一种同位素示踪与宏基因组相结合的解析氮限制生态系统的方法
本发明属于氮循环转化及生物信息学领域,更具体地,涉及一种同位素示踪与宏基因组相结合的解析氮限制生态系统的方法。
背景技术:
1、氮是生物体生长和发展所必需的关键元素之一,在生态系统中扮演着重要的生物地球化学角色。然而,当前许多生态系统面临氮限制的问题,即氮供应不足,无法满足生物体对氮的需求。据2020年《nature geoscience》的研究指出,全球自然陆地生态系统中有18%的区域面临较强的氮限制。这些氮限制生态系统广泛分布于河口、森林和海洋等自然环境中。氮的限制对生态系统的影响不仅仅局限于生物体的生长和代谢,还涉及物质循环、能量流动、微生物群落结构和物种组成的改变,以及生态系统的稳定性和功能。
2、在氮限制的生态系统中,氮的可利用性和循环转化主要受微生物活动的影响。微生物驱动的硝酸盐还原过程,包括反硝化、异化硝酸盐还原成铵(dnra)和厌氧氨氧化(anammox),被认为是调控氮保存和流失的关键过程。反硝化和厌氧氨氧化将no3-转化为n2,导致氮损失,而dnra通过将no3-转化为nh4+,实现生物可利用氮的内部再循环,在氮限制的生态系统中具有重要意义。目前,研究人员使用各种方法来了解氮限制生态系统中的氮循环转化过程。同位素示踪是一种常用技术,通过标记同位素来追踪氮的来源、转化和去向。该方法提供了关于氮输入、输出和转化速率的信息,有助于揭示生态系统中氮的动态变化。另一方面,宏基因组分析是一种研究微生物群落组成和功能的方法,通过分析环境样品中的微生物dna序列,可以了解微生物群落的多样性、结构和代谢潜能。这对于理解氮限制生态系统中微生物参与的氮循环过程同样至关重要。
3、然而,当前的研究面临一些挑战和限制。在氮限制的生态系统中,氮的循环转化过程非常复杂,同时受到了多种因素的影响,包括环境条件、微生物相互作用和底物供应等。传统的单一方法可能无法全面解析这些复杂的氮循环过程。因此,迫切需要建立一种基于多样化手段相结合的方法,以解析氮限制生态系统内部的动态过程。
技术实现思路
1、本发明要解决的技术问题是克服现有技术在解析氮限制生态系统中的复杂性、数据综合和定量分析方面的缺陷和不足,提供一种全面、准确且高效以解析氮循环和微生物动态过程的方法
2、本发明的目的是提供一种同位素示踪与宏基因组相结合的解析氮限制生态系统的方法。
3、本发明上述目的通过以下技术方案实现:
4、步骤一、选取拟研究的氮限制生态系统进行土壤或者沉积物样品的采集;
5、步骤二、采集完的样品进行物理化学指标的测定以及dna的提取;
6、步骤三、构建泥浆混合体系,添加15n进行同位素示踪实验,在一个体系内同时测定反硝化、dnra以及anammox过程的速率;
