基于深度模态数据融合的药物靶标亲和度预测方法
本发明涉及大数据,尤其涉及基于深度模态数据融合的药物靶标亲和度预测方法。
背景技术:
1、药物靶标亲和性(drug-target affinity,dta)预测在药物发现中发挥着重要作用,因为它能够从大量候选化合物中筛选潜在药物,并描述药物靶标对中相互作用的强度。然而,通过大规模化学或生物实验进行dta预测的工作大多需要消耗大量的时间、资源和成本。随着药物、靶点和相互作用数据的不断积累,通过计算机辅助药物设计技术,相关领域已经开发了诸多方法来预测dta。由于深度学习的突破和计算能力的巨大提升,基于深度学习的dta预测模型已逐渐应用于预测药物与靶标的结合亲和力。虽然部分模型已经取得了良好的预测性能,但是仍存在一些问题。例如,现有的计算模型在预测精度、鲁棒性和泛化能力方面仍有很大的改进空间;大多数用于dta预测的深度学习模型缺乏可解释性分析,这在一定程度上降低了它们在实际应用中的可信度。
技术实现思路
1、本发明所要解决的技术问题是针对背景技术的不足提供一种基于深度模态数据融合的药物靶标亲和度预测方法。
2、本发明为解决上述技术问题采用以下技术方案:
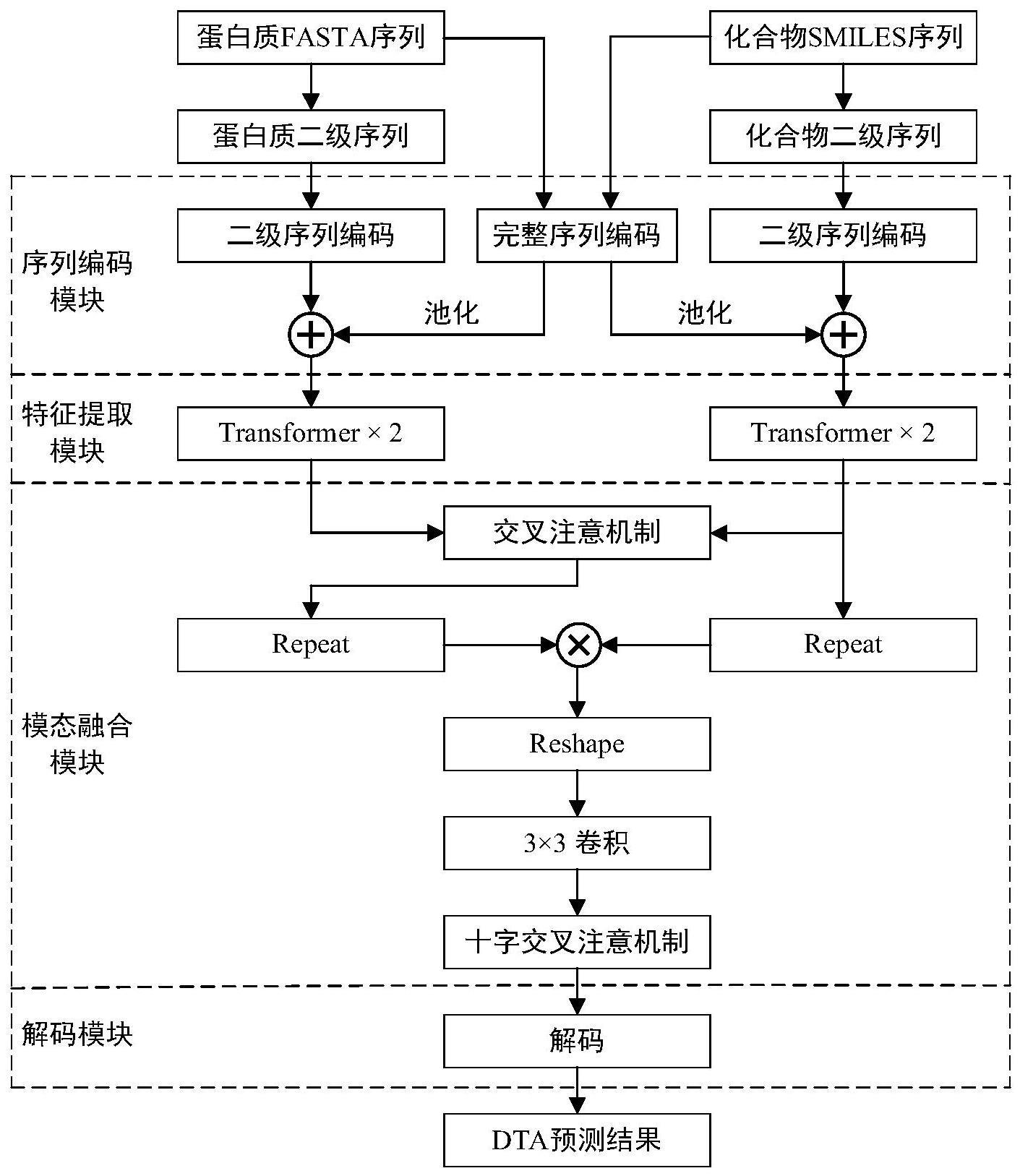
3、基于深度模态数据融合的药物靶标亲和度预测方法,包含序列编码模块、特征提取模块、模态融合模块、解码模块四部分,具体包含如下步骤,
4、步骤1,利用大量的无标签序列信息构造蛋白质与药物序列的二级序列,并对蛋白质与药物的完整序列和二级序列进行编码;
5、步骤2,利用transformer深度提取蛋白质与药物的完整序列及二级序列的特征,实现序列全局特征与局部特征的同步捕获;
6、步骤3,通过交叉注意机制和十字交叉注意机制进行模态融合,实现蛋白质序列与药物序列两种模态特征之间的双向交互,进而挖掘交互后的模态特征,提升药物靶标亲和度预测的精度;
7、步骤4,对模态融合结果进行解码,获得蛋白质和药物之间的结合亲和力的预测结果。
8、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤1中,分别基于fasta和smiles序列对蛋白质和药物的二级序列进行表达,基于bpe算法获得的二级序列词汇表,通过对完整序列进行one-hot编码的方式来构建蛋白质和药物的二级序列。
9、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤1中,利用bpe算法对序列进行分词处理,利用大量未标记序列构建蛋白质和药物二级序列词汇表,具体如下:
10、步骤1.1,从pubchem、chembl、drugbank、uniprot、pdbbind和bindingdb等诸多数据库搜集、下载蛋白质fasta序列和药物smiles序列,其中包括无标签数据;
11、步骤1.2,初始化由单个氨基酸字符或smiles字符组成的词汇表,使用bpe算法分别对所有的蛋白质和药物序列进行频繁连续二级序列挖掘,形成新的按照二级序列出现频率从高到底排序的蛋白质和药物词汇表;
12、步骤1.3,对于蛋白质,取前vs个词汇构成最终的词汇表vt;对于药物,取前qs个词汇构成最终的词汇表vd;
