α-色烯螺哌啶类化合物在制备5-羟色胺2C受体激动剂中的应用
α-色烯螺哌啶类化合物在制备5-羟色胺2c受体激动剂中的应用
技术领域
1.本发明涉及药物学领域,具体涉及α-色烯螺哌啶类化合物在制备5-羟色胺2c受体激动剂中的应用。
背景技术:
2.5-羟色胺2c(5-ht
2c
)受体是近年来被广泛研究的一种gpcr受体,属于5-羟色胺受体家族,是唯一经rna编辑的5-羟色胺受体亚型。因主要集中表达在中枢神经系统而产生外周副作用的风险低,因此5-ht
2c
受体成为了治疗中枢神经系统疾病的理想靶点,如儿童难治性癫痫dravet综合征、肥胖、精神分裂症、成瘾和药物滥用等。通常认为gpcr受体只通过g蛋白介导下游复杂的信号转导通路,但近来越来越多的研究发现,gpcr受体还可通过g蛋白偶联受体激酶(grk)和β-阻遏蛋白(β-arrestin)激活非g蛋白依赖的信号通路,介导gpcr的脱敏、内化和循环等。因gpcr受体激动剂不均匀地激活这两条信号通路,从而表现出不同程度的“偏向性(bias)”,即“g蛋白偏向性激动剂”和“β-arrestin偏向性激动剂”两种。其中,g蛋白偏向性激动剂因能避免β-arrestin通路介导的脱敏而诱发抗药性,显示出更长效的疗效,故而近来成为了研究的新重点。
3.与其他gpcr受体相比,靶向5-ht
2c
受体的偏向性激动剂研究尚处于初期阶段,直到2016年才有报道。一类是美国伊利诺伊大学芝加哥分校(uic)证实了(2-苯基环丙基)甲胺类g蛋白偏向性5-ht
2c
受体激动剂(+)-7e能使5-ht
2c
受体的脱敏反应延迟,延长受体介导的下游效应(j med chem,2016,59,9866-80)。另一类是本团队前期发现的一系列阿朴菲类完全g蛋白偏向性5-ht
2c
受体激动剂,其具有体内抗肥胖活性和抗精神分裂症活性(acs cent sci,2020,6,213-25;bioorganic chemistry 123(2022)105795)。
4.但现有g蛋白偏向性5-ht
2c
受体激动剂存在对5-ht
2c
受体选择性低[如(+)-7e(j med chem,2016,59,9866-80),(+)-15a和(+)-19(j med chem,2017,60,6273-88),11b和11f(bioorganic chemistry 123(2022)105795)],在激活5-ht
2c
受体的同时,也会激活其高度同源的5-ht2受体亚型,5-ht
2a
和5-ht
2b
,进而引发致幻作用,以及肺动脉高压和心脏瓣膜病的发生,从而可能导致副作用的发生。此外,化合物还存在偏向性差(如(+)-7e,(+)-19)、5-ht
2c
受体激动活性弱[如1857(acs cent sci,2020,6,213-25)]或herg抑制率较高[mq02-439(acs chem neurosci,2020,11,549-59)]等缺陷,从而限制了上述化合物进一步的临床发展。因而,仍需开发结构独特,具有高选择性、高偏向性、高活性和好的成药性的5-ht
2c
受体激动剂。
[0005]
α-色烯螺哌啶是在色满-4-酮的c-2位置引入螺哌啶环并将c-4位羰基还原为双键而得到的,其作为有机合成和药物设计中的重要结构骨架,目前已被广泛用于药物研发,通过作用于乙酰辅酶a羧化酶(acc)、瞬时受体电位trpm8、gpr119和5-ht
2a
等重要药物靶标(journal of medicinal chemistry,2015,26:539
–
563;bioorganic&medicinal chemistry,2020,28:115813),从而作为代谢调节药、抗炎、抗氧化药、抗糖尿病药、抗肿瘤
药、抗精神病药、抗癫痫药和抗痴呆药等用于疾病的潜在治疗。目前尚无α-色烯螺哌啶衍生物激动5-ht
2c
受体的报道。
技术实现要素:
[0006]
本发明要解决的技术问题是提供α-色烯螺哌啶类化合物在制备5-羟色胺2c受体激动剂中的应用,α-色烯螺哌啶类化合物表现出高5-ht
2c
受体选择性和完全g蛋白偏向性,且显著降低了herg抑制率。
[0007]
为解决上述技术问题,本发明提供以下技术方案:
[0008]
本发明提供了α-色烯螺哌啶类化合物或药剂学上可接受的盐在制备5-羟色胺2c受体激动剂中的应用,所述α-色烯螺哌啶类化合物的结构通式m如下所示:
[0009][0010]
其中,为芳基或杂芳基,所述芳基为苯基、萘基或联苯基,所述杂芳基为含氮芳杂环或稠芳环取代基,例如:吡啶环、吡咯环、吡唑环、噻唑环、喹啉、异喹啉、吲哚、苯并呋喃环、呋喃环、苯并呋喃环、苯并噻吩环或噻吩环;
[0011]
r选自氢、卤素、氰基、c
1-c8烷基、c
1-c8烷氧基、c
1-c8卤烷基中的一种或几种,或与形成二氢呋喃环;
[0012]
x为-o-、-s-或-so
2-;
[0013]
r1选自氢、氰基、c
1-c8烷基、芳基中的一种;
[0014]
m,n独立地选自0、1或2;
[0015]
r2、r3,r4,r5,r6,r7,r8,r9独立地选自氢、卤素、氰基、c
1-c8烷基、c
1-c8卤烷基、c
1-c8烷氧酰基、c
1-c8烷酰胺基、c
1-c8烷酰氧基、c
1-c8烷氧基中的一种,或任两个取代基形成并环或螺环。
[0016]
进一步地,所述药剂学上可接受的盐为通式m所示的化合物与无机酸或有机酸形成的盐,例如盐酸盐。
[0017]
进一步地,所述c
1-c8卤烷基为含有卤素的1-8个碳的烷基,优选为三氟甲基、二氟甲基、单氟甲基、三氟乙基中的一种。
[0018]
进一步地,所述c
1-c8烷基(结构式:r
10-*),c
1-c8烷氧基(结构式:r
10
o-*),c
1-c8烷
氧酰基(结构式:),c
1-c8烷酰胺基(结构式:或),c
1-c8烷酰氧基(结构式:),其中r
10
,r
11
为脂肪族烷基或芳香族烷基。
[0019]
进一步地,所述脂肪族烷基包括直链烷基、支链烷基、螺环烷基、桥环烷基、烯烷基、炔烷基、环烷基、环烯基、环炔基、烷氧烷基、烷氧酰基烷基、环烷基烷基中的一种,优选甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、环丙烷基、环丁烷基、环戊烷基、环己烷基、烯丙基、炔丙基、环丁烯基、环己烯基中的一种。
[0020]
进一步地,所述芳香族烷基为取代或未取代的芳烷基或杂芳烷基,包括取代或未取代的芳氧基、取代或未取代的苯甲基、取代或未取代的苯乙基。
[0021]
进一步地,优选为苯环;x为-o-;r选自卤素、c1-c8烷基、c
1-c8烷氧基,或与形成二氢呋喃环;r1选自氢、氰基、c
1-c8烷基中的一种;r2、r3,r4,r5,r6,r7,r8,r9独立地选自氢、c