7、步骤四:对提取的dna进行建库,并且使用illumina测序技术进行环境样品的宏基因组测序,通过metawrap等手段进行宏基因组数据的组装和分箱;
8、步骤五:使用kegg,ncycdb(n转化相关数据库)等数据库进行氮转化相关功能基因丰度的计算,并通过筛选出相关功能基因对应的序列,结合kraken2进行功能基因对应的微生物物种起源的探索;
9、步骤六:通过mantel test以及spearman相关性分析等将微生物学的信息,物理化学指标和氮转化的速率联系起来,得到影响氮转化过程的相关因子;
10、步骤七:将步骤六得到的影响因子,通过宏基因组分箱得到的氮转化相关mags的功能代谢分析去进一步解释影响的机制,最终更好解析氮限制生态系统内部的动态过程。
11、进一步的,所述步骤二中,采用冷冻研磨和十二烷基硫酸钠(sds)裂解法进行样品dna的初始提取,得到粗dna之后,进一步使用power soil dna纯化试剂盒进行dna的纯化,从而得到更高质量的dna。之后通过nanodrop评估dna的质量,其中260/280和260/230比例分别为1.8-1.9和1.7以上,并使用荧光法测定dna的浓度。
12、进一步的,所述步骤三中,将1.0g环境样品与6.0ml无菌水混合在一个12.0ml的顶空进样瓶内,然后,用高纯度的氦气(99.99%)通气30分钟,创造一个无氧的环境。并将该体系在没有任何外源底物的情况下摇床预培养72小时,以完全消耗体系中的no3-和no2-。同时为了抑制硝化作用,需要使用进样针向系统中加入50ul浓度为18.0g/l的烯丙基硫脲。之后,使用进样针向瓶内中添加100ul浓度为10mm的na15no3。将加入完同位素的泥浆混合体系在25℃以及200rpm的摇床速度下孵育,通过在0、6、12和24小时时注入100ul浓度为7mol/l的氯化锌来终止反应。使用gasbench-irms在不同的时间点测量29n2和30n2的产生量。由此得到反硝化和厌氧氨氧化的速率,计算公式如下:
13、
14、其中rdenitrification和ranammox分别为反硝化和厌氧氨氧化的速率,单位为nmol n g-1h-1。p30和p29分别是通过反硝化和厌氧氨氧化产生的30n2和29n2的量。
15、进一步的,所述步骤三中,测定完反硝化和厌氧氨氧化速率后剩下的泥浆混合液进行重新的30min氦气曝气,然后加入200ul的次溴酸碘溶液使dnra过程产生的15nh4+转化为n2时进行第二次的测定,计算公式如下:
16、
