一种肿瘤恶性细胞基因预后风险模型构建方法与流程
1.本发明涉及生物技术领域,具体涉及一种肿瘤恶性细胞基因预后风险模型构建方法。
背景技术:
2.恶性肿瘤是严重危及人类健康的一类疾病,根据恶性肿瘤的起源不同可分为不同类型,包括来源于上皮细胞的恶性克隆性增生、间叶源性的恶性肿瘤以及淋巴造血系统的恶性疾病,来源于上皮细胞的恶性克隆性增生称为癌症,如肺癌、胃癌、乳腺癌、食管癌等,来源于间叶源性的恶性肿瘤可以称为肉瘤,如脂肪肉瘤、纤维肉瘤、骨肉瘤、间皮瘤等,淋巴造血系统的恶性疾病起源于淋巴系统的恶性肿瘤以及部分血液系统疾病,如白血病、多发性骨髓瘤等。不同的恶性肿瘤存在着特异的分子亚型与临床表现,虽然肿瘤的早期治愈率很高,但由于大部分肿瘤早期症状不明显,患者就诊时往往已处于中晚期,生存预后情况也不容乐观,临床上常用的肿瘤生物标志物特异性并不高,在不同肿瘤、年龄、性别的患者间均有较大的差异,不利于诊断与治疗,寻找与某类肿瘤起源相关的某种特定类型细胞基因,有利于不同肿瘤诊断生物标志物的应用手册制定与靶向药物的设计,具有较大的现实意义和学术意义。
3.随着高通量测序技术的发展与应用,恶性肿瘤的发病机制探索拥有了新的手段与方法,大大加快了人类肿瘤分子特征研究的步伐。传统的全转录组测序虽然可以提供海量的基因表达谱数据,但无法区分不同细胞谱系与细胞相互作用之间的关系,单细胞转录组测序的出现弥补了全转录组测序的不足,它提供了一种可以表征单个细胞转录状态的方法,可以根据恶性肿瘤的起源或具体特征选择合适的细胞类型进行深入研究。
技术实现要素:
4.为此,本发明提供一种肿瘤恶性细胞基因预后风险模型构建方法,以联合单细胞转录组测序和全转录组测序数据寻找肿瘤恶性细胞基因并构建预后风险模型,为肿瘤的临床诊断与治疗提供新的思路。
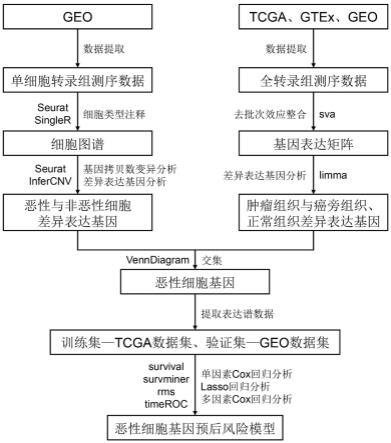
5.为了实现上述目的,本发明提供如下技术方案:一种肿瘤恶性细胞基因预后风险模型构建方法,所述方法包括:步骤一:从多个数据库中获取肿瘤患者的肿瘤组织、癌旁组织与正常组织的单细胞转录组测序数据集和全转录组测序数据集;步骤二:对单细胞转录组测序数据集进行预处理,筛选符合设定标准的细胞并获取其测序数据;步骤三:对预处理得到的单细胞转录组测序数据进行标准化和归一化,通过锚点整合并进行降维和聚类得到不同细胞群;步骤四:获取不同细胞类型的标志基因,根据标志基因在不同细胞群中的分布情况对不同细胞群进行细胞类型注释;
步骤五:将其中一种注释为设定细胞类型的细胞群提取出来,并区分为恶性与非恶性细胞,并对恶性与非恶性细胞进行差异表达基因分析,获得恶性与非恶性细胞的差异表达基因;步骤六:对全转录组测序数据集进行肿瘤组织与癌旁组织、正常组织的差异表达基因分析,然后进行取交集处理,获得肿瘤致病相关的恶性基因;步骤七:将得到的恶性与非恶性细胞的差异表达基因与获得的肿瘤致病相关的恶性基因取交集处理,获得肿瘤致病相关的恶性细胞基因;步骤八:对得到的肿瘤致病相关的恶性细胞基因进行单因素cox回归分析、lasso回归分析和多因素cox回归分析,筛选出与预后相关的恶性细胞基因,并构建预后风险模型。
6.进一步地,所述步骤一,具体包括:从geo数据库获得单细胞转录组测序数据集,从tcga、geo、gtex数据库获得全转录组测序数据集;纳入的数据集需满足以下条件:使用人类肿瘤组织样本,包括:人类正常组织样本、肿瘤组织样本和癌旁组织样本;每个数据集至少包含预设个数的样本。
7.进一步地,所述步骤一中,单细胞转录组测序数据集样本为具有配对原发性肿瘤和癌旁组织的患者样本;全转录组测序数据集包括:tcga-stad数据集,含有多个胃癌组织样本和多个癌旁组织样本;gtex-stomach数据集,含有多个正常胃组织样本;gse15459数据集,含有多个胃癌组织样本;gse29272数据集,含有多个胃癌组织样本和多个癌旁组织样本;gse57303数据集,含有多个胃癌组织样本;gse62254数据集,含有多个胃癌组织样本;gse66229数据集,含有多个胃癌组织样本和多个癌旁组织样本。