1-c8烷基、卤素,或任两个取代基形成并环或螺环。
[0022]
进一步地,所述α-色烯螺哌啶类化合物为以下结构式中的一种:
[0023][0024]
[0025]
进一步地,优选为苯环;x为-o-;r选自氢、卤素、c
1-c8烷基、c
1-c8烷氧基中的一种;m、n均为1;r1~r9均为氢。
[0026]
进一步地,所述α-色烯螺哌啶类化合物选自式s1、s2、s4、s5、s8、s16、s18所示化合物中的一种。
[0027]
进一步地,当为苯环;x为-o-;r选自氢、卤素、c
1-c8烷基、c
1-c8烷氧基,或与形成二氢呋喃环;r1~r5均为氢时;所述α-色烯螺哌啶类化合物的制备方法包括以下步骤:
[0028]
(1)将式m1所示的化合物与式m2所示的化合物在有机溶剂以及碱性条件下反应,制备得到式m3所示的化合物;
[0029]
(2)将式m3所示的化合物在有机溶剂、还原剂的存在下反应,得到式m4所示的化合物;将式m4所示的化合物在脱水剂、还原剂以及有机溶剂的存在下,脱去水与保护剂得到式m5所示的化合物;或,
[0030]
将式m3所示的化合物在碱性、三氟甲磺酰化试剂以及有机溶剂的存在下,异构化得到式m6所示的化合物;将式m6所示的化合物在金属催化剂、苯硼酸、碱性溶液以及有机溶剂的存在下,引入r1片段后脱保护,即得到式m7所示的化合物。
[0031]
上述m1~m7的结构式及反应路线如下所示:
[0032][0033]
其中,反应条件:(a)吡咯,乙醇,rt(室温);(b)nabh4(硼氢化钠),meoh(甲醇),0℃-rt;(c)tsoh(对甲苯磺酸),甲苯,130℃;(d)c6h5n(so2cf3)2((三氟甲磺酰基)苯胺),
lihmds(双-(三甲基硅基)胺锂),thf(四氢呋喃),-78℃-25℃;(e)苯硼酸,pd[p(c6h5)3]4,licl(氯化锂),aq.na2co3(碳酸钠),二甲醚,回流。
[0034]
进一步地,所述5-羟色胺2c受体激动剂用于预防和/或治疗以5-羟色胺2c受体为靶点的疾病。
[0035]
进一步地,所述疾病为肥胖、尿失禁、抑郁症、焦虑症、强迫症、癫痫、精神分裂症、疼痛、糖尿病以及药物成瘾中的一种或多种。
[0036]
本发明的有益效果在于:
[0037]
1.本发明设计具有通式m结构的α-色烯螺哌啶类化合物并发现具有此类结构的化合物对5-ht
2c
受体表现出优异的激活作用以及良好的受体选择性,且具有完全g蛋白信号通路偏向性以及低herg抑制活性,对β-arrestin信号通路无作用,副作用和耐药性较低。此外,本发明更加深入地研究了α-色烯螺哌啶类结构和活性效能的关系,为开发用于治疗和/或预防以5-ht
2c
受体为治疗靶点的疾病的药物提供了新的理论和技术支撑。
[0038]
2.本发明所述的α-色烯螺哌啶类化合物可作为活5-ht
2c
受体激动剂,用以预防或治疗以5-ht
2c
受体为靶点的疾病,例如肥胖、尿失禁、抑郁症、焦虑症、强迫症、癫痫、精神分裂症、疼痛、糖尿病以及药物成瘾等疾病。
附图说明
[0039]
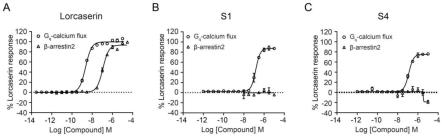
图1为氯卡色林与α-色烯螺哌啶类化合物对gq信号通路以及β-arrestin-2信号通路的剂量-效应曲线;图1a:氯卡色林,图1b:s1化合物,图1c:s4化合物。
具体实施方式
[0040]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0041]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0042]
本发明以下实施例中,化合物的结构是通过核磁共振(nmr)或质谱(ms)确定的。nmr是用安捷伦400mhz或600mhz仪测定,测定溶剂为氘代二甲亚砜(dmso-d6),氘代氯仿(cdcl3),内标为四甲基硅烷(tms),ms用gct premiertm(ci)质谱仪测定,除注明外均为ci源(70ev)。
[0043]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板的规格是0.15mm-0.2mm,柱层析一般使用烟台黄海100~200目或200~300目硅胶为载体。
[0044]
反应条件充氮气是指反应瓶连接一个约1l容积的氮气球。反应条件充氢气是指反应瓶连接一个约1l容积的氢气球。反应条件为室温(rt),温度范围是20-30℃。
[0045]
本发明以下实施例中,所有溶剂在使用前均经过重新蒸馏,所使用的无水溶剂均是按标准方法干燥处理获得。
[0046]
实施例1:化合物s1的合成
[0047]
化合物s1的合成路线如下:
[0048][0049]
其中,反应条件:(a)吡咯,乙醇,rt(室温);(b)nabh4(硼氢化钠),meoh(甲醇),0℃-rt;(c)tsoh(对甲苯磺酸),甲苯,130℃。
[0050]
化合物s1-3的合成:将0.25ml(3.25mmol)市售化合物2-羟基苯乙酮(s1-1)溶于20ml无水甲醇溶液,并加入345mg(4.87mmol)四氢吡咯,搅拌5min后,加入969.16mg(4.87mmol)市售化合物1-boc-4-哌啶酮(s1-2),室温下搅拌2h。减压浓缩甲醇,用二氯甲烷和饱和氯化钠溶液萃取分离,有机相用无水硫酸钠干燥,并在减压下浓缩。得到的粗品通过硅胶柱色谱(pe/ea=10/1)纯化,得到976mg白色固体。
[0051]
化合物s1-4的合成:将634mg化合物s1-3(2mmol)溶于15ml无水甲醇,在冰浴下向混合物中加入380mg(10mmol)nabh4,加完移至室温,2h后,将甲醇减压浓缩,用二氯甲烷和饱和氯化钠溶液萃取分离,有机相用无水硫酸钠干燥,减压浓缩即得粗品,得到的粗品通过硅胶柱色谱(pe/ea=4/1)纯化,得到437mg纯产物。
[0052]
化合物s1的合成:将319mg化合物s1-4(1mmol)溶于10ml甲苯,并加入209mg(1.1mmol)tsoh,130℃下回流过夜。降温后加入过量tea,浓缩混合物,得到粗品用碱性氧化铝柱色谱pe:ea=1:1,再用洗脱剂dcm:meoh:nh3·
h2o=70:1:0.1分离,得到140mg白色固体。1h nmr(400mhz,cdcl3)δ7.13(t,j=7.6hz,1h),6.99(d,j=7.2hz,1h),6.87(t,j=7.6hz,2h),6.38(d,j=9.8hz,1h),5.62(d,j=9.7hz,1h),3.13(t,j=11.4hz,2h),2.88(d,j=12.3hz,2h),2.20(s,1h),1.99(d,j=13.4hz,2h),1.72
–
1.59(m,2h).
