一种用于推断lncRNA-疾病关联的受校正相似性约束的概率矩阵分解方法
本发明涉及机器学习与生物基因结合领域,更具体地,涉及一种用于推断lncrna-疾病关联的受校正相似性约束的概率矩阵分解方法。
背景技术:
1、长非编码rna(lncrnas)与疾病之间的关系近年来引起了极大的兴趣。人们对lncrnas的理解尚处于起步阶段,但现在越来越多的证据表明lncrnas与某些人类疾病密切相关。在许多情况下,lncrnas被证明是基因表达的主要调节器,lncrna介导的基因表达涉及多种机制,如转录调节、翻译、蛋白质修饰以及rna蛋白或蛋白质复合物的形成。因此,它们在各种生物功能和疾病过程中起着关键作用,包括癌症;如乳腺癌、肺癌、乳腺癌、肾细胞癌、膀胱癌等。lncrnas与疾病之间的关联对于复杂人类疾病发病机制的研究具有重要意义,获取越多的lncrna-疾病关联对于了解疾病发病的病因,诊治与预防有着重要作用。但目前只有少数lncrna-疾病关联得到了实验验证,因为进行常规生物实验既昂贵又耗时。因此,迫切需要以有效的方式识别潜在的lncrna-疾病关联。随着技术的发展,越来越多的研究证实了通过构建计算模型可作为预测潜在的lncrna-疾病关联的有效工具。
2、近年来,有越来越多的计算方法被开发用来更好地推断潜在的lncrna-疾病关联。现有的计算方法大致可以分为三类:(1)基于机器学习的方法(2)基于网络的方法(3)基于矩阵分解的方法。
3、基于机器学习方法的模型,根据训练样本(已知的疾病相关lncrna)和未标记的样本(没有任何已知关联证据的lncrna-疾病对)来预测潜在的lncrna-疾病关联。其中用的比较多的是监督分类器如:支持向量机(svm)和朴素贝叶斯分类器等。但其局限性也是比较突出,由于lncrna-疾病关联数据集很难获得可靠的负样本信息,于是总是随机选择未标记的lncrna-疾病对作为阴性样本,这将严重影响预测性能。
4、基于构建网络的方法则不需要考虑负样本信息问题。基于网络的模型倾向于整合已知的lncrna-疾病关联网络,疾病语义/表型相似性网络,以及从已知的lncrna-疾病关联或lncrna-mirna相互作用中获得的lncrna功能相似性网络,利用已知的其它数据如lncrna-基因关联,lncrna-蛋白质,疾病-药物关联等来获取更多lncrna和疾病的相似性网络。然后在架构的网络上利用传播算法等做预测。但大多数这些方法的重要缺点是它们可能无法获得新疾病和/或新lncrna的预测结果。此外,lncrna-mirna相互作用网络、蛋白质相互作用网络和lncrna-疾病网络的不完整覆盖可能会对具有更多已知相关疾病的lncrna或与更多疾病相关的mirna相互作用伙伴产生一些偏倚的预测。
