基于机器学习算法的物种鉴定模型、物种鉴定方法和物种鉴定系统与流程
本发明属于生物信息学,涉及一种基于机器学习算法的物种鉴定模型、物种鉴定方法和物种鉴定系统。
背景技术:
1、在过去的几十年中,高通量测序技术(例如扩增子测序和宏基因组测序)的进步极大的提升了人类对微生物的理解,并广泛和显著的促进了食品工业、农业、环境修复、药物开发、人体健康等相关领域的发展。病毒的传播和传染疾病在不断的提醒着人类,先发制人地检测致病微生物的重要性毋庸置疑。目前,高通量测序技术越来越多的被用于临床样本的微生物检测,从而解决传统微生物诊断方法如体外培养、血清抗体检测和pcr的局限性,且高通量测序技术已经在确定抗生素耐药性、传染病暴发和癌症诊疗等方面发挥了关键作用。
2、在使用高通量测序技术进行微生物研究中,利用生物信息学工具来准确鉴定物种和评估其丰度对于解构高通量微生物组测序数据至关重要。然而,目前的生物信息学工具却在解析微生物数据时引入了大量的假阳性信号,极大的干扰了微生物的鉴定,以及相关下游分析。工欲善其事必先利其器,可以说好的物种分类器对于发展基于高通量测序数据的微生物研究至关重要。
技术实现思路
1、为了解决现有技术存在的不足,本发明的目的是提供一种基于机器学习算法的物种鉴定模型,对微生物组高通量测序数据进行准确的物种鉴定,从而解决高通量测序数据分析过程中假阳性和假阴性物种的鉴定问题,并服务于微生物组大数据分析以及临床致病菌的检测。
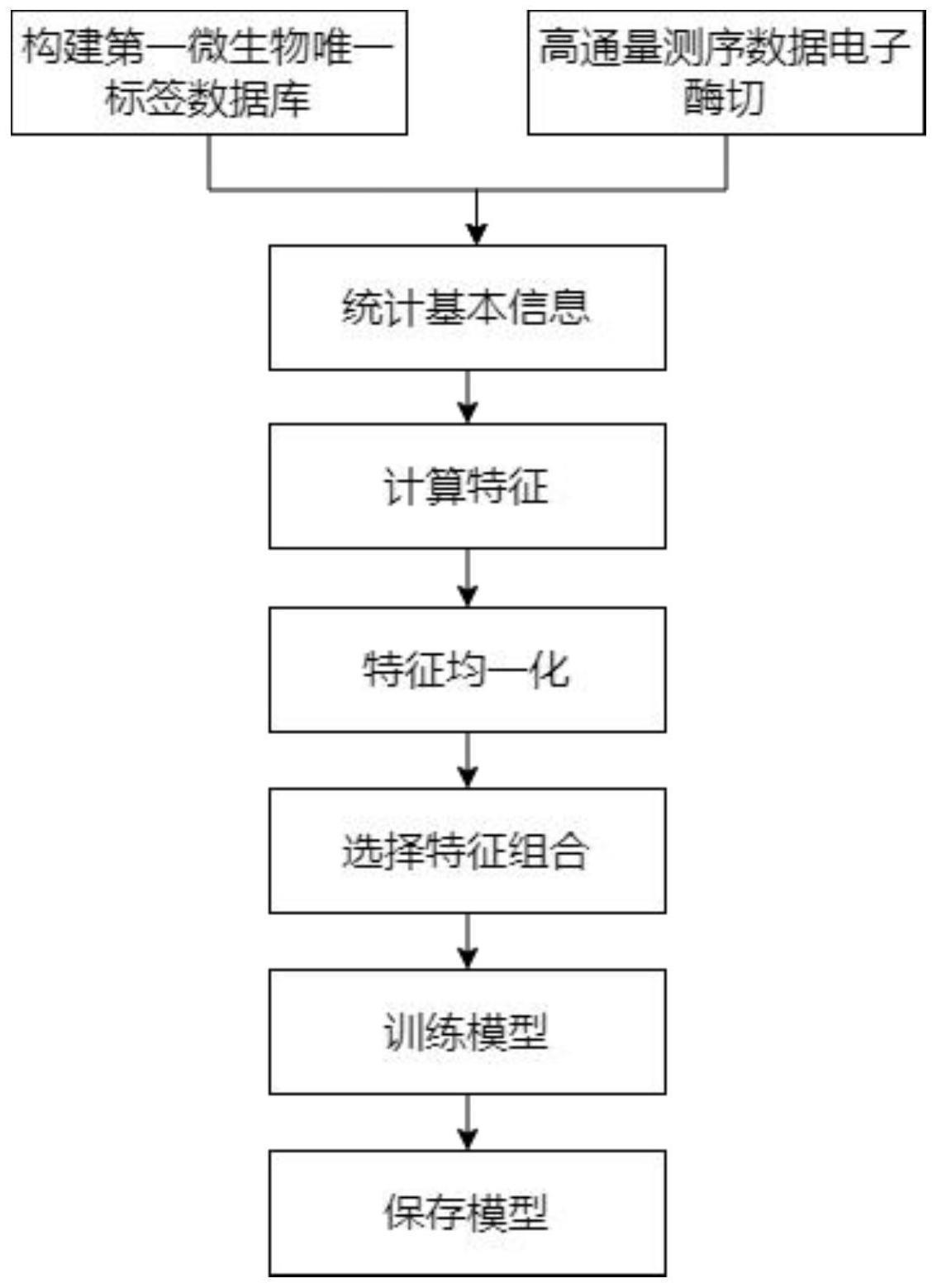
2、本发明提出了一种基于机器学习算法的物种鉴定模型,所述模型基于经电子酶切获取的标签序列与第一微生物唯一标签数据库进行比对,获得包括物种信息、实际测得的该物种唯一标签种类数sequenced tag num、实际测得的该物种唯一标签数量sequencedreads num和该物种的理论唯一标签种类数theoretical tag num在内的信息,基于上述信息获取基础特征和复合特征并进行处理,通过选择进行过处理的至少两个特征构成特征组合,对待构建的机器学习模型进行训练,将训练完毕后的机器学习模型保存为文件即获得所述的物种鉴定模型。
3、本发明提出了一种基于机器学习算法的高通量测序物种鉴定方法,包括如下步骤:
4、步骤一、构建机器学习模型,具体包括以下步骤:
5、步骤1.1、基于公开发表的基因组(例如包括但不限于从gtdb数据库、refseq数据库、ensembl数据库等下载)构建第一微生物唯一标签数据库(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。
6、步骤1.2、将已知真实物种组成的高通量测序数据进行电子酶切,获取iib标签序列,将标签序列比对到第一微生物唯一标签数据库,并统计比对上的物种信息、实际测得的该物种唯一标签种类数(sequenced tag num)、实际测得的该物种唯一标签数量(sequenced reads num)和该物种的理论唯一标签种类数(theoretical tag num)等信息(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。步骤1.2中比对获得的物种信息,含有大量的假阳性,同时步骤1.2中使用的是已知真实物种组成的数据,因此步骤1.2中还会同时标注物种的存在情况,使用0、1对物种是否真实存在进行标记,0表示不存在,1表示存在,用作后续模型训练的判别标签。将步骤1.2中获得的物种信息、实际测得的该物种唯一标签种类数(sequencedtagnum)、实际测得的该物种唯一标签数量(sequenced reads num)和该物种的理论唯一标签种类数(theoretical tag num)以及表示物种是否存在的标签保存成以制表符分隔的文本文件。