[0053]
实施例2:化合物s2的合成
[0054]
化合物s2与化合物s1合成方法相同,不同之处在于化合物s2起始原料为2羟基-3-氯-苯乙酮。1h nmr(400mhz,dmso)δ7.13(d,j=7.9hz,1h),6.84(d,j=7.2hz,1h),6.74(t,j=7.6hz,1h),6.31(d,j=9.8hz,1h),5.59(d,j=9.8hz,1h),3.17(t,j=11.8hz,2h),2.84(d,j=12.0hz,2h),1.98(s,2h),1.95(s,1h),1.66
–
1.54(m,2h).
13
c nmr(101mhz,cdcl3)δ148.27(s),130.73(s),129.59(s),124.67(s),123.40(s),122.71(s),121.72(s),121.19(s),76.47(s),41.61(s),36.08(s).
[0055]
实施例3:化合物s3的合成
[0056]
化合物s3与化合物s1合成方法相同,不同之处在于化合物s3的起始原料为2-羟基-5-氯-苯乙酮。1h nmr(400mhz,cdcl3)δ7.02(d,j=8.5hz,1h),6.92(s,1h),6.73(d,j=8.5hz,1h),6.26(d,j=9.8hz,1h),5.62(d,j=9.8hz,1h),3.04(t,j=11.4hz,2h),2.85
–
2.77(m,2h),1.91(d,j=13.5hz,2h),1.85(s,1h),1.65
–
1.55(m,2h).
13
c nmr(101mhz,
cdcl3)δ151.01(s),131.01(s),128.63(s),125.95(s),125.59(s),123.26(s),122.26(s),117.70(s),75.55(s),41.64(s),36.20(s).
[0057]
实施例4:化合物s4的合成
[0058]
化合物s4与化合物s1合成方法相同,不同之处在于化合物s4的起始原料为4-氯-2-羟基-苯乙酮。1h nmr(400mhz,cdcl3)δ6.80(d,j=9.2hz,2h),6.74(d,j=7.9hz,1h),6.23(d,j=9.8hz,1h),5.52(d,j=9.8hz,1h),2.99(t,j=11.2hz,2h),2.75(d,j=12.4hz,2h),1.86(d,j=13.4hz,2h),1.64(s,1h),1.59
–
1.51(m,2h).
13
c nmr(101mhz,cdcl3)δ153.21(s),133.81(s),129.78(s),127.03(s),122.25(s),120.99(s),120.47(s),116.82(s),75.77(s),41.62(s),36.32(s).
[0059]
实施例5:化合物s5的合成
[0060]
化合物s5与化合物s1合成方法相同,不同之处在于化合物s5的起始原料为2-羟基-6-氯-苯乙酮。1h nmr(400mhz,cdcl3)δ7.01(t,j=8.0hz,1h),6.89(d,j=7.9hz,1h),6.78
–
6.69(m,2h),5.71(d,j=9.9hz,1h),3.07(t,j=10.9hz,2h),2.88
–
2.82(m,2h),1.99(s,1h),1.95(d,j=13.6hz,2h),1.70
–
1.62(m,2h).
13
c nmr(101mhz,cdcl3)δ153.53(s),131.20(s),130.83(s),129.08(s),121.75(s),120.20(s),119.61(s),115.25(s),75.25(s),41.66(s),36.08(s).
[0061]
实施例6:化合物s6的合成
[0062][0063]
其中,反应条件:(a)吡啶,乙酰氯,二氯甲烷,氮气,rt(室温);(b)三氯化铝,150℃。
[0064]
化合物s6-2的合成:将1.6g(10mmol)市售的s9-1溶于45ml无水二氯甲烷后,室温下搅拌下加入6.8ml(85mmol)无水吡啶,5min后加入0.86ml(12mmol)乙酰氯,充入n2,室温下搅拌。将混合物减压蒸馏,加入1n稀盐酸,用二氯甲烷和饱和氯化钠溶液分离,有机相用无水硫酸钠干燥,减压浓缩即得粗品,得到的粗品用硅胶柱色谱纯化,得到白色固体1.7g。
[0065]
化合物s6-3的合成:将650mg化合物s6-2(3mmol)与600mg(4.5mmol)alcl3混合,150℃下搅拌1h后固化。降至室温后,用乙酸乙酯溶解混合物,并加入适量1n稀盐酸,用乙酸乙酯和饱和氯化钠溶液分离,有机相用无水硫酸钠干燥,减压浓缩即得粗品,得到的粗品用硅胶柱色谱纯化,得到白色固体450mg。
[0066]
化合物s6与化合物s1的合成方法相同,不同之处在于s6的起始原料为s6-3。1h nmr(400mhz,cdcl3)δ6.96(t,j=12.3hz,1h),6.81(d,j=8.1hz,1h),6.31(d,j=9.8hz,1h),5.62(d,j=9.8hz,1h),3.18(t,j=11.7hz,2h),2.86(d,j=12.1hz,2h),1.98(d,j=13.3hz,2h),1.73(s,1h),1.68
–
1.58(m,2h).