8.进一步地,所述步骤二中,筛选标准包括:每个基因有三个或三个以上的细胞表达;每个细胞表达500-6000个基因;线粒体rna含量小于20%。
9.进一步地,所述步骤三具体还包括:对不同细胞群进行差异表达基因分析,获得不同细胞群的差异表达基因,其中差异表达基因是采用r软件中的“seurat”包来识别的。
10.进一步地,所述步骤四中具体包括:使用r软件中的“singler”包或通过文献检索获得不同细胞类型的标志基因,并将细胞群中表达最高的标志基因所对应的细胞类型注释为细胞群的细胞类型。
11.进一步地,所述步骤四中,不同细胞类型的标志基因具体包括,上皮细胞:cdh1;内皮细胞:plvap;成纤维细胞:fn1;t细胞:cd8a;b细胞:tnfrsf17;巨噬细胞:cd163;nk细胞:klrd1;肥大细胞:kit。
12.进一步地,所述步骤五中,采用r软件中的“infercnv”包来区分恶性与非恶性细胞,以癌旁组织细胞作为参照,对肿瘤组织细胞的基因拷贝数变异情况进行分析,采用kmeans算法聚类并计算基因拷贝数变异得分值,将高于平均得分值的类群定义为恶性细胞,不高于平均得分值的类群定义为非恶性细胞。
13.进一步地,所述步骤六和步骤七中,采用r软件中的“venn”包进行取交集处理。
14.进一步地,所述步骤八中,以tcga-stad数据集作为训练集,gse62254数据集作为验证集,所构建的预后风险模型在训练集与验证集的预测能力评价标准roc大于设定值,证
明模型性能良好。
15.本发明具有如下优点:本发明提出的一种肿瘤恶性细胞基因预后风险模型构建方法,应用生物信息学,选择肿瘤患者群体作为研究对象,通过整合单细胞转录组测序数据集和全转录组测序数据集识别肿瘤致病相关的恶性细胞预后基因,并构建预后风险模型,有利于不同肿瘤诊断生物标志物的应用手册制定与靶向药物的设计,为肿瘤的临床诊断与治疗提供新的思路。
附图说明
16.为了更清楚地说明本发明的实施方式或现有技术中的技术方案,下面将对实施方式或现有技术描述中所需要使用的附图作简单地介绍。显而易见地,下面描述中的附图仅仅是示例性的,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图引伸获得其它的实施附图。
17.图1为本发明实施例提供的一种肿瘤恶性细胞基因预后风险模型构建方法的流程示意图;图2为本发明实施例提供的一种肿瘤恶性细胞基因预后风险模型构建方法中得到的胃癌致病相关的恶性基因示意图。
具体实施方式
18.以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
19.如图1所示,本发明实施例提供了一种基于单细胞和全转录组测序数据的胃癌恶性上皮细胞基因预后风险模型构建方法,该方法具体包括:从肿瘤基因组图谱(tcga, https://portal.gdc.cancer.gov/)基因表达综合数据库(geo, https://www.ncbi.nlm.nih.gov/geo/)和基因型组织表达数据库(gtex, https://gtexportal.org/home/)中获得肿瘤患者的肿瘤组织、癌旁组织与正常组织的单细胞和全转录组测序数据集,包括tcga-stad、gtex-stomach、gse15459、gse29272、gse57303、gse62254、gse66229和gse183904。
20.单细胞转录组测序数据集选择9名具有配对原发性肿瘤和癌旁组织的患者共18个样本进行深入研究,全转录组测序数据集tcga-stad含有375个胃癌组织样本和32个癌旁组织样本,gtex-stomach含有217个正常胃组织样本,gse15459含有192个胃癌组织样本,gse29272含有134个胃癌组织样本和134个癌旁组织样本,gse57303含有70个胃癌组织样本,gse62254含有300个胃癌组织样本,gse66229含有300个胃癌组织样本和100个癌旁组织样本。
