一种VOCs吸附强度的预测方法
一种vocs吸附强度的预测方法
技术领域
1.本发明涉及吸附vocs材料技术领域,具体涉及一种vocs吸附强度的预测方法。
背景技术:
2.挥发性有机污染物气体具有一定的毒性,一旦超标对人体和环境都存在一定的危害。因此,对上述气体进行有效的监测和检测是非常有必要的。
3.单层mos2和掺杂fe原子单层mos2对挥发性有机污染物气体有较强吸附能力,但是,吸附强度不同,如果能够准确比较单层mos2和掺杂fe原子单层mos2对挥发性有机污染物气体的吸附强度,则可以根据吸附强度的结果来针对性应用单层mos2或金属掺杂的mos2。
4.现有技术中,针对单层mos2或掺杂fe原子单层mos2对不同挥发性有机污染物气体吸附强度的测试,通常需要先制备得到单层mos2或掺杂fe原子单层mos2,然后,利用得到的单层mos2或掺杂fe原子单层mos2进行不同挥发性有机污染物气体的吸附试验。然而,目前,单层mos2或掺杂fe原子单层mos2的合成十分困难,以至其对不同的挥发性有机污染物气体的吸附强度的测试也十分困难。所以,准确比较单层mos2和掺杂fe原子单层mos2对挥发性有机污染物气体的吸附强度仍是一大难题。
技术实现要素:
5.有鉴于此,本发明提供了一种vocs吸附强度的预测方法,本发明提供的预测方法可以准确比较单层mos2或掺杂fe原子单层mos2对vocs的吸附强度。
6.为了实现以上目的,本发明提供了一种vocs吸附强度的预测方法,包括以下步骤:
7.采用第一性原理,对所述本征mos2单层原胞模型、fe掺杂mos2单层超胞模型以及vocs分子原胞模型进行几何优化,分别得到稳定态的本征mos2单层原胞模型、稳定态的fe掺杂mos2单层超胞模型和稳定态的vocs分子原胞模型;
8.将所述稳定态的vocs分子原胞模型分别添加到稳定态的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型表面,分别得到吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型;
9.对所述吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型进行几何优化,分别得到稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型;
10.通过广义梯度近似法计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea;
11.比较稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea绝对值,吸附能ea绝对值大的对应的模型,则说明该模型对vocs分子的吸附强度高;
12.优选地,所述构建fe掺杂mos2单层超胞模型包括以下步骤:采用第一性原理,在垂直于本征mos2单层原胞模型方向胞上添加一个fe原子,得到fe掺杂mos2单层超胞模
型。
13.优选地,所述构建本征mos2单层原胞模型包括以下步骤:对mos2原胞模型沿0001晶面进行切割,然后,在垂直于z轴方向添加的真空层,得到所述本征mos2单层原胞模型。
14.优选地,所述vocs分子包括苯、环氧乙烷、氯乙烯或乙醛。
15.优选地,所述吸附时,vocs分子原胞模型与稳定态的本征mos2单层原胞模型或稳定态的fe掺杂mos2单层超胞模型表面的距离为
16.优选地,所述吸附能ea通过式i计算得到:
17.ea=e
system-e
mos2-e
vocs 式i;
18.e
system
为稳定态的吸附vocs的本征mos2单层原胞模型或稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能量;
19.e
mos2
为稳定态的本征mos2单层原胞模型或稳定态的fe掺杂mos2单层超胞模型的能量;
20.e
vocs
表示稳定态的vocs分子原胞模型的能量;
21.所述e
system
为稳定态的吸附vocs的本征mos2单层原胞模型的能量时,所述e
mos2
为稳定态的本征mos2单层原胞模型的能量;
22.所述e
system
为稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能量时,所述e
mos2