13
c nmr(151mhz,cdcl3)δ149.61(s),132.54(s),130.54(s),124.37(s),122.11(s),122.31
–
119.29(m),120.80(s),120.80(s),77.43
–
77.02(m),76.91(d,j=32.0hz),41.54(s),36.09(s),35.37(s).
[0067]
实施例7:化合物s7的合成
[0068]
化合物s7与化合物s1的合成方法相同,不同之处在于s7的原料通过同s6-3的方法合成。1h nmr(400mhz,dmso)δ6.91(s,1h),6.78(s,1h),6.66(d,j=10.0hz,1h),5.72(d,j=10.0hz,1h),3.05(t,j=11.0hz,2h),2.90
–
2.80(m,2h),1.93(d,j=13.3hz,2h),1.70
–
1.57(m,3h).
13
c nmr(151mhz,cdcl3)δ153.95(s),133.80(s),131.52(s),130.85(s),121.61(s),118.85(d,j=8.3hz),115.81(s),77.21(s),76.93(d,j=20.9hz),76.79(s),76.10(s),41.63(s),36.18(s).
[0069]
实施例8:化合物s8的合成
[0070]
化合物s8与化合物s1合成方法相同,不同之处在于化合物s8起始原料为2-羟基-3-氟-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.17
–
7.08(m,3h),6.31
–
6.25(m,1h),5.78(d,j=9.3hz,1h),3.17(p,j=3.7hz,1h),2.92
–
2.78(m,4h),2.06
–
2.00(m,1h),2.03
–
1.98(m,1h),1.94(ddd,j=12.5,5.7,3.3hz,2h).
[0071]
实施例9:化合物s9的合成
[0072]
化合物s9与化合物s1合成方法相同,不同之处在于起始原料为2-羟基-4-氟-苯乙酮。1h nmr(400mhz,cdcl3)δ7.53(ddd,j=8.4,5.1,0.7hz,1h),7.00(td,j=8.2,2.4hz,1h),6.57(dd,j=8.0,2.3hz,1h),6.32(dd,j=9.4,0.7hz,1h),5.80(d,j=9.3hz,1h),3.17(q,j=3.7hz,1h),2.92
–
2.79(m,4h),2.01(ddd,j=12.5,5.7,3.4hz,2h),1.92(ddd,j=12.4,5.7,3.4hz,2h).
[0073]
实施例10:化合物s10的合成
[0074]
化合物s10与化合物s1合成方法相同,不同之处在于化合物s10起始原料2-羟基-5-氟-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.13(dd,j=8.0,2.2hz,1h),7.02(td,j=8.0,2.3hz,1h),6.79(dd,j=7.9,5.0hz,1h),6.28(d,j=9.1hz,1h),5.80(d,j=9.2hz,1h),3.17(p,j=3.7hz,1h),2.92
–
2.79(m,4h),2.01(ddd,j=12.5,5.7,3.4hz,2h),1.91(ddd,j=12.5,5.6,3.4hz,2h).
[0075]
实施例11:化合物s11的合成
[0076]
化合物s11与化合物s1合成方法相同,不同之处在于化合物s11起始原料2-羟基-6-氟-苯乙酮。1h nmr(500mhz,chloroform-d)δ7.28(ddd,j=8.1,7.1,4.9hz,1h),7.00(td,j=8.0,1.1hz,1h),6.72(dd,j=7.1,1.1hz,1h),6.63(d,j=9.2hz,1h),5.90(d,j=9.2hz,1h),3.17(p,j=3.7hz,1h),2.92
–
2.79(m,4h),2.01(ddd,j=12.5,5.7,3.4hz,2h),1.92(ddd,j=12.4,5.7,3.4hz,2h).
[0077]
实施例12:化合物s12的合成
[0078]
化合物s12与化合物s1合成方法相同,不同之处在于化合物s12起始原料为2-羟基-3-甲基-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.33
–
7.28(m,1h),7.11
–
7.03(m,2h),6.23
–
6.18(m,1h),5.77(d,j=9.3hz,1h),3.20
–
3.14(m,1h),2.91
–
2.79(m,4h),2.05(ddd,j=12.5,5.7,3.4hz,2h),1.94(ddd,j=12.3,5.7,3.4hz,2h).
[0079]
实施例13:化合物s13的合成
[0080]
化合物s13与化合物s1合成方法相同,不同之处在于化合物s13起始原料为2-羟基-4-甲基-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.38(dd,j=8.2,0.7hz,1h),6.84(ddd,j=8.5,1.9,0.8hz,1h),6.61(dd,j=2.2,0.7hz,1h),6.32(dd,j=9.3,0.7hz,1h),
5.79(d,j=9.3hz,1h),3.20
–
3.14(m,1h),2.91
–
2.79(m,4h),2.33(s,2h),2.01(ddd,j=12.3,5.7,3.4hz,2h),1.92(ddd,j=12.4,5.7,3.4hz,2h).
[0081]
实施例14:化合物s14的合成
[0082]
化合物s14与化合物s1合成方法相同,不同之处在于化合物s14起始原料为2-羟基5-甲基-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.16
–
7.12(m,1h),7.09
–
7.04(m,1h),6.78(d,j=7.8hz,1h),6.20(d,j=9.3hz,1h),5.76(d,j=9.3hz,1h),3.20
–
3.14(m,1h),2.91
–
2.79(m,4h),2.38(s,2h),2.01(ddd,j=12.5,5.7,3.4hz,2h),1.91(ddd,j=12.5,5.6,3.4hz,2h).
[0083]
实施例15:化合物s15的合成
[0084]
化合物s15与化合物s1合成方法相同,不同之处在于化合物s15起始原料为2-羟基-6-甲基-苯乙酮。1h nmr(400mhz,chloroform-d)δ7.12(dd,j=7.9,7.1hz,1h),6.87
–
6.81(m,1h),6.72(dd,j=7.1,1.1hz,1h),6.56(d,j=9.1hz,1h),5.82(d,j=9.1hz,1h),3.17(p,j=3.7hz,1h),2.92
–
2.79(m,4h),2.47(d,j=0.7hz,3h),2.01(ddd,j=12.3,5.7,3.4hz,2h),1.92(ddd,j=12.4,5.7,3.4hz,2h).