5、第三种方法是基于矩阵分解的方法来预测潜在的lncrna-疾病关联。标准矩阵分解的目的是找到两个低等级的潜在特征矩阵,其乘积为一个权重矩阵用于拟合原始矩阵。到目前为止,矩阵分解技术在生物信息领域得到了广泛的应用,因为矩阵分解不仅可以降低计算复杂度,而且相比前两种方法矩阵分解可以在解决矩阵稀缺性问题方面取得良好的性能,且不需要负样本。概率矩阵分解是在标准矩阵分解基础上假设用于拟合原始矩阵的潜在矩阵具有高斯噪声的正分布情况然后优化求解找到两个更合适的低等级的潜在特征矩阵。但传统的概率矩阵分解模型并没有考虑太多的生物信息如lncrna和疾病的相似性,因此在性能还有一定的上升空间。
6、到目前为止所有的预测方法尽管都使用了lncrna和疾病的相似信息来辅助关联预测,然而并没有考虑到原有的相似性矩阵中存在的弱相关项会给预测过程带来噪音。同时传统的概率矩阵分解模型仍有一定的改进空间,它只是利用带有高斯噪声的概率线性模型来模拟lncrna与疾病的相互作用,没有利用好相似的lncrna(疾病)通常与对应的疾病(lncrna)存在相互关联这一条件,且没有利用好更多的生物信息(即lncrna和疾病的相似性)。因此,在这方面的工作仍有性能空间需要提升。
技术实现思路
1、本发明提供一种用于推断lncrna-疾病关联的受校正相似性约束的概率矩阵分解方法。
2、为解决上述技术问题,本发明的技术方案如下:
3、一种用于推断lncrna-疾病关联的受校正相似性约束的概率矩阵分解方法,包括以下步骤:
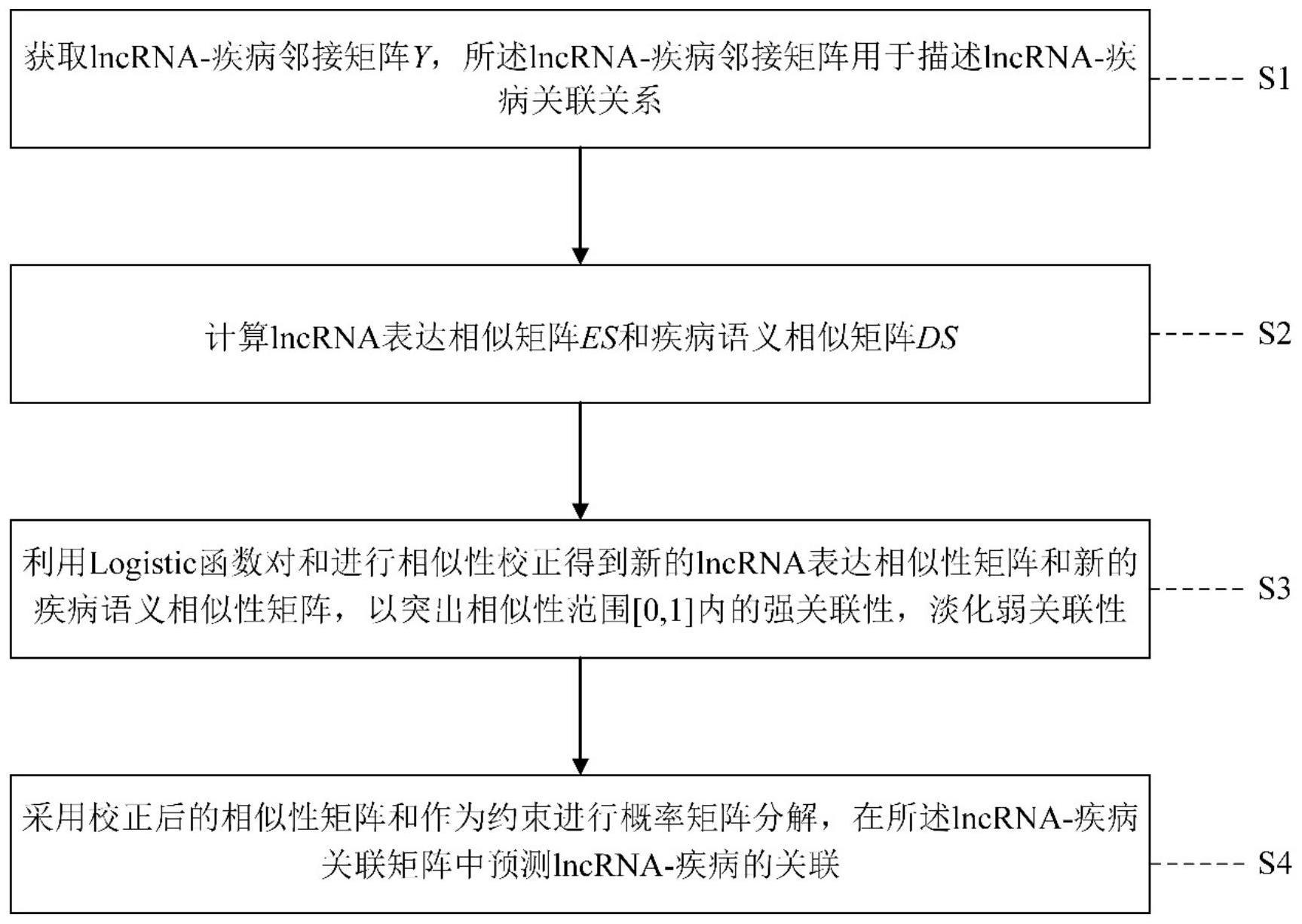
4、s1:获取lncrna-疾病邻接矩阵y,所述lncrna-疾病邻接矩阵用于描述lncrna-疾病关联关系;
5、s2:计算lncrna表达相似矩阵es和疾病语义相似矩阵ds;
6、s3:利用logistic函数对es和ds进行相似性校正得到lncrna校正相似性矩阵le和疾病校正相似性矩阵ld,以突出相似性范围[0,1]内的强关联性,淡化弱关联性;
7、s4:采用校正相似性矩阵le和ld作为约束进行概率矩阵分解,在所述lncrna-疾病关联矩阵中预测lncrna-疾病的关联。
8、优选地,步骤s1中获取lncrna-疾病邻接矩阵y,具体为:
9、从lncrnadisease数据库下载了一个lncrna-疾病关联数据集,删除了一些重复的lncrna和疾病以及非人类的数据。最后,在447种lncrnas和218种疾病之间得到了1690对实验验证的lncrna-疾病关系。通过建立lncrna-疾病邻接矩阵y∈rnl×nd来描述lncrna-疾病的关系。其中变量nl和nd分别代表lncrna和疾病的数量。矩阵y定义如下:
10、
11、如果一个lncrna li被证实与疾病dj相关,则y(i,j)被设置为1。否则,y(i,j)为0。