为稳定态的fe掺杂mos2单层超胞模型的能量。
23.优选地,还包括测量稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附高度d。
24.优选地,还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能带结构。
25.优选地,还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的态密度。
26.优选地,还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的密立根电荷布居。
27.本发明提供了一种vocs吸附强度的预测方法,包括以下步骤:采用第一性原理,构建本征mos2单层原胞模型、fe掺杂mos2单层超胞模型和vocs分子原胞模型;采用第一性原理,对所述本征mos2单层原胞模型、fe掺杂mos2单层超胞模型以及vocs分子原胞模型进行几何优化,分别得到稳定态的本征mos2单层原胞模型、稳定态的fe掺杂mos2单层超胞模型和稳定态的vocs分子原胞模型;将所述稳定态的vocs分子原胞模型分别添加到稳定态的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型表面,分别得到吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型;对所述吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型进行几何优化,分别得到稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型;通过广义梯度近似法计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea;比较稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea绝对值,吸附能ea绝对值大的对应的模型,则说明该模型对vocs分子的吸附强度高。本发明无需通过复杂的试验过程,通过上述步骤的设定,
可以准确比较单层mos2或掺杂fe原子单层mos2对vocs的吸附强度。
附图说明
28.图1为vocs气体分子和稳定态的fe-mos
2-ml构型图;
29.图2为实施例1~4以及对比例1~3所得吸附模型的吸附能图;
30.图3为实施例1~4所得到的吸附模型的能带结构图;
31.图4为实施例1~4所得到的吸附模型的态密度图;
32.图5为实施例1~4所得吸附模型中的差分电荷密度图。
具体实施方式
33.本发明提供了一种vocs吸附强度的预测方法,包括以下步骤:
34.采用第一性原理,构建本征mos2单层原胞模型、fe掺杂mos2单层超胞模型和vocs分子原胞模型;
35.采用第一性原理,对所述本征mos2单层原胞模型、fe掺杂mos2单层超胞模型以及vocs分子原胞模型进行几何优化,分别得到稳定态的本征mos2单层原胞模型、稳定态的fe掺杂mos2单层超胞模型和稳定态的vocs分子原胞模型;
36.将所述稳定态的vocs分子原胞模型分别添加到稳定态的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型表面,分别得到吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型;
37.对所述吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型进行几何优化,分别得到稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型;
38.通过广义梯度近似法计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea;
39.比较稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea绝对值,吸附能ea绝对值大的对应的模型,则说明该模型对vocs分子的吸附强度高。
40.本发明采用第一性原理,构建本征mos2单层原胞模型(记为mos
2-ml)、fe掺杂mos2单层超胞模型(记为fe-mos
2-ml)和vocs分子原胞模型。
