基于密度泛函理论指导混合多胺CO2吸收剂搭配设计的方法
本发明属于co2捕集领域,尤其涉及基于密度泛函理论指导混合多胺co2吸收剂搭配设计的方法,属于量子化学计算范畴。
背景技术:
1、co2的过度排放问题一直是全球关注的焦点,近年来co2捕集技术已经在火电、化工等领域得到了广泛的研究。其中,燃烧后化学吸收碳捕集技术发展迅速,有机胺吸收剂是当前应用最为广泛的碳捕集材料,以单胺溶液为吸收剂的碳捕集系统在国际上已经有了一定规模的应用。
2、近年来,已经有大量的示范产业和中试实验证明了一些单胺可以用于工业碳捕集,比如mea、mdea;但是,存在解吸能耗高、粘度大、稳定性不高等问题,阻碍了其大规模工业应用。大量的实验研究证明,混合胺在吸收过程中集中了不同胺的优点,在应用中具有很大的优势,它们可以同时达到吸收速率快、吸收容量大、再生能耗低的目的。
3、然而,目前大多数关于混合胺的研究仅限于某一特定搭配的实验研究,因此在特定情况下筛选合理的搭配是耗时耗力的。同时,在理论计算上,目前更先进的方法(机器学习、qsar)专注于设计具有优异性能的吸收材料,很少有关于混合胺搭配设计的研究报道。找到可用的描述符可以在理论上快速确定特定环境下的配对。但很少有研究从微观参数来描述反应能垒的差异,以指导配对。
4、因此,从微观角度找到能够影响反应能垒的参数,成为亟待解决的问题之一,针对以上现状,迫切需要开发基于密度泛函理论指导混合多胺co2吸收剂搭配设计的方法,以克服当前实际应用中的不足。
技术实现思路
1、本发明实施例的目的在于提供基于密度泛函理论指导混合多胺co2吸收剂搭配设计的方法,基于密度泛函理论,从氢键强度出发,可以对混合胺的能垒提供较为准确的预测,结合实际需求,合理地搭配混合多胺,大大减少了在混合多胺搭配筛选上投入大量实验耗费的人力、物力和财力,降低了时间成本。
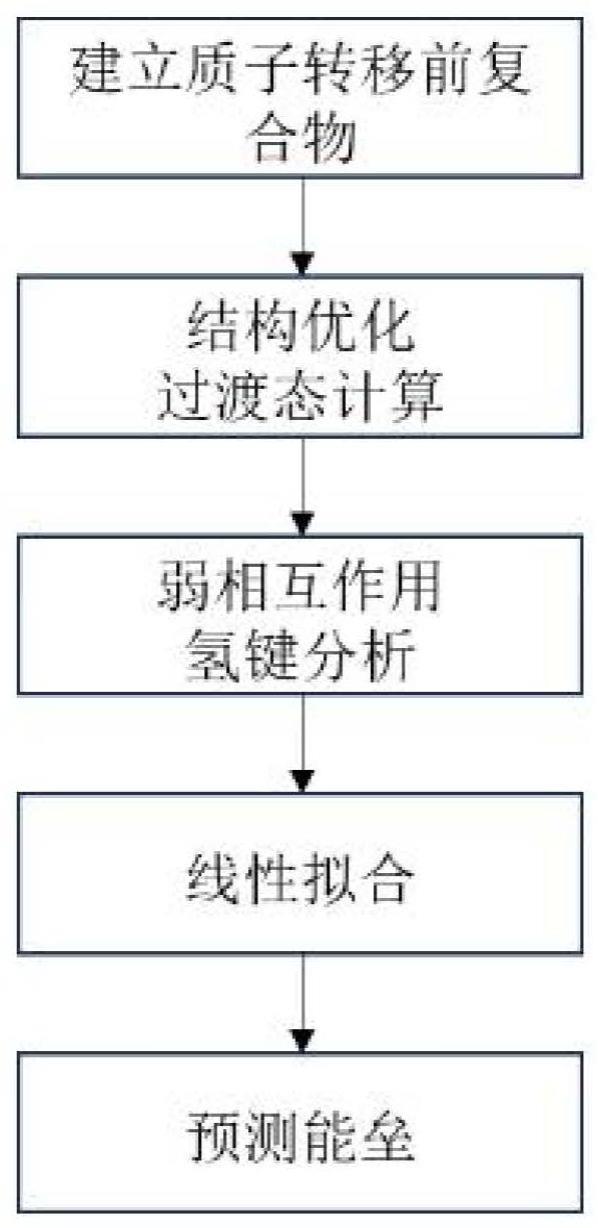
2、本发明实施例是这样实现的,基于密度泛函理论指导混合多胺co2吸收剂搭配设计的方法,包括以下步骤:
3、步骤1、单胺的选择
4、根据需求选择具有不同优势的胺为研究对象,优化其分子单体和吸收co2后不稳定的中间产物结构;
5、步骤2、结构优化
6、基于隐式溶剂化模型,结合质子转移方向,建立并优化质子转移前后的复合物结构,并进行频率分析;
7、步骤3、过渡态计算
8、在同计算水平下计算质子转移过程的过渡态,得到电子能垒,在高精度泛函下进行溶剂下的溶质自由能修正,得到自由能垒;
9、步骤4、氢键计算
10、利用波函数分析程序,基于步骤1得到的转移前复合物的结构和波函数文件,计算其相互作用强度和氢键强度参数;
11、步骤5、数据统计和能垒预测
12、将能垒数据和氢键强度参数进行线性拟合,发现极好的相关性,证明可以利用氢键强度预测反应能垒。
13、进一步的技术方案,在步骤1和步骤2中,结构优化采用b3lyp泛函结合ma-def2-tzvp计算基组与dft-d3色散校正,并对优化后结构进行频率计算,检测结构是否存在虚频,如果存在虚频,需要重新优化,由于体系存在明显的弱相互作用及阴离子,基组需要引入弥散函数;复合物构型搭建采用gaussview5.0,结构优化和频率分析及后续单点能计算采用orca5.0.3软件;隐式溶剂化模型采用smd模拟体系处于溶剂环境。