13、步骤1.4,分别利用二级序列词汇表vt和vd对蛋白质fasta序列和药物smiles序列进行二级序列表达,获得蛋白质的二级序列st和药物的二级序列sd。
14、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤1中,序列编码模块的输入为蛋白质fasta序列和药物smiles序列的完整序列和二级序列,分别对完整序列和二级序列进行编码,获得编码输出;以蛋白质的编码过程为例,对序列编码模块的工作过程进行具体介绍:
15、(1)完整序列编码
16、蛋白质fasta序列由不同的氨基酸组成,将由氨基酸构成的原始完整序列t表示为
17、t={t1,t2,…,ti,…,tn},ti∈nt (1)
18、ti表示蛋白质序列中第i个氨基酸,nt表示氨基酸集合,包含了常见的25个氨基酸,n为输入蛋白质的长度,定义最大蛋白质的长度为l,对完整序列进行编码,包括嵌入和位置编码;
19、嵌入:以序列t为输入,对蛋白质完整序列进行嵌入,获得输出该过程为线性转换过程,嵌入层具有可训练权重其中v表示上述氨基酸集合的大小,e表示氨基酸嵌入的大小;
20、位置编码:为了添加蛋白质t中每个氨基酸的相对或绝对位置信息,还需要进行位置编码;以序列t为输入,对蛋白质完整序列进行位置编码,输出为表示了t中所有氨基酸的位置编码,定义为
21、
22、
23、其中,i是位置,j是维度,d是氨基酸位置编码的大小;pet(i,:)是矩阵pet的第i行,表示蛋白质t中第i个氨基酸的位置编码;当蛋白质的长度n<l时,n+1至l的部分为0;这里设置位置编码大小等于嵌入大小,即d=e;因此可以直接令pet和et两者相加;
24、定义xwt为完整序列编码的输出,结果可表示为
25、xwt=et+pet (4)
26、(2)二级序列编码
27、在二级序列编码模块中,以蛋白质的二级序列st为输入,对二级序列进行嵌入和位置嵌入;
28、嵌入:对二级序列st进行编码,获得矩阵其中lt为最大蛋白质的二级序列的长度,vs为蛋白质二级序列词汇表vt的大小;以mst为输入,对二级序列进行嵌入操作,输出为其中es为每个二级序列嵌入的大小;嵌入层具有可训练权重其中vs表示上述蛋白质二级序列词汇表的大小;
29、位置嵌入:对蛋白质的二级序列进行单独的hot vector位置编码,结果表示为ist;以ist为输入,对二级序列进行位置嵌入,输出pest可表示为
30、
31、其中为位置嵌入层的位置查询词典;
32、定义xst为二级序列编码的输出,结果可表示为
33、xst=est+pest (6)
34、(3)池化
35、对完整序列的编码结果xwt进行池化操作,使其维度与二级序列编码结果xst的维度相同;定义xwt池化的结果为xwt1,将该结果与xst相加,获得蛋白质的序列编码模块的输出xt,表示为
36、
37、(4)药物的序列编码
38、药物smiles序列的序列编码过程与蛋白质fasta序列的类似;将药物smiles序列d的数学表达式表示为
39、d={d1,d2,…,di,…,dm},di∈nd (8)
40、di表示药物序列中第i个smiles字符;nd表示包含62个smiles字符的smiles集合;药物d的smiles序列长度m是不固定的。定义最大药物的长度为z,因此m≤z;