41.在本发明中,所述mos
2-ml的构建,优选包括以下步骤:对mos2原胞模型沿0001晶面进行切割,然后,在垂直于z轴方向添加的真空层,得到mos
2-ml。在本发明中,所述mos2原胞模型优选从第一性原理的模拟软件优选为materials studio软件,所述所述mos2原胞模型优选从materials studio软件的数据库中获取,在本发明中,所述mos2原胞模型具体优选为4
×4×
1的mos2的原胞模型。
42.在本发明中,所述vocs分子原胞模型中的vocs分子优选包括苯、环氧乙烷、氯乙烯或乙醛。
43.所述构建fe-mos
2-ml,优选包括以下步骤:采用第一性原理,在垂直于本征mos2单层原胞模型方向胞上添加一个fe原子,得到fe-mos
2-ml。
44.得到mos
2-ml、fe-mos
2-ml和vocs分子原胞模型后,本发明采用第一性原理,对所述
本征mos2单层原胞模型、fe掺杂mos2单层超胞模型以及vocs分子原胞模型进行几何优化,分别得到稳定态的本征mos2单层原胞模型(记为稳定态的mos
2-ml)、稳定态的fe掺杂mos2单层超胞模型(稳定态的fe-mos
2-ml)和稳定态的vocs分子原胞模型。
45.在本发明中,所述几何优化优选包括对mos
2-ml和fe-mos
2-ml的晶格常数和原子位置同时进行优化。
46.在本发明中,所述优化的步骤,优选包括以下步骤:
47.1)对所需进行待优化模型进行迭代,并计算所需优选的模型的总势能,然后利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
48.2)对势能面求最低极小值,若最低极小值大于收敛精度,则该步迭代结束,改变优化的模型的晶格常熟或待优化模型的各原子位置,继续进行下一步迭代
49.重复步骤1)和步骤2),直至低极小值小于收敛精度,优化结束。
50.在本发明中,当对mos
2-ml、fe-mos
2-ml进行优化时,所述优化的参数优选为:k空间网格优选为4
×4×
1;布里渊区k点的路径优选为:γ-m-k-γ,平面波能量精度为1
×
10-5
ev/atom,原子之间最大作用力为最大内应力0.05gpa,最大位移为平面波截断能量ecut为330ev,自洽收敛误差为1
×
10-6
ev/atom;当对vocs分子原胞模型进行优化时,所述k空间网格为2
×2×
2;布里渊区k点的路径优选同对mos
2-ml、fe-mos
2-ml进行优化时的设置相同,不再赘述。在本发明中,所述迭代次数优选为100。
51.得到稳定态的mos
2-ml、稳定态的fe-mos
2-ml和稳定态的vocs分子原胞模型后,本发明将所述稳定态的vocs分子原胞模型分别添加到稳定态的本征mos2单层原胞模型和稳定态的fe掺杂mos2单层超胞模型表面,分别得到吸附vocs的本征mos2单层原胞模型(记为vocs/mos
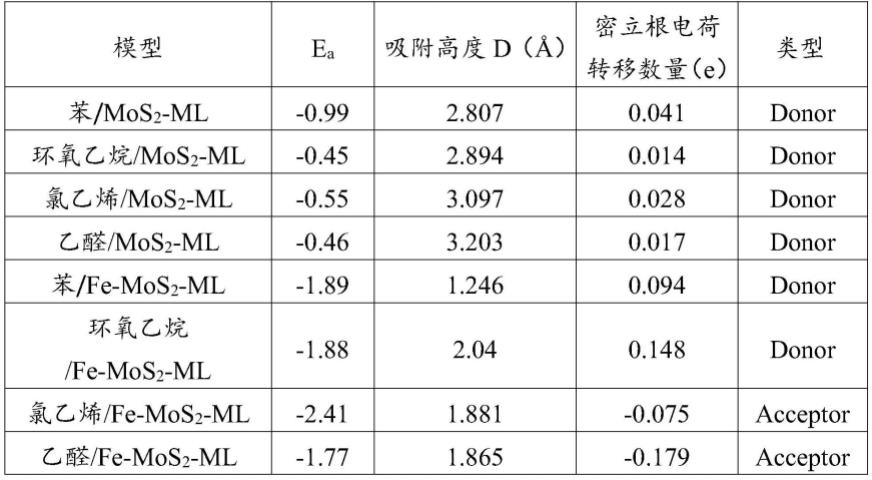
2-ml)和吸附vocs的fe掺杂mos2单层超胞模型(记为vocs/fe-mos
2-ml)。
52.在本发明中,所述稳定态的vocs分子原胞模型与稳定态的mos
2-ml或稳定态的fe-mos
2-ml表面的距离优选为
53.得到vocs/mos
2-ml和vocs/fe-mos
2-ml后,本发明对吸附vocs的本征mos2单层原胞模型和吸附vocs的fe掺杂mos2单层超胞模型进行几何优化,分别得到稳定态的吸附vocs的本征mos2单层原胞模型(记为稳定态的vocs/mos
2-ml)和稳定态的fe掺杂mos2单层超胞模型(记为稳定态的vocs/fe-mos
2-ml)。
54.得到vocs/mos
2-ml和vocs/fe-mos
2-ml后,本发明通过广义梯度近似法计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea;
55.在本发明中,所述吸附能ea通过式i计算得到:
56.ea=e
system-e
mos2-e
vocs 式i;
57.e
system
为为稳定态的吸附vocs的本征mos2单层原胞模型或稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能量;
58.e
mos2