7、步骤1.3、计算特征,并将计算得到的特征增加到步骤1.2中所述的文本文件中。所述特征包括但不限于如下所列举,其中(1)-(4)为基础特征,即直接从步骤1.2所述的文本文件中获取,或者经过简单的加和计算得到(例如实际测得的全部物种的唯一标签种类数的和(total reads num));(5)-(23)为复合特征,即使用多个基础特征经过运算得到,或者为某个基础特征或复合特征排序后的次序信息。
8、步骤1.3中提出的特征具体如下:
9、(1)sequenced tag num:实际测得的属于某物种的唯一标签种类数;
10、(2)sequenced reads num:实际测得的属于某物种的唯一标签数量;
11、(3)theoretical tag num:某物种的理论唯一标签种类数;
12、(4)total reads num:实际测得的全部物种的唯一标签种类数的和;
13、(5)coverage:覆盖度,即实际测得的属于某物种的唯一标签种类数除以该物种的理论唯一标签种类数,公式为该复合特征描述说明了某一微生物基因组在测序数据中的相对完整性;覆盖度越高,则相对完整性越好;
14、(6)theoretical reads num:理论测序量,通过基础特征(例如“某物种的理论唯一标签种类数”)复合得到,即实际测得的属于某物种的唯一标签数量除以实际测得的属于该物种的唯一标签种类数,然后乘以该物种的理论唯一标签种类数,公式为该复合特征描述说明了某一微生物唯一标签在测序中的推测数量;
15、(7)tag depth:第一标签深度,即实际测得的属于某物种的唯一标签数量除以实际测得的属于该物种的唯一标签种类数,公式为
16、(8)tag depth2:第二标签深度,即实际测得的属于某物种的唯一标签数量除以实际测得的属于该物种的唯一标签种类数,然后再除以该实际测得的全部物种的唯一标签种类数的和,公式为
17、(9)species depth:物种深度,即实际测得的属于某物种的唯一标签数量除以该物种的理论唯一标签种类数,公式为
18、(7)-(9)复合特征从不同角度描述说明了某一微生物的测序深度;某一微生物的测序深度即指测序得到的碱基总量与微生物基因组大小的比值;
19、(10)reads dtr:物种测序量,即实际测得的属于某物种的唯一标签数量除以实际测得的全部物种的唯一标签种类数的和,公式为
20、(11)reads dtr sqrt:物种测序量的算数平方根,即实际测得的属于某物种的唯一标签数量除以实际测得的全部物种的唯一标签种类数的和,最后取算数平方根,公式为
21、(12)theoretical reads dtr sqrt:物种理论测序量的算数平方根,即实际测得的属于某物种的唯一标签数量除以实际测得的属于该物种的唯一标签种类数,然后乘以该物种的理论唯一标签种类数,再除以实际测得的全部物种的唯一标签种类数的和,最后取算数平方根,公式为
22、(10)-(12)复合特征从不同角度描述说明了某一微生物的在测序中的dna含量;
23、(13)g-score:g分数,即实际测得的属于某物种的唯一标签种类数乘以实际测得的属于该物种的唯一标签数量,然后取算数平方根,公式为该复合特征是一个经验值,与物种是否存在呈高度正相关;
24、(14)g-score rank:g分数的秩,即将g分数按照从大到小的顺序排列,取排序后的次序;
25、(15)coverage log:覆盖度的对数,即对所述特征(5)取自然对数,公式为
26、(16)theoretical reads num log:理论测序量的对数,即对所述特征(6)取自然对数,公式为
27、(17)tag depth log:第一标签深度的对数,即对所述特征(7)取自然对数,公式为
28、(18)tag depth2 log:第二标签深度的对数,即对所述特征(8)取自然对数,公式为
29、(19)species depth log:物种深度的对数,即对所述特征(9)取自然对数,公式为
