一种识别circRNA蛋白结合位点的新方法
本方案属于生物信息学,提出了一种识别circrna蛋白结合位点的新方法。
背景技术:
1、circrna蛋白结合位点是指循环rna(circular rna)与蛋白质之间的结合区域。circrna是一种形成闭环结构的非编码rna,与蛋白质相互作用可以调控基因表达、细胞信号传导等生物学过程。
2、通过分析circrna与蛋白质的相互作用网络、研究其调控机制以及探究其在疾病中的作用,我们可以更好地理解circrna与蛋白质之间的关系,并为进一步揭示其功能提供重要的思路。通过识别circrna与蛋白质的结合位点可以预测其结合模式和可能的功能,对于深入了解细胞调控机制、疾病的发生发展以及新药物的开发具有重要的意义。
3、现有技术用于识别circrna蛋白结合位点的方式普遍采用的是卷积神经网络和循环神经网络、自注意力机制等提取特征的常规有监督分类方法。尽管现有的有监督学习算法在识别任务上已经取得了不错的性能,但都是基于有监督学习的,换句话说,算法需要大量的样本标签进行训练。虽然获得了不错的性能,但这大大限制了算法针对未知circrna-rbp相互作用机制的探索。
技术实现思路
1、本方案的目的是针对前人在circrna蛋白结合位点预测任务中需要大量标签进行训练这一弊端,提供一种基于自监督学习的识别circrna蛋白结合位点的新方法。
2、一种识别circrna蛋白结合位点的新方法,其特征在于,该方法包括:
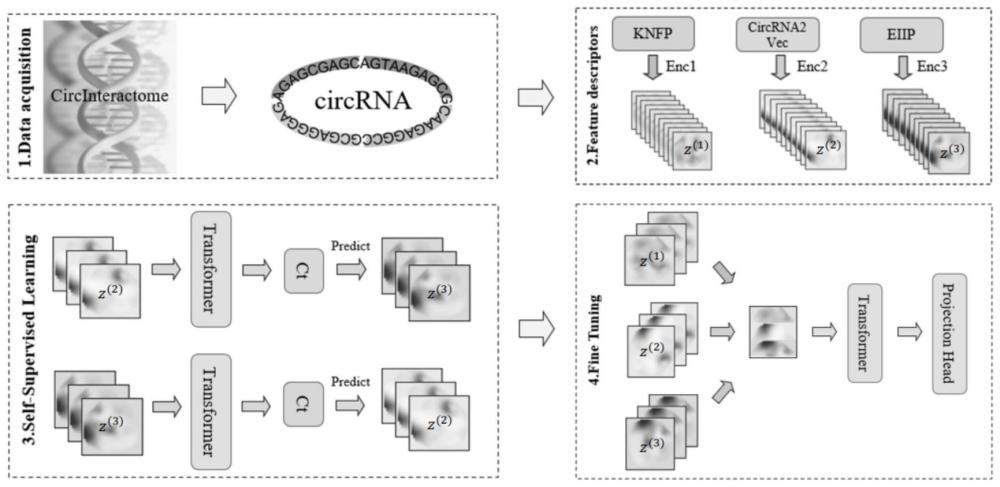
3、s1.使用两种特征提取技术分别对无标签样本进行特征提取得到两种特征子集x(2),x(3);当然,理论上,这里的样本也可以是带标签样本,只是训练过程中不需要使用到标签,即实质上仍然是无标签样本,故此种情况仍然在本专利的保护范围内。
4、s2.将两种特征子集分别用编码器(如一维卷积神经网络)进行编码得到两种编码特征z(2),z(3);
5、s3.通过跨视图相互预测的方式训练预测模型的上下文提取能力;
6、s4.使用三种特征提取技术分别对带标签样本进行特征提取并进行编码得到联合编码特征z(all);
7、s5.基于对比预测结果和真实标签差异的交叉熵损失训练预测模型的circrna蛋白结合位点识别能力。
8、提出利用三种特征提取技术实现针对序列的多种编码特征,由跨视图预测和微调两部分构成预测模型,利用其中两种编码特征对预测模型进行无监督的跨视图预测(自监督)以训练预测模型的上下文提取能力,同时进一步利用全部三种特征提取技术基于有标签样本得到的融合编码特征对预测模型进行微调,最终实现通过少量带标签样本实现具有circrna蛋白结合位点识别能力且具有更佳的综合能力、更高的预测性能的预测模型。
9、在上述的识别circrna蛋白结合位点的新方法中,所述的预测模型包括第一模型和第二模型,步骤s3中训练第一模型的上下文提取能力;
10、步骤s5中,训练由第一模型和第二模型构成的预测模型的circrna蛋白结合位点识别能力。
11、在上述的识别circrna蛋白结合位点的新方法中,步骤s5中,预测模型训练过程中,首先由第一模型基于编码特征z(all)提取序列的上下文信息c(all),然后通过第二模型处理后输出预测标签,最后通过交叉熵损失调整预测模型参数。
12、在上述的识别circrna蛋白结合位点的新方法中,步骤s3具体为:
13、预测模型对编码特征z(2)预测上下文c(2),对编码特征z(3)预测上下文c(3);
14、使用上下文c(2)预测编码特征z(3),使用上下文c(3)预测编码特征z(2);
15、通过上述相互预测的方式训练预测模型的上下文提取能力。
16、在上述的识别circrna蛋白结合位点的新方法中,步骤s3中,训练预测模型上下文提取能力的损失函数如下:
17、
18、
19、
20、目的是使编码特征z(2)的上下文c(2)对编码特征z(3)的逐一分量预测结果与z(3)原来值差距最小;
21、目的是使编码特征z(3)的上下文c(3)对编码特征z(2)的逐一分量预测结果与z(2)原来值差距最小;
22、其中,n表示特征序列长度,t表示序列各个元素的索引值,表示将空间c投影到空间z的线性转换矩阵。
23、在上述的识别circrna蛋白结合位点的新方法中,所述的第一模型结构为针对rna序列改进的transformer算法rna_transformer,依次包括输入层、线性层、归一化层、多头注意力机制、归一化层、前馈神经网络和输出层,输入层用于输入编码特征,输出层用于输出上下文信息;
24、所述的第二模型包括projection_head层和softmax层;
25、编码特征输入至第一模型结构后得到上下文信息,上下文信息经过第二模型输出预测标签。
