度上缓解了黑箱效应,使得模型从序列中学习到的信息具备可解释性。图4展示短程耦合信息处理模块中的第二个senet块的输出,其中横坐标表示蛋白质序列中的位置,纵坐标表示其中一个训练子集中的样本。每个元素表示的是senet输出的权重,颜色越深表示该位置的权重越大。从图中可以看出,在预测未知的ptm位点时,deepptm更加关注潜在位点附近的残基,从而更符合生物学认知。
[0065]
4、ptm位点预测模型的训练
[0066]
利用adam优化器对deepptm模型的参数进行学习更新,采用模型输出与位点标签的交叉熵作为损失函数。为了防止模型过拟合并具有更好的泛化能力,训练过程中采用早停法(early stopping),将给定迭代范围内验证集准确率不再提升时的模型参数作为最优参数进行保存。采用bootstrap重采样和stacking集成学习相结合的策略来解决数据集中正负样本数量不平衡的的问题,将负样本随机划分为n个部分,并与同等数量的正样本相结合构建平衡数据集进行训练,得到n次的训练结果,最后通过集成学习得到训练后的deepptm模型。具体步骤如下:
[0067]
(1)在模型训练过程中,使用adam优化器对deepptm进行梯度更新,并采用标准交叉熵作为损失函数,将验证集表现最优时的参数作为最优参数。使用早停法防止模型过拟合并具有更好的泛化能力。
[0068]
(2)针对不平衡数据集,采用一种结合bootstrap重采样和集成学习(ensemble learning)的非平衡数据集策略,最后得到预测的结果。
[0069]
具体地,首先将负样本随机分为n个部分,每个部分有m个样本,其中m等于正样本的数量。然后,将各部分负样本与所有正样本相结合,构建平衡数据集。接下来使用n个模型在这些平衡数据集上进行训练。最后,使用集成学习将n个预测结果进行组合并再次进行学习以获得最终预测。
[0070]
(3)使用模型预测进行ptm位点预测。将待测蛋白质序列输入模型,得到ptm位点预测结果。
[0071]
具体地,本发明以磷酸化位点预测为例,将本发明的deepptm模型与其它四种常用的磷酸化位点预测方法使用tp(ture positive),tn(ture negative),fp(false positive)和fn(false negative)等统计方法来测试数据集中两类样本被正确和错误分类的数量,用精确率pr(precision),召回率re(recall),f1值(f1 score),相关系数mcc(matthews correlation coefficient),接收者工作特征曲线roc(receiver operating characteristic curve),接收者工作特征曲线下面积auroc(area under the receiver operating characteristic curve),精确率召回率曲线下面积auprc(area under the precision recall curve)等指标上行了比较。
[0072]
在pre、re、f1-score、mcc以及auroc、auprc曲线的指标上,根据图5以及表1所示,deepptm在auroc和auprc预测值上总体表现更好。
[0073]
表1
[0074][0075]
notes:auroc:areas under the roc;auprc:areas under the prc;
[0076]
为了展示deepptm在预测真实ptm位点方面的优势,本发明使用最近发布的蛋白质序列(uniprot id:a0a2r8y619,于2020年6月17日发布)作为测试数据,将deepptm与其他三种提供web服务的系统进行比较。本发明将置信度大于50%的位点视为阳性位点,测试蛋白质序列的真实磷酸化位点为33、81、112,将测试蛋白质序列输入各模型进行测试,测试结果分别为:musite预测的位点为2、18、19、24、33、116、119;phospred-rf预测的位点为18、19、33、85、93;musitedeep预测的位点为2、33、112;deepptm预测的位点为2、33、35、81、109、112,与其他系统对比,可以正确预测出了所有磷酸化位点,这一结果也表明本发明能准确预测ptm位点并非依赖于数据。
[0077]
5、在线交互系统的实现
[0078]
使用python的flask框架作为后端,html与javascript作为项目前端,将deepptm的预测模块封装成接口对外暴露提供服务,用户只需要输入fasta格式的蛋白质序列,即可在线或通过邮件获取预测结果。
[0079]
具体的,在上述步骤4的基础上,利用python和javascript将deepptm模型部署到云平台上,得到在线的ptm位点预测系统。利用python中的flask构建系统的后端框架,使用html与javascipt技术构建系统的前端框架。用户预测新蛋白质序列的ptm位点时,只需向系统提交fasta格式的蛋白质序列文件,系统计算后返回相应的预测结果,包括蛋白质序列的信息、位点位置、ptm位点种类等信息。
[0080]
综上,本发明中提出一种新的深度神经网络模型deepptm,并开发一个基于deepptm模型的蛋白质翻译后修饰位点预测的在线预测技术系统,结果证明,deepptm可以有效地提高ptm位点的预测性能。希望本发明能够帮助研究人员发现新的位点,为生物医学研究提供便利,并为未来的医学研究提供有用的帮助。
[0081]
以上所述,仅为本发明较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1