30、(20)reads dtr log:物种测序量的对数,即对所述特征(10)取自然对数,公式为
31、(21)reads dtr sqrt log:物种测序量的算数平方根的对数,即对所述特征(11)取自然对数,公式为
32、(22)theoretical reads dtr sqrt log:物种理论测序量的算数平方根的对数,即对所述特征(12)取自然对数,公式为
33、(23)g-score log:g分数的对数,即对所述特征(13)取自然对数,公式为
34、步骤1.4、对所述步骤1.3中提到的基本特征和复合特征进行数据处理,所述数据处理是指根据在基础特征或复合特征的获取过程中,是否经过对数处理,判断是否需要对获得的基础特征或复合特征进行数据处理;若未经过对数处理,则需要对对应的基础特征或复合特征进行均一化处理,若已经过对数处理,则不做任何处理;均一化处理包括但不限于robustscaler、minmaxscaler、normalizer、z-score等,所述均一化处理能够消除原始特征的尺度和数量级差异大的影响,使得各维特征对目标函数有相同权重的影响。
35、步骤1.5、从步骤1.4的所述数据处理后的特征中选择至少两个特征构成特征组合,使用机器学习算法(包括但不限于随机森林(random forest)、逻辑回归(logisticregression)、支持向量机(support-vector machines)、朴素贝叶斯(naive bayes)、k近邻(k-neighbors)、自适应增强(adaboost)、梯度提升(gradient boosting)等算法)进行模型训练,并将训练得到的模型保存为文件,作为后续调用的用于物种鉴定的模型。
36、步骤二、基于高通量测序数据进行物种鉴定并计算物种的相对丰度,包括以下步骤:
37、步骤2.1、对待鉴定的高通量测序数据进行电子酶切,获取标签序列,将标签序列比对到第一微生物唯一标签数据库,并统计比对上的物种信息、测得的该物种唯一标签种类数(sequenced tag num)、测得的该物种唯一标签数量(sequenced reads num)和该物种的理论唯一标签种类数(theoretical tag num)等信息,保存到文件中(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。
38、步骤2.2、计算与模型构建过程中相同的所有的基础特征和复合特征,并将计算得到的特征增加到步骤2.1所述文件中。
39、步骤2.3、对所述步骤2.2中提到的基础特征或复合特征进行数据处理,数据处理方法需要和步骤1.4所述方法一致。
40、步骤2.4、使用步骤1.5所述的机器学习算法,选择步骤1.5所述特征组合,使用步骤1.5所述保存的物种鉴定模型对步骤2.2中所述文件中的物种是否存在进行判定,存在记为1,不存在记为0。
41、步骤2.5、从步骤1.1的所述基因组中调取步骤2.4所述判定为存在的物种下的基因组,进行电子酶切,并构建第二微生物唯一标签数据库(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。
42、步骤2.6、将步骤2.1所述标签序列比对到步骤2.5所述第二微生物唯一标签数据库,并根据物种丰度计算公式计算得到物种丰度(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。
43、基于以上方法,本发明还提出了一种对高通量测序数据进行物种鉴定的系统。
44、所述物种鉴定系统包括:数据预处理模块、定性模块、定量模块和多酶切结果定性/定量合并模块;