17、其中,rdnra为dnra过程的速率,p29’和p30’是由dnra过程产生的15nh4+经过氧化剂化学反应后产生的29n2和30n2的量。同时所述29n2和30n2的产生量的测量需要依次对每一个密闭的顶空样品瓶进行480s的he气(纯度>99.999%,流速100ml/min)排空处理,以便去除瓶内空气对氮气同位素比值测定的影响。在经过排空处理后,取0.1ml的样品气注入排空的样品瓶中,用pal自动进样器加定量环(100ul)进样,高纯氦气和n2混合气经75℃的气相色谱柱而与其他杂质气体得到分离。分离后的n2由氦气带入delta v检测器,高能电子束轰击离子化,经过加速电场,不同质荷比(m/z28、m/z29、m/z30)气态离子进入磁场分离成不同的离子束,进入接收器并转换为电信号,测定氮同位素比值。
18、进一步的,所述步骤四中,将宏基因组测序得到的原始数据使用metawrap流程中read_qc功能进行了原始序列的质量过滤,获得高质量的双端序列。使用megahit将经质量过滤后的序列组装成contigs,然后使用metabat2和maxbin2进行binning得到组装后的基因组mags。进一步使用bin_refinement和reassemble_bins功能对得到的mags进行了处理和优化。使用drep对这些mags进行ani 99%的去冗余处理,然后使用checkm对这些基因组的质量进行评估,选取完整度≥50%和污染率<10%的mags进行下游分析。
19、其中,下游分析中包含了使用metawrap quant bins功能计算mags的丰度。使用gtdb-tk获得mags的分类学信息,并使用metabolic v4.0找出具有与n代谢相关基因的mags进行进一步分析。通过prodigal得到这些mags的开放阅读框(orfs),并使用kaas(keggautomatic annotation server)对预测的基因进行进一步注释,以探索与微生物相关的代谢功能。
20、进一步的,所述步骤五中,通过对组装得到的contigs的orfs使用diamond与kegg以及ncycdb数据库进行比对,并结合salmon计算得到的结果,获得n转化相关基因的丰度。对于氮转化相关功能基因的物种起源,使用pear工具将原始的序列进行双端的合并,合并后的序列使用diamond与kegg以及ncycdb数据库进行比对。使用seqtk提取与反硝化(nirs,nirk,norb,norc和nosz),dnra(nrfa),anammox(hzsa)相关的功能序列,并通过kraken2对这些序列进行分类注释。
21、进一步的,所述步骤六中,结合氮转化相关功能基因和mags的丰度,氮转化速率以及一系列物理化学指标进行mantel test以及spearman相关性分析,得到可能影响到氮转化过程的相关因子。
22、进一步的,所述步骤七中,通过步骤六得到的相关因子,寻找相应的数据库进行mags功能代谢的注释,或者是按照kass注释得到结果进行进一步的探究。此外,可以使用diamond makedb功能,在ncbi上下载需要研究的相关功能的蛋白序列集合,构成一个独特的数据库,再进行其功能的比对,得到需要的氮转化相关mags的功能代谢特征,以解释其对氮转化过程的影响。