[0085]
实施例16:化合物s16的合成
[0086]
化合物s16与化合物s1合成方法相同,不同之处在于化合物s16起始原料为2-羟基-4-甲基-苯乙酮,并且再将产物成盐,方法是将其溶于盐酸的乙酸乙酯溶液中,减压浓缩,重复三次,得到化合物盐酸盐s16。1h nmr(500mhz,chloroform-d)δ7.39(dd,j=9.0,0.7hz,1h),6.71(dd,j=9.0,2.4hz,1h),6.38(d,j=2.4hz,1h),6.32(dd,j=9.4,0.8hz,1h),5.78(d,j=9.3hz,1h),3.82(s,2h),3.17(p,j=3.7hz,1h),2.92
–
2.79(m,4h),2.01(ddd,j=12.3,5.7,3.4hz,2h),1.91(ddd,j=12.4,5.7,3.5hz,2h).
[0087]
实施例17:化合物s17的合成
[0088]
化合物s17与化合物s16合成方法相同,不同之处在于化合物s17起始原料为2-羟基-5-甲氧基-苯乙酮。1h nmr(400mhz,chloroform-d)δ6.84
–
6.79(m,1h),6.78
–
6.73(m,2h),6.30(d,j=9.1hz,1h),5.80(d,j=9.2hz,1h),3.81(s,3h),3.20
–
3.14(m,1h),2.85(ddt,j=10.9,5.6,3.6hz,4h),2.05
–
1.97(m,2h),1.96
–
1.88(m,2h).
[0089]
实施例18:化合物s18的合成
[0090]
化合物s18与化合物s16合成方法相同,不同之处在于化合物s18起始原料为2-羟基-6-甲氧基-苯乙酮。1h nmr(500mhz,chloroform-d)δ7.09
–
7.03(m,1h),6.75
–
6.68(m,2h),6.62(d,j=9.5hz,1h),5.81(d,j=9.3hz,1h),3.88(s,2h),3.20
–
3.14(m,1h),2.89
–
2.82(m,2h),2.03
–
1.98(m,1h),1.92(dd,j=5.7,3.5hz,1h).
[0091]
实施例19:化合物s19的合成
[0092][0093]
其中,反应条件:(a)吡咯,甲醇,rt(室温);(b)溴化苄,碳酸钾,dmf(n,n-二甲基甲酰胺);(c)硼氢化钠,甲醇,0℃-rt;(d)tsoh(对甲苯磺酸),甲苯,130℃。
[0094]
化合物s19与化合物s1的合成方法相同,不同之处在于起始原料s19-3的合成。
[0095]
化合物s19-3的合成:将化合物s19-2溶于dmf溶液,加入k2co3后,混合物移至0℃下搅拌并滴加溴化苄,加完后移入70℃下回流搅拌过夜。降至室温后,浓缩并用乙酸乙酯和饱和氯化钠溶液分离,有机相用无水硫酸钠干燥,减压浓缩即得粗品,得到的粗品用硅胶柱色谱纯化,即得纯品。
[0096]
化合物s19之后的合成方法与化合物s1的合成方法相同。1h nmr(400mhz,chloroform-d)δ7.45
–
7.40(m,1h),7.35(d,j=6.9hz,1h),6.92(s,1h),6.43(s,1h),6.34(d,j=9.1hz,1h),5.79(d,j=9.2hz,1h),5.14(d,j=1.1hz,2h),3.88(s,2h),3.17(d,j=3.8hz,1h),2.85(dd,j=10.9,5.7hz,2h),2.00(dd,j=5.7,3.3hz,1h),1.95
–
1.90(m,1h).
[0097]
实施例20:化合物s20的合成
[0098]
化合物s20与化合物s1合成方法相同,不同之处在于化合物s20起始原料为2-羟基-4-氯-苯乙酮和3-氮芥酮。1h nmr(400mhz,chloroform-d)δ7.49(dd,j=8.4,0.6hz,1h),7.25(dd,j=8.4,2.2hz,1h),6.93(d,j=2.3hz,1h),6.36
–
6.31(m,1h),5.89(d,j=9.2hz,1h),3.90(dd,j=12.4,3.8hz,2h),3.69(dd,j=12.4,3.8hz,2h),3.34(p,j=3.7hz,1h).
[0099]
实施例21:化合物s21的合成
[0100]
化合物s21与化合物s1合成方法相同,不同之处在于化合物s21起始原料为2-羟基-4-氯-苯乙酮和3-吡咯烷酮。1h nmr(400mhz,chloroform-d)δ7.49(dd,j=8.4,0.6hz,1h),7.25(dd,j=8.4,2.2hz,1h),6.92(d,j=2.2hz,1h),6.35
–
6.30(m,1h),5.77(d,j=9.3hz,1h),3.74(p,j=3.4hz,1h),3.37
–
3.27(m,2h),2.94(q,j=3.6hz,2h),2.11(dt,j=12.5,3.4hz,1h),2.07
–
2.00(m,1h).
[0101]
实施例22:化合物s22的合成
[0102]
化合物s22与化合物s1合成方法相同,不同之处在于化合物s22起始原料为2-羟基-4-氯-苯乙酮和3-哌啶酮。1h nmr(400mhz,chloroform-d)δ7.49(dd,j=8.4,0.6hz,1h),7.25(dd,j=8.4,2.2hz,1h),6.91(d,j=2.2hz,1h),6.35
–
6.30(m,1h),5.74(d,j=9.3hz,1h),3.31
–
3.22(m,3h),2.85
–
2.78(m,2h),2.01(dt,j=7.0,5.7hz,2h),1.69
–
1.60(m,2h).
[0103]
实施例23:化合物s23的合成
[0104]
化合物s23与化合物s1合成方法相同,不同之处在于化合物s23起始原料为2-羟基-4-氯-苯乙酮和去甲托品-3-酮。1h nmr(400mhz,chloroform-d)δ7.49(dd,j=8.4,0.6hz,1h),7.25(dd,j=8.4,2.2hz,1h),6.91(d,j=2.3hz,1h),6.33(dd,j=9.9,0.7hz,1h),5.76(d,j=9.9hz,1h),3.48(t,j=5.0hz,1h),3.22(dddd,j=6.8,4.5,2.5,1.4hz,2h),1.98(dd,j=12.4,3.9hz,2h),1.87
–
1.81(m,1h),1.78
–
1.70(m,2h),1.64
–
1.57(m,2h).