41、药物完整序列由完整序列编码模块进行编码后得到输出表示为其中f表示smiles字符嵌入的大小;此处设置氨基酸和smiles字符具有相同的嵌入大小,即f=e;药物二级序列由二级序列编码模块进行编码后得到的输出表示为其中ld为最大药物的二级序列的长度,且ld<z,fs为药物二级序列嵌入的大小;此处设置氨基酸和smiles字符二级序列具有相同的嵌入大小,即fs=es;对药物完整序列编码结果xwd进行池化操作,结果为xwd1,其维度与二级序列encoding结果xsd的维度相同,即最终获得药物smiles序列的序列编码模块输出xd,表示为
42、
43、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤2中,特征提取模块主要由两个transformer组成,其输入为已编码的蛋白质和药物xt和xd,经过transformer特征提取后获得该模块的输出xat和xad,该模块的transformer编码器主要包含了两个子层,分别是多头注意力层和前馈层;
44、(1)多头注意力层
45、特征提取模块的transformer工作机理主要是基于自注意力机制进行特征提取;自注意力模块主要包括线性转换层,带缩放点积注意力层与合并层;对输入信号xt(xd)进行线性转换,获得矩阵ql=kl=vl,dk=dv,其中n为蛋白质或药物的最大长度n=ltarget或n=ldrug;将ql、kl以及vl输入到带缩放点积注意力层,对ql和kl进行点积计算并将结果除以然后使用softmax获取vl的权重,最终获得带缩放点积注意力层的输出,表示为
46、
47、采用多头注意力机制来共同关注不同位置的不同表达子空间的信息,多头注意力层包含h个平行运行的带缩放点积注意力层;线性转换层的输入q=k=v是transformer编码模块的输入xt或xd;
48、将es维矩阵q、k和v进行线性投影h次,分别获得h个ql矩阵、h个kl矩阵和h个vl矩阵;
49、利用带缩放点积注意力层处理ql、kl和vl,获得第i个带缩放点积注意力层的输出headi,i=1,2,...,h。
50、
51、其中,为线性投影矩阵;
52、将带缩放点积注意力层的输出进行连接并传递至线性转换层,得到多头注意力层的输出,表示为
53、multihead(q,k,v)=concat(head1,...,headh)wo (12)
54、其中,wo为线性投影矩阵;
55、(2)前馈层
56、前馈层由2个线性变换和relu激活组成,连接顺序为线性变换——relu激活——线性变换;多头注意力层的输出结果multihead(q,k,v)在输入至前馈层获得输出;另外,在多头注意力层和前馈层两个子层的每一层周围都应用了一个残差连接和层规范化,以解决多层网络训练问题和加速收敛;
57、经过两次transformer特征提取处理后,获得特征提取模块的输出,即蛋白质特征xat和药物特征xad。
58、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤3中,构建了一个模态融合模块,由交叉注意机制和十字交叉注意机制实现蛋白质特征与药物特征之间的交叉融合,将蛋白质特征xat和药物特征xad输入模态融合模块,利用交叉注意模块实现药物对蛋白质的关注,经由repeat、reshape和卷积操作后馈入十字交叉注模块,输出模态融合的结果;具体过程如下:
59、首先,将蛋白质特征xat和药物特征xad输入交叉注意模块,其中,查询query、键值key和值value的计算方法与上述特征提取模块中标准注意力机制的方法相同,query为蛋白质特征xat,key和value为蛋白质特征xad,从而实现药物到蛋白质的交叉关注;对输入xat和xad进行线性投影,分别得到矩阵qca和kca=vca,其中dk1=dv1=ed/h;给定qca、kca和vca,应用带缩放点积注意力机制来获得交叉注意模块的输出xat1;
60、分别对d2t融合特征xat1和药物特征xad进行repeat操作,获得特征矩阵xat2和xad2;将xat2和xad2按元素相乘并进行reshape处理和3x3卷积计算,获得特征矩阵x;
61、将矩阵x输入到十字交叉注意模块,对x进1×1卷积计算和线性转换,分别生成两个特征矩阵qcc和kcc;对qcc和kcc进行相关度计算:在qcc空间维度的每个位置u,获得一个向量qu,然后通过与位置u在同一行或列中的kcc提取特征向量,构成集合ωu;对相关度计算进行表示,如式(13)所示
62、
63、其中,ωi,u是ωu的第i个元素,bi,u∈b是ωu和ωi,u之间的相关度,i=[1,…,(ld+lt-1)];在此基础上,对矩阵b进行softmax处理,计算得出注意力矩阵a;
64、再对x进行1×1的卷积计算和线性转换,获得特征自适应矩阵vcc;在vcc空间维度的每个位置u,可以获得一个向量vu和一个集合φu,其中集合φu是矩阵vcc中以u为中心的十字交叉结构的特征向量集合,在此基础上进行邻居节点间信息聚合计算,计算公式如下:
65、
66、其中,x'是十字交叉注意模块的输出,即为模态融合模块的输出,x′u是x'在位置u处的特征向量,ai,u是注意力矩阵a中位置u处的第i个标量值;通过上述十字交叉注意模块的处理,同一行和同一列中的邻居信息均被添加到特征x中,从而实现蛋白质与药物之间的特征交互融合。
67、作为本发明基于深度模态数据融合的药物靶标亲和度预测方法的进一步优选方案,在步骤4中,对模态融合的输出x'进行解码,以获得dta预测结果,解码器模块由3个前馈层和1个线性层组成;
68、其中,第一个前馈层包含线性层和relu激活,接下来的两个前馈层包含线性层、relu激活和层规范化,将x'发送到解码模块,以获得dta模型的输出y,该输出y即为蛋白质t和药物d之间的dta预测得分,用于评价蛋白质与药物之间的相关性程度。
69、本发明采用以上技术方案与现有技术相比,具有以下技术效果:
70、本发明基于深度模态数据融合的药物靶标亲和度预测方法,利用大量的无标签序列信息构造蛋白质与药物序列的二级序列,并对蛋白质与药物的完整序列和二级序列进行编码;利用transformer深度提取蛋白质与药物的完整序列及二级序列的特征,实现序列全局特征与局部特征的同步捕获;通过交叉注意机制和十字交叉注意机制进行模态融合,实现蛋白质序列与药物序列两种模态特征之间的双向交互,进而更准确的挖掘交互后的模态特征,提升药物靶标亲和度预测的精度;对模态融合结果进行解码,获得dta预测结果。
- 还没有人留言评论。精彩留言会获得点赞!