12、优选地,骤s2中lncrna相似矩阵es,具体为:
13、本研究从arrayexpress数据库中获取并使用rna测序技术生成的lncrna表达谱。计算其表达谱之间的spearman相关系数可以得到lncrnas之间的表达相似性。我们使用es来表示lncrna li和lncrna lj之间的表达相似性,其中es(li,lj)∈[0,1]。
14、优选地,步骤s2中疾病语义相似矩阵ds,具体为:
15、基于医学图书馆(mesh)(https://www.nlm.nih.gov/mesh/meshhome.html)的描述符信息,我们构建了一个有向无环图(dag)来计算疾病之间的语义相似性。一个疾病d由dag(d)=(d,v(d),e(d))描述,其中v(d)和e(d)分别是dag的顶点集和边集。根据疾病d的dag图结构,我们可以计算出疾病m对疾病d的语义价值(s)。的语义值(s)如下。
16、
17、根据疾病的dag图,疾病的语义值被定义为该疾病祖先节点和该疾病对其本身的语义贡献值之和,公式表示如下:
18、td=∑m∈v(d)td(m)
19、基于上述步骤,我们可以构建语义相似度矩阵ds以表示疾病di和疾病dj之间的语义相似度:
20、
21、优选地,步骤s3中计算lncrna校正相似性矩阵le,具体为:
22、相似性校正方法被采用来降低矩阵分解过程中lncrna和疾病相似性矩阵所带来的噪声影响。通过logistic函数转换,以突出相似性范围[0,1]内的强关联性,淡化弱关联性的方式去减少相似性矩阵中存在的噪声。在疾病相关基因的研究中已经使用过这种方法。logistic函数定义如下。
23、
24、当x∈[0,0.3]时l(x)≈0;当x∈[0.6,1]时l(x)≈1。这意味着处于[0,0.3]范围内的弱相似系数是损失的信息,强相似系数值在在[0.6,1]的范围内,通常显示出显著的共同表达的关系。这意味着l(0)需要接近0,于是我们设置l(0)=0.0001可以得到d=log(9999)。而c作为校正程度系数参与模型的参数调节。因此我们可以得到lncrna校正相似性矩阵le如下所示:
25、
26、优选地,步骤s3所述疾病校正相似性矩阵ld的计算方法与lncrna校正相似性矩阵le的计算方法相同。
27、优选地,步骤s4中采用校正相似性矩阵le和ld作为约束进行概率矩阵分解,具体为:
28、设w∈rk×nl和d∈rk×nd为lncrna和疾病特征矩阵,列向量wi和dj分别代表lncrna专用和疾病专用潜在特征向量。然后,我们的目标是找到lncrna和疾病的潜在模型(w∈rk×nl和d∈rk×nd),其积(wtd)可以重建交互矩阵y。从概率学的角度来看,观察到的相互作用的条件分布y∈{0,1}如下所示:
29、