为稳定态的本征mos2单层原胞模型或稳定态的fe掺杂mos2单层超胞模型的能量;
59.e
vocs
表示稳定态的vocs分子原胞模型的能量;
60.所述e
system
为稳定态的吸附vocs的本征mos2单层原胞模型的能量时,所述e
mos2
为稳
定态的本征mos2单层原胞模型的能量;
61.所述e
system
为稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能量时,所述e
mos2
为稳定态的fe掺杂mos2单层超胞模型的能量。
62.比较稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附能ea绝对值,吸附能ea绝对值大的对应的模型,则说明该模型对vocs分子的吸附强度高。
63.在本发明中,当吸附能ea为正值时,表示吸附过程是吸热的,气体分子不能稳定吸附在材料的表面;当吸附能ea为负值时,表示吸附过程是放热的,气体分子能够稳定吸附在材料的表面;对于吸附在二维纳米材料上的气体分子,其吸附能大于-0.80ev的为物理吸附,小于-0.80ev的为化学吸附;
64.在本发明中,优选还包括测量稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的吸附高度d。在本发明中,所述吸附高度d的测量优选用第一性原理模拟软件materials studio。在本发明中,所述吸附高度d越低,说明吸附强度越大。
65.在本发明中,优选还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的能带结构。在本发明中,所述能带结构的计算优选用第一性原理模拟软件materials studio。
66.在本发明中,优选还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的态密度。在本发明中,所述态密度的计算优选用第一性原理模拟软件materials studio。
67.在本发明中,优选还包括计算稳定态的吸附vocs的本征mos2单层原胞模型和稳定态的吸附vocs的fe掺杂mos2单层超胞模型的密立根电荷布居。在本发明中,所述密立根电荷布居的结果可以获得模型中各原子在原子轨道上的电荷分布。在本发明中,所述密立根电荷转移数量越大,说明vocs气体分子与模型表面的电子转移相互作用越大。在本发明中,所述密立根电荷布居的计算优选用第一性原理模拟软件materials studio。
68.下面结合实施例对本发明提供的技术方案进行详细地说明,但是不能把它们理解为对本发明保护范围的限定。
69.实施例1
70.(1)构建mos
2-ml、fe-mos
2-ml、苯原胞模型
71.采用第一性原理模拟软件materials studio,构建4
×4×
1的mos2原胞模型,沿(001)极性面进行切片,得到厚度为1个mo-s的mos
2-ml。
72.采用第一性原理模拟软件materials studio,在垂直于mos
2-ml方向胞上添加一个fe原子,得到fe-mos
2-ml;
73.采用第一性原理模拟软件materials studio,在坐标原点插入一个c原子,按照理论的c-h键长度确定h原子坐标后,插入c原子,对h原子和c原子进行连接,得到苯原胞模型。
74.(2)优化mos
2-ml、fe-mos
2-ml和苯原胞模型
75.采用广义梯度近似法对mos
2-ml进行几何优化,具体优化的参数为:k空间网格为4
×4×
1,布里渊区k点的路径设置为:γ-m-k-γ,平面波能量精度为1
×
10-5
ev/atom,原子之间最大作用力为最大内应力为0.05gpa,最大位移为平面波截断能量ecut为330ev,自洽收
敛误差为1
×
10-6
ev/atom。
76.对mos
2-ml的晶格常数和原子位置同时进行优化,计算mos
2-ml的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,第一步迭代的最低极小值大于收敛精度,继续进行第二步迭代,第二步迭代改变mos
2-ml的晶格常数或移动mos
2-ml中各原子位置进行,然后重新计算总势能和最低极小值,直至最低极小值小于设置的收敛精度,优化结束;迭代次数为100次。
77.所述fe-mos
2-ml的优化与mos
2-ml的优化相同;所述vocs分子原胞模型与所述mos