[0105]
实施例24:化合物s24的合成
[0106]
化合物s24与化合物s1合成方法相同,不同之处在于化合物s24起始原料为2-羟基-4-氯-苯乙酮和4-酮氮杂卓。1h nmr(400mhz,chloroform-d)δ7.49(dd,j=8.4,0.6hz,1h),7.25(dd,j=8.4,2.2hz,1h),6.91(d,j=2.3hz,1h),6.32(dd,j=9.3,0.7hz,1h),5.78(d,j=9.3hz,1h),3.76(tt,j=4.9,3.5hz,1h),2.87
–
2.75(m,4h),1.98(dt,j=12.3,6.1hz,1h),1.90(dt,j=12.5,6.1hz,1h),1.89
–
1.74(m,2h),1.70
–
1.61(m,2h).
[0107]
实施例25:化合物s25的合成
[0108][0109]
其中,反应条件:(a)四氢呋喃,三乙胺,回流,5h;(b)nabh4,etoh;(c)tsoh,甲苯,回流,72h;(d)6n hcl,90℃,12h。
[0110]
化合物s25的合成通过参考j.med.chem.2002,45,492-503的方法合成。
[0111]
化合物s25-3的合成;化合物s25-3的合成通过参考patent ep 431943,june12,
1991的方法合成。将化合物s25-2(1eq)溶于四氢呋喃中,加入三乙胺(0.5eq),加热回流5小时。用0.5n hcl和乙酸乙酯萃取分离,有机层用氯化钠洗涤,用无水硫酸钠干燥,减压浓缩。用乙酸乙酯结晶得到化合物s25-3。
[0112]
化合物s25-4的合成:将化合物s25-3溶于甲醇中,在n2下加入nabh4,常温下搅拌12h。用饱和氯化铵和二氯甲烷萃取分离,有机层用饱和氯化钠洗涤,用无水硫酸钠干燥,减压浓缩,得到化合物s25-4。
[0113]
化合物s25-5的合成;将化合物s25-4溶于甲苯中,加入对甲苯磺酸,回流72h。之后用饱和碳酸钠和二氯甲烷萃取分离,有机层用饱和氯化钠洗涤,用无水硫酸钠干燥,减压浓缩得到化合物s25-5。
[0114]
化合物s25的合成;将化合物s25-5溶于乙醇中,加入6n hcl,90℃回流12h。冷却至室温,混合物与甲苯共沸,然后用甲醇纯化得到白色固体s25。1h nmr(400mhz,chloroform-d)δ7.27(s,1h),7.05(d,j=2.8hz,1h),6.89
–
6.84(m,1h),6.36(d,j=8.2hz,1h),6.18(d,j=8.2hz,1h),3.81(s,3h),2.87(q,j=3.7hz,4h),2.07
–
1.97(m,4h).
[0115]
实施例26:化合物s26的合成
[0116][0117]
其中,反应条件:(a)30%h2o2(过氧化氢),hoac(乙酸),90℃,2h。
[0118]
化合物s26的合成:化合物s26的合成方法与s25相同,不同之处在于中间体化合物s26-1的合成,化合物s26的合成通过参考j.med.chem.2002,45,492-503的方法合成。
[0119]
化合物s26-1的合成:将化合物s25-1溶解于冰醋酸中,然后加入30%的h2o2,将混合物加热到90℃,搅拌2h。反应完成后,冷却并浓缩至含适量的溶剂,用水和二氯甲烷萃取分离,有机层用氯化钠洗涤,用无水硫酸钠干燥,减压浓缩,得化合物s26-1。
[0120]
化合物s26之后的合成方法与化合物s25相同。1h nmr(400mhz,chloroform-d)δ7.76(d,j=8.8hz,1h),7.11(d,j=2.7hz,1h),6.98(dd,j=8.8,2.8hz,1h),6.74(d,j=9.6hz,1h),5.99(d,j=9.7hz,1h),3.81(s,2h),3.17
–
3.12(m,1h),2.88(dddd,j=5.6,4.6,3.8,0.8hz,4h),2.12(td,j=5.2,3.8hz,4h).
[0121]
实施例27:化合物s27的合成
[0122]
化合物s27的合成与化合物s25的合成方法相同,不同之处在于原料为2-巯基-5-氯-苯乙酮。1h nmr(500mhz,chloroform-d)δ7.47(d,j=2.3hz,1h),7.34(d,j=7.2hz,1h),7.30(dd,j=7.2,2.1hz,1h),6.42(d,j=8.2hz,1h),6.18(d,j=8.2hz,1h),2.87(q,j=3.7hz,4h),2.45
–
2.40(m,1h),2.09
–
1.97(m,4h).
[0123]
实施例28:化合物s28的合成
[0124]
化合物s28的合成与化合物s26的合成方法相同,不同之处在于原料为2-巯基-5-氯-苯乙酮。1h nmr(500mhz,chloroform-d)δ7.87(d,j=8.8hz,1h),7.63(d,j=2.2hz,1h),7.46(dd,j=8.8,2.2hz,1h),6.80(d,j=9.6hz,1h),5.99(d,j=9.7hz,1h),3.14(q,j=3.7hz,1h),2.88(dddd,j=5.6,4.6,3.7,0.8hz,4h),2.18
–
2.08(m,4h).