45、所述数据预处理模块,包括:数据质控单元和电子酶切单元(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法);用于对每个基因组采用iib型限制性内切酶进行电子酶切,对酶切片段测序结果进行数据质控,获得质控后的测序数据;
46、所述定性模块包括:第一微生物唯一标签数据库和物种鉴定模型判别单元;主要用于对物种是否存在进行判定;
47、其中,
48、第一微生物唯一标签数据库:将电子酶切单元获得标签,在界、门、纲、目、科、属、种、株不同物种分类水平上判断上述每个标签的唯一性,输出不同物种分类水平、每个基因组的唯一标签,这些唯一标签组成第一微生物唯一标签数据库(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法);
49、物种鉴定模型判别单元:包含本发明构建的物种鉴定模型,将样品的高质量测序数据和第一微生物唯一标签数据库比对,即通过构建哈希表的方式,检测高质量测序数据在第一微生物唯一标签数据库中是否有序列完全一致的标签,获得不同物种分类水平鉴定到的唯一标签,统计比对上的物种信息、实际测得的该物种唯一标签种类数(sequencedtag num)、实际测得的该物种唯一标签数量(sequenced reads num)和该物种的理论唯一标签种类数(theoretical tag num)等信息,并通过对实际测得的该物种唯一标签种类数(sequenced tag num)、实际测得的该物种唯一标签数量(sequenced reads num)、该物种的理论唯一标签种类数(theoretical tag num)和实际测得的全部物种的唯一标签种类数的和(total reads num)四个基础特征进行组合,生成复合特征,进一步进行包括均一化在内的数据处理,使用训练好的物种鉴定模型对物种是否存在进行判定;
50、所述基础特征为直接对比对结果进行统计,或者经过简单的统计加和计算得到(例如实际测得的全部物种的唯一标签种类数的和(total reads num));
51、所述复合特征为使用多个基础特征经过运算得到,或者为某个特征排序后的次序信息;
52、所述训练好的物种鉴定模型为使用本发明物种鉴定方法中“步骤一、构建机器学习模型”所述方法构建的模型;
53、所述定量模块包括:第二微生物唯一标签数据库和定量分析单元(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法);用于根据物种丰度计算公式计算得到物种丰度;
54、所述第二微生物唯一标签数据库为:通过对候选微生物进行假阳性过滤,然后对过滤后微生物基因组信息通过所述电子酶切单元获得标签,在界、门、纲、目、科、属、种、株不同物种分类水平上判断每个标签的唯一性并输出不同物种分类水平、每个基因组的唯一标签,该唯一标签组成第二微生物唯一标签数据库;
55、所述多酶切结果定性/定量合并模块,包括:多酶切结果定性合并单元和多酶切结果定量合并单元(专利号:zl202011355328.7,专利名称:基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法)。用于判断将一种以上iib型限制性内切酶的不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度进行累加合并,判断是否检测到某微生物的唯一标签,并重新计算微生物定性信息,然后将一种以上iib型限制性内切酶酶切的定量结果进行累加合并,作为最终的相对定量结果。
56、该系统的运行需要提前基于步骤1.1所述方法构建一个第一微生物唯一标签数据库,基于步骤一所述方法构建一个物种鉴定模型。
57、本发明的有益效果包括:相较于目前其他同类主流生物信息学工具能够极为精准的进行物种鉴定并对其进行相对丰度估计,从而有效的避免了假阳性物种鉴定结果对下游分析的影响,这是其他工具难以实现的。
- 还没有人留言评论。精彩留言会获得点赞!