2-ml的优化的不同在于:k空间网格为2
×2×
2。
78.(3)将优化有的苯原胞模型分别添加至稳定态的mos
2-ml和稳定态的fe-mos
2-ml中,所述苯原胞模型位置与mos
2-ml中mo的距离为得到苯/mos
2-ml和苯/fe-mos
2-ml,所述苯原胞距离mos
2-ml和稳定态的fe-mos
2-ml的距离为
79.(4)将苯/mos
2-ml和苯/fe-mos
2-ml进行几何优化,优化方法和参数同苯原胞模型的优化。
80.(5)通过广义梯度近似法计算稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的吸附能。
81.(6)通过广义梯度近似法测量稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的吸附高度d。
82.(7)通过广义梯度近似法计算稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的能带结构。
83.(8)通过广义梯度近似法计算稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的态密度。
84.(9)通过广义梯度近似法计算稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的密立根电荷布居。
85.实施例2
86.与实施例的区别在于vocs分子为:环氧乙烷
87.所述环氧乙烷原胞模型的构建方法包括:在坐标原点插入一个c原子,按照理论的h-c键长和c-o键长确定h原子,o原子坐标后,插入h原子和o原子对其进行连接,得到环氧乙烷原胞模型。
88.实施例3
89.与实施例的区别在于vocs分子为:氯乙烯
90.在坐标原点插入一个c原子,按照理论的cl-c键长和c-h键长确定h原子,cl原子坐标后,插入h原子和cl原子对其进行连接,得到氯乙烯原胞模型。
91.实施例4
92.与实施例的区别在于vocs分子为:乙醛
93.在坐标原点插入一个c原子,按照理论的h-c键长和c-o键长确定h原子,o原子坐标后,插入h原子和o原子对其进行连接,得到乙醛原胞模型。
94.对比例1
95.①
吸附衬底替换为al-g,vocs分子为苯;
96.②
吸附衬底替换为al-g,vocs分子为环氧乙烷;
97.③
吸附衬底替换为al-g,vocs分子为氯乙烯;
98.④
吸附衬底替换为al-g,vocs分子为乙醛。
99.对比例2
100.①
吸附衬底替换为tm-石墨烯,vocs分子为苯;
101.②
吸附衬底替换为tm-石墨烯,vocs分子为环氧乙烷;
102.③
吸附衬底替换为tm-石墨烯,vocs分子为氯乙烯;
103.④
吸附衬底替换为tm-石墨烯,vocs分子为乙醛。
104.对比例3
105.①
吸附衬底替换为黑磷,vocs分子为苯;
106.②
吸附衬底替换为黑磷,vocs分子为环氧乙烷;
107.③
吸附衬底替换为黑磷,vocs分子为氯乙烯;
108.④
吸附衬底替换为黑磷,vocs分子为乙醛。
109.本发明中对实施例1~4中所得到的吸附能ea、吸附高度d和密立根电荷转移数量进行了汇总,见表1。
110.表1吸附能ea、吸附高度d和电荷转移量q
[0111][0112]
从表1中可以看出,vocs分子吸附在mos
2-ml和fe-mos
2-ml上的吸附能都是负的,这表示mos
2-ml和fe-mos
2-ml吸附vocs分子的过程是放热的,vocs能够稳定吸附在mos
2-ml和fe-mos
2-ml表面。
[0113]
稳定态的苯/mos
2-ml、稳定态的环氧乙烷/mos
2-ml、稳定态的氯乙烯/mos
2-ml和稳定态的乙醛/mos
2-ml的吸附能在-0.45~-0.99ev,属于弱物理吸附范围;稳定态的苯/fe-mos
2-ml、稳定态的环氧乙烷/fe-mos
2-ml、稳定态的氯乙烯/fe-mos
2-ml和稳定态的乙醛/fe-mos
2-ml的吸附高度在范围之间,相较于初始设置的吸附高度出现很大程度的减小。
[0114]
稳定态的苯/mos
2-ml、稳定态的环氧乙烷/mos
2-ml、稳定态的氯乙烯/mos
2-ml和稳
定态的乙醛/mos
2-ml的密立根电荷转移数量均小于0.1e,而稳定态的苯/fe-mos
2-ml、稳定态的环氧乙烷/fe-mos