[0125]
实施例29:化合物s29的合成
[0126][0127]
其中,反应条件:(a)乙酸铵,水,室温;(b)吡咯,乙醇,rt(室温);(c)c6h5n(so2cf3)2(n-苯基双(三氟甲烷磺酸亚胺)),lihmds(双胺基锂),thf(四氢呋喃),-78℃-rt;(d)4-乙基苯硼酸,pd[p(c6h5)3]4(四(三苯基膦)钯),licl(氯化锂),aq na2co3(碳酸钠),dme(二甲醚),回流;(e)tsoh,甲苯,130℃。
[0128]
化合物s29-1的合成:化合物s29-1通过参考j.org.chem.2008,73,6,2090-2095的方法合成。将市售的2,4-戊二酮(1eq)溶于水(5ml)中,加入醋酸铵(5eq),缓慢滴加市售的苯乙二醛一水合物(1eq),室温搅拌。原料反应完全后,产生固体,过滤,滤饼用水洗涤三次,用乙醇纯化得到化合物s29-1。
[0129]
化合物s29-2的合成:化合物s29-2的合成与化合物s1-3的合成过程相同。
[0130]
化合物s29-3的合成:化合物s29-3通过参考j.med.chem.2009,52,18,5685
–
5702的方法合成。将化合物s29-2(1eq)溶于四氢呋喃中,充入氮气,然后置于-78℃下,将1n的lihmds(1.2eq)用四氢呋喃稀释后缓慢滴入,搅拌1h。滴入c6h5n(so2cf3)2(1.2eq)的四氢呋喃溶液,缓慢升至室温,搅拌12h。反应完成后,将混合物倒入冰水中,有机相分别用1n的盐酸水溶液、1n的氢氧化钠水溶液、氯化钠溶液洗涤,然后用无水硫酸钠干燥,浓缩,用硅胶柱色谱法纯化得到化合物s29-3。
[0131]
化合物s29的合成:化合物s29是通过suzuki偶联反应合成的,参考j.org.chem.2008,73,6,2090
–
2095。将s29-3溶于dme中,向溶液中加入pd[p(c6h5)3]4(0.02eq),2n na2co3(3eq),licl(3eq),4-乙基苯硼酸(1.1eq)。充入氮气,回流12h。冷却至室温,用水和乙酸乙酯萃取,有机层用盐水洗涤,并用无水硫酸钠干燥。粗品用柱色谱法纯化得到化合物s29。1h nmr(400mhz,chloroform-d)δ8.70(s,1h),7.44
–
7.39(m,2h),7.35
–
7.30(m,3h),7.26
–
7.21(m,1h),5.71(s,1h),2.90
–
2.83(m,2h),2.69(d,j=7.3hz,1h),2.07(dd,j=6.0,3.8hz,1h),1.97(dd,j=6.1,3.9hz,1h),1.23(t,j=7.2hz,2h).
[0132]
实施例30:化合物s30的合成
[0133][0134]
其中,反应条件:(a)2-羰基丙醛,乙酸,水,室温;(b)乙二醛,水,回流。
[0135]
化合物s30与化合物s29合成方法相同,不同之处在于原料s30-1的合成。
[0136]
化合物s30-1的合成:化合物s30-1通过参考j.med.chem.2012,55,2,935
–
942的方法合成。将乙酸缓慢滴入肼(1eq)的水溶液中,然后缓慢滴加2-羰基丙醛(1eq),将混合物室温下搅拌过夜。向混合物中加入二氯甲烷,并萃取分离,有机相干燥浓缩,用柱色谱纯化得到化合物腙s30-1。将其配置成40%的腙(1eq)水溶液,缓慢加入乙二醛(1eq),回流1h。用二氯甲烷萃取分离,有机层用无水硫酸钠干燥,浓缩,使用柱色谱纯化。
[0137]
化合物s30的合成:化合物s30的合成过程与s29相同。1h nmr(400mhz,chloroform-d)δ7.33
–
7.22(m,3h),6.26(s,1h),3.80(s,2h),3.17(d,j=3.8hz,1h),2.90
–
2.83(m,2h),2.69(d,j=7.3hz,1h),2.08(dd,j=6.0,3.8hz,1h),1.98(dd,j=6.0,3.8hz,1h),1.23(d,j=14.5hz,1h),1.23(s,1h).
[0138]
实施例31:化合物s31的合成
[0139][0140]
其中,反应条件:(a)bnoh(苄醇),nah(氢化钠),dmf,0℃,72%;(b)naome,meoh,toluene,70℃,56%;(c)n-buli(正丁基锂),thf,-75℃,acetaldehyde(乙醛),65%;(d)dmp(戴斯马丁试剂),dcm,75%;(e)h2,cat.pd/c,etoac,88%。
[0141]
化合物s31与化合物s29合成方法相同。不同之处在于原料s31-6的合成,通过参考j.med.chem.2020,63,5879-5955的方法合成。
[0142]
化合物s31-2的合成:将市售的2,4-二氯吡啶(1eq)用dmf溶解,置于0℃下,在n2保护条件下加入nah(1.1eq),在该温度下搅拌20min。然后加入苯甲醇(1eq),混合物继续在该温度下搅拌1h。反应结束后,倒入冰水中,过滤取滤饼,然后用水洗涤。将固体在正己烷中搅拌,再次收集固体,干燥后得到化合物s31-2。
[0143]
化合物s31-3的合成:将化合物s31-2(1eq)溶于甲苯中,加入将naome(2eq)的甲醇溶液,在60℃搅拌5h。反应后除去溶剂,用乙酸乙酯和水萃取分离,有机层用氯化钠洗涤并用无水硫酸钠干燥,减压浓缩,使用正己烷结晶得到白色固体s31-3。
[0144]
化合物s31-4的合成:将化合物s31-3(1eq)溶于四氢呋喃中,置于-78℃下,然后加入正丁基锂(1.2eq)的己烷溶液,保持该温度搅拌30min。加入乙醛,继续在该温度下搅拌2h。反应完成后,用饱和氯化铵溶液淬灭反应,用乙酸乙酯萃取,有机相用氯化钠洗涤,然后用无水硫酸钠干燥,减压浓缩,然后使用柱色谱纯化得到白色固体s31-4。
[0145]
化合物s31-5的合成;在室温下用二氯甲烷溶解化合物s31-4(1eq),滴加dmp(1.2eq)后,滴加水,搅拌10min。反应完成后,混合物用硅藻土过滤,滤饼用二氯甲烷洗涤,滤液加饱和碳酸氢钠萃取,有机相用饱和氯化钠洗涤并用无水硫酸钠干燥,浓缩后用柱色谱纯化得到化合物s31-5。
[0146]
化合物s31-6的合成:将化合物s31-5溶于乙酸乙酯中,加入pd/c,将空气置换为n2,然后置换为h2,室温下搅拌3h。将混合物用硅藻土过滤,滤饼用乙酸乙酯洗涤,然后滤液减压浓缩,用柱色谱纯化得到化合物s31-6。
[0147]
化合物s31的合成:化合物s31的合成过程与s29相同,不同之处在于原料为s31-6。1h nmr(400mhz,chloroform-d)δ8.31(d,j=1.8hz,1h),7.62(d,j=1.8hz,1h),7.37
–
7.32(m,2h),7.28(dt,j=8.1,1.0hz,2h),5.86(s,1h),3.20
–
3.14(m,1h),2.87(ddt,j=9.8,6.0,3.7hz,4h),2.73
–
2.65(m,2h),2.06(ddd,j=12.4,6.1,3.8hz,2h),1.95(ddd,j=12.3,6.0,3.9hz,2h),1.23(t,j=7.2hz,3h).