14、进一步的技术方案,在步骤3中,采用neb-ts方法计算过渡态,过渡态是否正确的验证方法是,频率分析结果有且仅有一个虚频,虚频振动方向连接反应物和生成物;通过过渡态计算得到电子能垒eb表征反应速率,eb采用公式1进行计算:
15、eb=ets-eis (1)
16、其中,ets为过渡态能量,eis为反应物能量;
17、通过过渡态计算得到自由能垒gb表征反应发生的难易程度,gb采用公式2进行计算:
18、gb=gts-gis (2)
19、其中,gts为过渡态自由能,gis为反应物自由能;
20、在溶剂化条件下需要对溶质的自由能进行修正,具体做法是先获得步骤1结构优化后的液相下自由能修正量gco,然后在pwpb95 d3 def2-tzvpp水平下获得结构的气相单点能,在wb97m-v def2-tzvp水平下分别获得结构液相、气相下的单点能,修正自由能g由公式3进行计算:
21、g=(e-gas-1+gco)+(e-sol-e-gas-2)+1.89 (3)
22、其中,e-gas-1是pwpb95水平下的气相单点能,e-sol和e-gas-2分别是wb97m-v水平下的液相单点能和气相单点能,二者的差值为溶质的溶解自由能。
23、进一步的技术方案,在步骤4中,采用independent gradient model based onhirshfeld partition(igmh)方法分析复合物的相互作用,atoms in molecules(aim)方法计算氢键强度,分析软件用multiwfn波函数分析程序,可视化用软件vmd;以上分析方法均使用xxx.molden文件,需要把包含波函数信息的xxx.gbw文件转化为xxx.molden文件,具体操作为在文件所在位置下输入命令orca_2mkl xxx-molden。
24、进一步的技术方案,在步骤4中,采用aim分析得到的n-h…n型氢键的(3,-1)点的电子密度值和能量密度值参数代表氢键强度;其中,(3,-1)点对应实空间函数的二阶鞍点,函数在一个方向曲率为正,另两个方向为负,对于电子密度函数,通常出现在有相互作用的两个原子之间,也被称为键临界点(bcp)。
25、进一步的技术方案,在步骤5中,对于混合二胺,采用唯一氢键的参数进行拟合,用该结果去预测其他混合二胺的反应能垒;对于混合三胺,基元反应包括两个质子转移,采用较弱氢键的参数去进行拟合,用得到的结果去预测其他混合三胺、四胺的反应能垒。
26、本发明实施例提供的基于密度泛函理论指导混合多胺co2吸收剂搭配设计的方法,从氢键的角度出发,运用密度泛函理论结合波函数分析进行计算。对质子转移反应的反应物生成物进行计算,并计算反应过渡态。通过提取出反应物的波函数信息,运用multiwfn软件对反应物结构进行弱相互作用分析和拓扑分析,利用得到的氢键强度参数与能垒数据进行拟合,得到拟合公式,可以通过该公式从氢键强度去预测反应能垒。
27、本发明在胺溶液研究开发领域创新性的提出了从氢键强度的角度出发去预测质子转移反应能垒的方法,为实验和工程上混合胺溶液的搭配设计提供了理论依据。该方法的优势:计算结果和预测效果准确可靠,可以降低实验筛选过程耗费的时间、人力成本。
- 还没有人留言评论。精彩留言会获得点赞!