2-ml、稳定态的氯乙烯/fe-mos
2-ml和稳定态的乙醛/fe-mos
2-ml的密立根电荷转移数量显著增大,这表明fe的引入增强了vocs气体分子与mos
2-ml表面的电子转移相互作用,进一步证实了fe可以有效改善mos
2-ml的气敏性能。
[0115]
图1为vocs气体分子(苯、环氧乙烷、氯乙烯或乙醛)和稳定态的fe-mos
2-ml,从图1可知:vocs气体分子、fe-mos
2-ml的原子结构关系。
[0116]
图2为实施例1~4以及对比例1~3所得吸附模型的吸附能,从图2可知:fe原子掺杂mos
2-ml后的吸附能提升。
[0117]
本发明利用模拟软件对实施例1~4所得到的吸附模型的能带结构进行了计算,能带结构数值图见图3,如图3可知,vocs吸附后会导致吸附体系(fe-mos
2-ml或mos
2-ml)的禁带宽度eg增大,当应用于气体传感器中时,带隙的微小变化会导致电导率的显著变化,从而影响气体传感中信号的灵敏度。
[0118]
本发明利用模拟软件对实施例1~4所得到的吸附模型的态密度进行计算,绘制其态密度图如图4,图4中(a)为稳定态的苯/mos
2-ml和稳定态的苯/fe-mos
2-ml的态密度图;图4(b)为稳定态的环氧乙烷/mos
2-ml和稳定态的环氧乙烷/fe-mos
2-ml的态密度图;图4(c)为稳定态的氯乙烯/mos
2-ml和稳定态的氯乙烯/fe-mos
2-ml的态密度图;图4(d)为稳定态的乙醛/mos
2-ml和稳定态的乙醛/fe-mos
2-ml的态密度图;
[0119]
从图4可知,vocs气体分子的加入并没有改变mos
2-ml的半导体性质,在费米表面附近也没有明显的变化。稳定态的vocs/fe-mos
2-ml的态密度图表明,fe的镶嵌整体向左波形变化,导致在其费米能级发生重大变化,从半导体金属属性的属性,但由于引入了vocs气体分子,fe-mos
2-ml的电子属性受到影响,在费米能级附近有一个明显的变化,从金属性质到半导体性质,表明vocs气体分子的引入与fe-mos
2-ml之间存在一定的相互作用。
[0120]
为了更清晰的了解vocs分子与mos
2-ml和fe-mos
2-ml衬底之间的相互作用,分别计算实施例1~4吸附模型体系中各稳定结构的差分电荷密度δρ,用来分析体系的电荷转移情况:
[0121]
所述差分电荷密度δρ的计算公式为:δρ=ρ
total-ρ
vocs-ρ
mos2
[0122]
所述ρ
total
表示整个吸附体系的电荷密度;
[0123]
ρ
vocs
表示单个vocs分子的电荷密度;
[0124]
ρ
mos2
表示mos
2-ml或fe-mos
2-ml的电荷密度。
[0125]
实施例1~4所得吸附模型中的差分电荷密度图,见图5。从图5(a)为稳定态的苯/mos
2-ml的差分电荷密度图;图4(b)为稳定态的环氧乙烷/mos
2-ml的差分电荷密度图;图4(c)为稳定态的氯乙烯/mos
2-ml的差分电荷密度图;图4(d)为稳定态的乙醛/mos
2-ml的差分电荷密度图;图5(e)为稳定态的苯/fe-mos
2-ml的差分电荷密度图;图5(f)为稳定态的环氧乙烷/fe-mos
2-ml的差分电荷密度图;图5(g)为氯乙烯/fe-mos
2-ml的差分电荷密度图;图5(h)为稳定态的乙醛/fe-mos
2-ml的差分电荷密度图。从图5(a)-(d)可以看出,mos
2-ml和vocs分子之间没有电荷交叠,表明vocs分子与mos
2-ml表面的吸附为弱物理吸附。从图5(e)-(h)可以看出,vocs分子与fe-mos
2-ml衬底之间有明显的电荷交叠,说明vocs分子与fe-mos
2-ml的相互作用要比vocs吸附在mos
2-ml上强得多,表明vocs与fe-mos
2-ml之间的吸附是化学吸附,电荷密度变化主要集中在金属fe原子和vocs分子以及vocs分子和它所接触
的衬底表面区域。
[0126]
综上,vocs在fe-mos
2-ml上吸附时具有更小的吸附能,更小的吸附高度,以及更多的电荷转移数,表明vocs吸附在fe-mos
2-ml上时是化学吸附,而在本征mos
2-ml上是弱物理吸附,fe-mos
2-ml对vocs的吸附更为敏感。
[0127]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1