[0148]
实施例32:钙离子流的测定
[0149]
选择以上实施例中的部分化合物以及式b1~b3所示的对照组化合物作为实验对象,式b1~b3的结构式如下所示:
[0150][0151]
测试上述化合物对5ht受体钙离子流的活性,具体操作如下:在钙流测定实验8小时之前,将稳定转染的5-ht
2a/2b/2c
受体的hek 293t细胞以15,000个细胞/孔的密度接种在384孔板中,孔板中含有1%透析的fbs的dmem。除去培养基后,然后将细胞(20μl/孔)在37℃下用在flipr缓冲液(19hbss,2.5mmol/l丙磺舒和20mmol/l hepes,ph 7.4)中重建的fluo-4直接染料(invitrogen)孵育1小时。加载染料后,将细胞置于fliprtetra荧光成像板读数器(molecular devices)中;在flipr缓冲液中以3倍浓度制备的药物稀释液,并等分到384孔板中,也加入到flipr中。fliprtetra的流体模块和平板读数器被编程为读取基线荧光10秒(1读/秒),然后加入10μl药物/孔并读取6分钟(1读/秒)。将每个孔中的荧光归一化至起始前10个度数的平均值(即,基线荧光)。然后,测定药物添加后60s内发生的最大倍数增加,超过载体或药物引起的基线荧光。
[0152]
表1代表性α-色烯螺哌啶衍生物及对比化合物对5ht2受体钙离子流活性测定结果
[0153][0154][0155]
na:表示无活性,nd:表示没有进行活性测定。
[0156]
与目前现有的两类g蛋白偏向性5-ht
2c
受体激动剂相比,α-色烯螺哌啶结构独特,为本发明首次报道。代表性化合物如化合物s1,s2,s4,s5和s8活性比阿朴菲类1857[ec
50
(e
max
)=308nm(86.1%),acs cent sci,2020,6,213-25]和18b[ec
50
(e
max
)=103nm(95.9%),acs chem neurosci,2020,11,549-59;zl201910594756.6]相当;其5-ht
2c
受体选择性明显优于阿朴菲类11b[5-ht2c:ec
50
(e
max
)=51nm(93.6%);5-ht2b:ec
50
(e
max
)=794.3nm(25.4%);5-ht2a:ec
50
(e
max
)=317.7nm(55.2%)]和11f[5-ht2c:ec
50
(e
max
)=23.6nm(101.8%);5-ht2b:ec
50
(e
max
)=278.4nm(63.5%);5-ht2a:ec
50
(e
max
)=596.4nm(44.9%)](bioorganic chemistry 123(2022)105795;zl201910594756.6),对5-ht
2a
和/或5-ht
2b
受体几乎无激动活性。
[0157]
与曾批准上市的5-ht
2c
受体激动剂氯卡色林(lorcaserin)相比,氯卡色林对5-ht
2a
、5-ht
2c
受体均有较强的活性,对5-ht
2b
受体的活性达到了136nm,且为完全激动剂,本技术所筛选的部分α-色烯螺哌啶类化合物(如s1、s2、s4、s5、s8等)虽然对5ht
2c
受体的活性弱于氯卡色林,但对5ht
2b
受体具有较弱的部分活性甚至无活性,因此安全性更好。
[0158]
此外,α-色烯螺哌啶环的双键被还原的对照化合物b1~b3,对5-ht
2c
受体均无活性,由此可知,α-色烯螺哌啶环中的双键对5-ht
2c
受体的活性有一定的相关性。
[0159]
实施例33:tangoβ-arrestin募集实验的测定
[0160]
选择以上化合物s1、s4以及氯卡色林作为实验对象,研究不同化合物对β-arrestin信号通路的影响,采用5-ht
2c tango质粒的htla细胞进行β-arrestin-2募集活性的检测。具体操作如下:细胞按照钙流实验相同的方法进行铺板(40μl/孔),然后加入与钙流实验一致的待测化合物溶液并孵育20小时(37℃,5%co2)。之后将化合物溶液倾出,加入bright-glo试剂(20μl/孔)并孵育20分钟。最后采用envision计数仪(perkinelmer)检测发光。同样地,5-ht的活性强度标准化为100%,剂量-效应曲线采用graphpad prism 7.0进行拟合。所有化合物的数据至少独立测试2次取平均值。
[0161]
测试结果如图1所示(图1a:氯卡色林、图1b:s1化合物、图1c:s4化合物),氯卡色林对gq信号通路以及β-arrestin信号通路均有激活作用,而化合物s1和s4完全偏向性激活gq信号通路,而对β-arrestin信号通路无作用,即α-色烯螺哌啶衍生物为完全g蛋白偏向性5-ht
2c
受体选择性激动剂。
[0162]
实施例34:herg钾离子通道抑制率实验的测定
[0163]
采用全自动电生理膜片钳qpatch(sophion,丹麦)检测化合物s4对herg钾离子通道的抑制作用。具体操作如下:本试验所用的细胞为转染有herg cdna与稳定表达herg通道的cho细胞系(由丹麦sophion bioscience公司提供),细胞代数为p30。在初始阶段达成破膜的全细胞配置状态后,细胞记录至少120秒,以达到稳定。然后在整个过程中,细胞钳制在-80mv的电压下。细胞钳制电压去极化到+20mv以激活herg钾通道,2.5秒后再钳制到-50mv以消除失活并产生外向尾电流。上述的电压模式每15秒被应用到细胞。以上参数阈值记录中只有稳定细胞被允许进入药物处置的过程。含0.1%二甲基亚砜(溶剂)的外液应用到细胞,建立基线,再允许电流稳定3分钟。化合物溶液加入后细胞保持在测试环境中,直至该化合物的效果达到了稳定状态或以4分钟为限。在化合物不同浓度梯度的测试实验中,化合物由低到高浓度加至所钳制的细胞上。完成化合物测试后用外液清洗细胞直到电流恢复到稳定的状态。阳性对照(西沙必利)用于实验中以确保细胞反应正常及细胞质量的可靠。如无特殊说明,实验都是在常规室温下进行(~25℃)。试验数据由sophion提供的qpatch分析软件,excel等进行分析。
[0164]
测试结果显示α-色烯螺哌啶衍生物s4在10μm浓度下对herg钾离子通道抑制率仅43.9%的抑制率,这也说明α-色烯螺哌啶衍生物s4的心脏毒性风险较低,显示较好的心脏安全性。
[0165]
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1