26、在上述的识别circrna蛋白结合位点的新方法中,步骤s5中,训练预测模型circrna蛋白结合位点识别能力的交叉熵损失函数如下:
27、
28、
29、表示预测正样本的概率,表示预测负样本的概率y表示真实标签,m表示批量大小,i表示一个批量中样本的索引,c(all)表示融合特征z(all)的上下文信息,bp表示projection_head层的权重(weights)和偏置(bias),σ表示非线性激活函数。
30、在上述的识别circrna蛋白结合位点的新方法中,步骤s1中,所使用的两种特征提取技术为circrna2vec提取算法和eiip提取算法。
31、在上述的识别circrna蛋白结合位点的新方法中,步骤s4中,所使用的三种特征提取技术为circrna2vec提取算法、eiip提取算法和knfp提取算法。
32、在上述的识别circrna蛋白结合位点的新方法中,经过步骤s1-s5训练得到预测模型以后,通过如下方式对待预测样本进行circrna蛋白结合位点的识别:
33、使用三种特征提取技术分别对待测样本进行特征提取;
34、使用编码器对三种特征子集进行编码并将三种编码结果进行融合得到联合编码特征z(all);
35、将联合编码特征z(all)输入至预测模型,预测模型基于联合编码特征z(all)输出circrna蛋白结合位点识别结果。
36、本方案的优点在于:
37、通过先使用跨视图的序列预测学习序列的整体表征,再根据样本标签微调网络,减少了算法对监督信息的依赖,且可获得卓越的识别性能;
38、设计一种基于transformer的rna序列特征提取算法rna_transformer,提高了算法的并行性和稳定性,有利于算法捕捉到适合分类的特征。
技术特征:
1.一种识别circrna蛋白结合位点的新方法,其特征在于,该方法包括:
2.根据权利要求1所述的识别circrna蛋白结合位点的新方法,其特征在于,所述的预测模型包括第一模型和第二模型,步骤s3中训练第一模型的上下文提取能力;
3.根据权利要求2所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s5中,预测模型训练过程中,首先由第一模型基于编码特征z(all)提取序列的上下文信息c(all),然后通过第二模型处理后输出预测标签,最后通过交叉熵损失调整预测模型和编码器参数。
4.根据权利要求2所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s3具体为:
5.根据权利要求4所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s3中,训练预测模型上下文提取能力的损失函数如下:
6.根据权利要求5所述的识别circrna蛋白结合位点的新方法,其特征在于,所述的第一模型结构为针对rna序列改进的transformer算法rna_transformer,依次包括输入层、线性层、归一化层、多头注意力机制、归一化层、前馈神经网络和输出层,输入层用于输入编码特征,输出层用于输出上下文信息;
7.根据权利要求3所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s5中,训练预测模型circrna蛋白结合位点识别能力的交叉熵损失函数如下:
8.根据权利要求1所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s1中,所使用的两种特征提取技术为circrna2vec提取算法和eiip提取算法。
9.根据权利要求3所述的识别circrna蛋白结合位点的新方法,其特征在于,步骤s4中,所使用的三种特征提取技术为circrna2vec提取算法、eiip提取算法和knfp提取算法。
10.根据权利要求1-9任意一项所述的识别circrna蛋白结合位点的新方法,其特征在于,经过步骤s1-s5训练得到预测模型以后,通过如下方式对待预测样本进行circrna蛋白结合位点的识别:
技术总结
本方案公开了一种识别c ircRNA蛋白结合位点的新方法,该方法提出利用三种特征提取技术实现针对序列的多种编码特征,由跨视图预测和微调两部分构成预测模型,利用其中两种编码特征对预测模型进行无监督的跨视图预测以训练预测模型的上下文提取能力,同时进一步利用全部三种特征提取技术基于有标签样本得到的融合编码特征对预测模型进行微调,最终实现通过少量带标签样本实现具有c ircRNA蛋白结合位点识别能力且具有更佳的综合能力、更高的预测性能的预测模型。
技术研发人员:邹权,曹超,王春宇,丁漪杰,周通
受保护的技术使用者:电子科技大学长三角研究院(衢州)
技术研发日:
技术公布日:2024/3/5
- 还没有人留言评论。精彩留言会获得点赞!