23、进一步的,上述方法在红树林生态系统研究中的应用。
24、本发明具有以下有益效果:
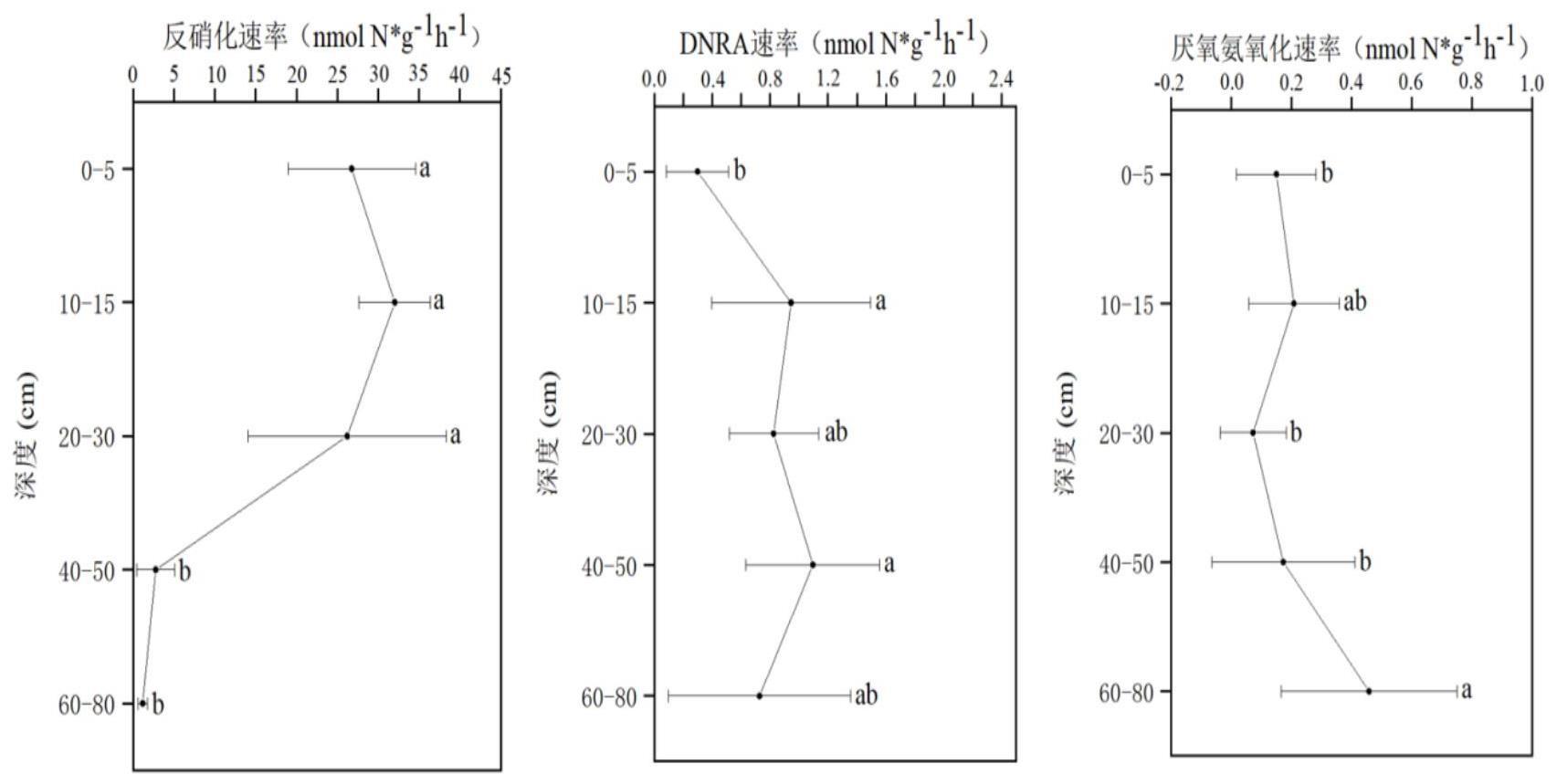
25、本发明一种同位素示踪与宏基因组相结合的解析氮限制生态系统的方法,通过结合同位素示踪和宏基因组的分析,克服了传统单一方法的局限性,能够提供更全面和准确的研究结果。
26、(1)数据分析的复杂性:氮限制生态系统的研究涉及大量的数据收集和分析,包括同位素数据和宏基因组数据。传统的数据处理方法常常面临复杂的数据整合和解读问题。本方法通过一系列相关性的分析研究,有效地整合和分析多源数据,提供了更准确和可靠的结果解读。
27、(2)研究深度和分辨率的提升:由于同位素示踪和宏基因组分析的高分辨率特性,本方法能够提供对氮限制生态系统更深入和详细的研究。传统方法常常无法捕捉微生物群落的多样性和功能特征,而本方法通过结合多样化的分析手段,以及相关数据库的选择或是个性化数据库的构建,能够全面揭示微生物的物种组成、代谢潜能和不同的环境适应性,提供更丰富的研究结果。
28、(3)应用广泛性:本方法适用于多种氮限制生态系统,包括不同环境中的河口、湿地、森林和海洋等。这使得研究人员可以在不同的生态系统中应用本方法,揭示氮循环的共性和差异,为生态系统管理和环境保护提供科学依据。
29、(4)具体而言,我们创新性地应用了一种同位素示踪与宏基因组相结合的方法,针对红树林生态系统中的氮限制进行深入解析。通过利用同位素示踪技术,我们能够准确追踪氮的转化过程,并了解不同深度沉积物中氮限制的动态变化。同时,宏基因组分析为我们提供了宝贵的生态因子信息,包括微生物群落结构、功能基因丰度等,帮助我们深入研究微生物参与氮循环的生态学机制。
30、在对红树林生态系统的进行研究后,我们发现这种同位素示踪与宏基因组相结合的方法具有显著的优势。
31、首先,我们能够对红树林生态系统中的氮转化过程进行全面而精确的定量分析,揭示了深度与氮限制之间的密切关系。其次,通过深入挖掘微生物的功能潜力,我们发现了一些关键的生态因子,如特定功能基因(如反硝化(nirs,nirk,norb,norc和nosz),dnra(nrfa),anammox(hzsa)相关的功能基因序列)丰度的变化和微生物群落的结构变化,这些因子可能在氮限制调控中发挥着重要作用。
32、这种创新的方法为我们提供了新的视角和全面的认识,有助于加深我们对红树林生态系统中氮限制的理解。同时,这些研究成果对于未来红树林保护与管理的决策制定也具有重要的参考价值。总体而言,这项专利在环境生态学领域具有巨大的潜力,为生态学和环境保护领域的研究提供了有力的支持。
- 还没有人留言评论。精彩留言会获得点赞!