21.对单细胞转录组测序数据集进行质量控制,选择符合标准的细胞用于后续研究,质量控制标准如下:1)每个基因有三个或三个以上的细胞表达;2)每个细胞表达500-6000个基因;3)线粒体rna含量小于20%,用于后续研究的细胞共49994个。
22.使用r软件中的“seurat”包对预处理后的单细胞数据进行标准化和归一化,通过
锚点整合并进行降维聚类,降维时所采用的pc数为10,聚类时所采用的分辨率为1.0,共降维聚类得到33个细胞群(cluster)。对不同细胞群进行差异表达基因分析,获得不同细胞群的差异表达基因,差异表达基因的|log2fc| 》 0.25且adj.p 《 0.05。
23.通过文献检索获得不同细胞类型的标志(marker)基因,根据标志基因在不同细胞群中的分布情况对不同细胞群进行细胞类型注释,不同细胞cluster类型注释时的marker基因需在该cluster中表达最高才可注释为该marker基因所对应的细胞类型。
24.不同细胞类型的标志基因具体如下:1)上皮细胞:cdh1;2)内皮细胞:plvap;3)成纤维细胞:fn1;4)t细胞:cd8a;5)b细胞:tnfrsf17;6)巨噬细胞:cd163;7)nk细胞:klrd1;8)肥大细胞:kit,共注释得到8种类型的细胞。
25.将其中注释为上皮细胞的细胞群提取出来,使用r软件中的“infercnv”包区分恶性与非恶性上皮细胞,以癌旁组织细胞作为参照,对肿瘤组织细胞的基因拷贝数变异情况进行分析,采用kmeans算法聚类并计算基因拷贝数变异得分值,将高于平均得分值0.00124的类群定义为恶性上皮细胞,然后使用r软件中的“seurat”包对恶性与非恶性上皮细胞进行差异表达基因分析,获得恶性与非恶性上皮细胞的差异表达基因,差异表达基因的|log2fc| 》 0.25且adj.p 《 0.05,排行前五的恶性与非恶性上皮细胞差异表达基因如下:1)恶性上皮细胞:capn8、cldn4、cyp3a5、phgr1和plec;2)非恶性上皮细胞:igfbp2、lipf、pga3、pga4和pga5。
26.对tcga-stad和gtex-stomach数据集以及gse15459、gse57303和gse66229数据集进行去批次效应整合,得到基因表达矩阵,对多个全转录组测序数据集使用r软件中的“limma”包进行肿瘤组织与癌旁组织、正常胃组织的差异表达基因分析,差异表达基因的|log2fc| 》 1.0且adj.p 《 0.05,然后使用r软件中的“venn”包进行取交集处理,获得胃癌致病相关的恶性基因,共获得92个上调基因和75个下调基因,如图2所示。
27.使用r软件中的“venn”包将单细胞转录组测序数据获得的恶性上皮细胞基因与胃癌致病相关的恶性基因进行取交集处理,获得胃癌致病相关的恶性上皮细胞基因共146个。
28.以tcga-stad数据集作为训练集,gse62254数据集作为验证集,使用r软件中的“survival”包、“survminer”包、“rms”包和“timeroc”包对胃癌致病相关的恶性上皮细胞基因进行单因素cox回归分析、lasso回归分析和多因素cox回归分析,筛选出与预后相关的恶性上皮细胞基因,构建预后风险模型,最终构建出一个包括10个胃癌致病相关的恶性上皮细胞基因预后风险模型,基因具体如下:akr1b1、cfdp1、impact、prr15l、pttg1ip、slc17a9、stx10、trim25、upp1和vcan,所构建的预后风险模型在训练集与验证集的预测能力评价标准roc均大于0.5,模型性能良好。
29.虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1