30、其中f(x|μ,σ2)是高斯正态分布的概率密度函数,其平均值为μ,方差为σ2,iij是指标函数,如果lncrna li与疾病dj有关,则等于1,否则等于0。因此,p(y|w,d,σ2)为我们提供了关联矩阵y的概率表示。作为lncrna和疾病潜伏模型的生成模型,我们在lncrna和疾病特征向量上放置零均值球面高斯先验,如下所示:
31、
32、
33、其中i是一个k维的身份对角矩阵。那么,lncrna和疾病特征的后验分布为:
34、
35、此后,我们取上式的对数并将其转化为:
36、
37、其中,c是一个常数。在超参数保持不变的情况下,对lncrna和疾病特征的对数后验最大化相当于最小化带有二次正则化项的平方误差之和目标函数。
38、
39、其中表示范式。
40、优选地,步骤s4中将校正后的相似性作为约束加入到常规概率矩阵分解中。具体为:
41、基于相似的lncrna通常与对应的疾病存在相互关联的假设,反之亦然。我们将更多的生物信息(即lncrna和疾病的相似性)考虑在内。因此,我们提出一个新的目标函数如下:
42、
43、其中wi代表lncrna的k维潜在特征向量,wtw是lncrna加权相似度矩阵,dtd是疾病加权相似度矩阵。这里,我们利用梯度下降算法来解决方程(11)中的优化问题。首先,方程(11)的相应拉格朗日函数γf定义为:
44、
45、其中tr(·)表示矩阵的迹;和ψ=[ψjk]分别是约束条件wik≥0和djk≥0的拉格朗日乘子。那么,w和d的偏导数为:
46、
47、利用karush-kuhn-tucker(kkt)条件和ψjkdjk=0,可以得到以下关于wik和djk的方程式:
48、(i×(-dyt+ddtw))ikwik+(λww)ikwik
49、+(2λ1(-w(ld)+wwtw))ikwik=0
50、(i×(-wy+wwtd))jkdjk+(λdd)jkdjk
51、+(2λ2(-d(le)+ddtd))jkdjk=0
52、因此,我们可以得到如下的更新规则:
53、
54、
55、矩阵w和d根据上述公式进行更新,直到目标函数的局部最小值。最后,得到预测的lncrna-疾病相互作用矩阵为y*=wtd。一般来说,y*的第j列表示疾病dj与lncrna之间的相互作用分数,分数越大,越有意义。
56、与现有技术相比,本发明技术方案的有益效果是:
57、本发明引入logistic函数来重构lncrna和疾病的相似度矩阵,建立一个更好的度量方式来精确描述lncrna与疾病之间的相似关系。然后基于相似的lncrna通常与对应的疾病存在相互关联的假设,在原有概率矩阵分解算法的基础上引入了更多的生物信息(即lncrna和疾病的相似性)加以限制,实现了更加准确的lncrna-疾病的关联预测。
- 还没有人留言评论。精彩留言会获得点赞!