一种基于全基因组测序的寄生虫虫种鉴定分析方法和设备与流程
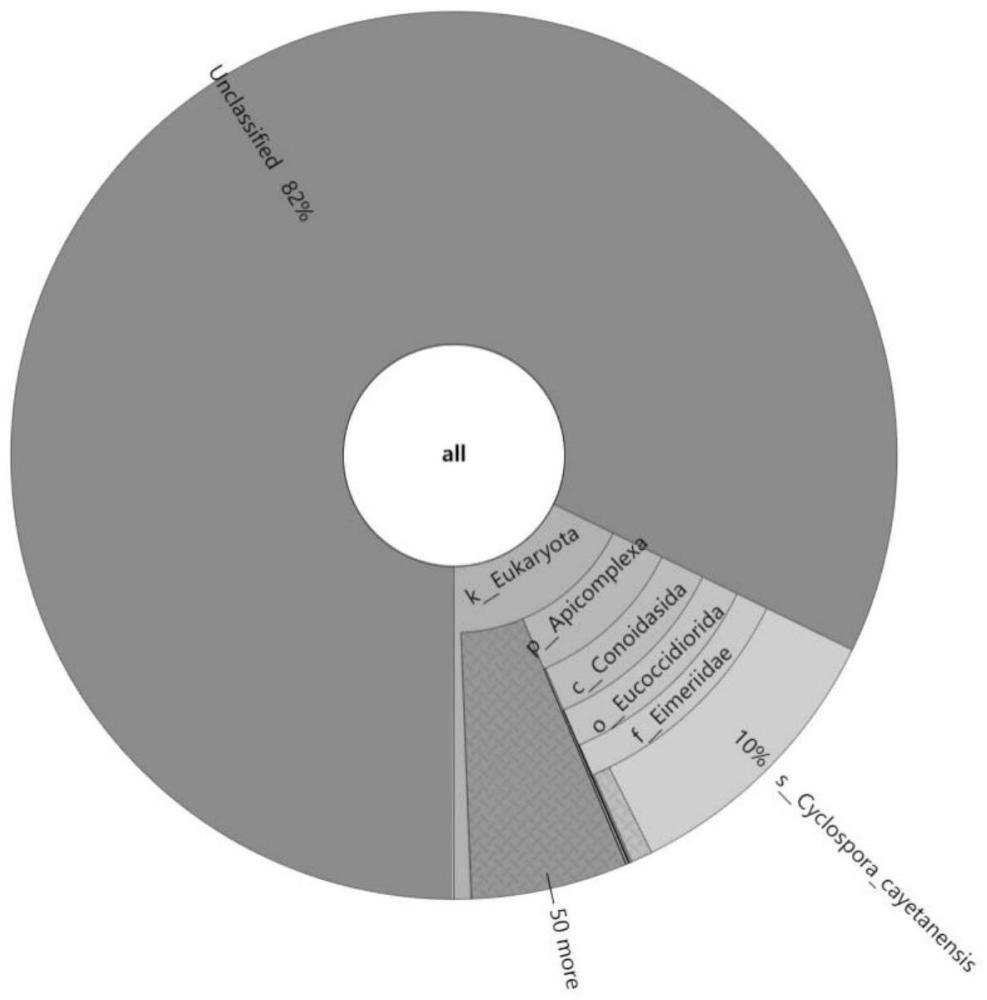
本发明涉及生物种类的鉴别领域,具体地,涉及一种基于全基因组测序的寄生虫虫种鉴定分析方法和设备。
背景技术:
1、近年来,随着全球一体化和人员的交流频繁,人兽共患寄生虫病的发病率显著上升,特别是临床上罕见寄生虫病的发现和报道、出入境动植物的检疫发现的入侵寄生虫物种,都极大影响到公共卫生、畜牧业、养殖业的发展,而对于这些寄生虫病的防控和治疗,鉴定是首要关卡。
2、虽然,目前全基因组测序技术发展迅速,且在寄生虫检测的应用愈来愈多,但该技术在寄生虫测序的最大挑战主要是缺乏一个针对性的数据库,测序得到的数据后分析难度大。目前的市面上的匹配数据库均是系统性的数据库,如ncbi真核生物,原核生物的数据库,范围大,匹配度极低,无法对寄生虫进行精准快速的鉴定。
3、此外,全基因组测序产生的数据量大,在数据存储和分析方面存在一定的技术门槛,需要生物信息学的专业支撑。大多数寄生虫基因组测序研究都集中在由数百万个细胞组成的大量样本上,大量样本的分析偏向于显性基因型,隐藏了细胞间变异和罕见变异。
技术实现思路
1、本发明旨在克服上述缺陷,针对寄生虫虫种鉴定这一技术难题,将处理后的全基因组序列通过与基于本发明方法构建的寄生虫虫种数据库进行匹配,可以在短时间内获知寄生虫虫种的相关序列,从而进行相关虫种的鉴定,提供了一种快速精准的寄生虫虫种鉴定技术。
2、本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于,包含如下步骤:
3、s1.获取原始数据;
4、s2.对原始数据进行数据质控和数据统计后,输出fastq序列,
5、所述数据质控,通过排除数据中的低质量碱基序列,获得优化后的序列;
6、s3.去除宿主人源基因组序列;
7、s4.将经s3处理后的测序数据与数据库进行比对分析,从而输出基于k-mer的所有基因组的最低共同祖先lca,对每条基因组序列进行分类,并按照门纲目科属和种给出对应的统计信息。
8、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
9、步骤s2中数据质控的过程包括:
10、s2a.去掉测序数据中可能存在的adaptors序列;
11、s2b.去掉测序数据中碱基质量低于20的碱基;
12、s2c.去掉测序数据中的重复序列;
13、s2d.去掉质控后测序长度低于36bp的序列;
14、s2e.去掉低复杂度的序列;所谓的低复杂度的序列为非简单重复序列;
15、s2f.获得高质量测序数据,即为a-e步骤的输出结果。
16、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
17、步骤s2中数据统计,包含对质控前后的数据进行如下内容的统计:测序的总reads数目、总碱基数、gc含量、q20、q30。
18、值得注意的是,质控前是为了评估测序数据量有多少,质控是为了评估测序有效的高质量测序数据量。
19、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
20、步骤s3中去除宿主人源基因组序列的步骤包括:
21、s3-1.选取基于千人基因组的人源基因组序列;
22、关于该千人基因组序列链接:https://github.com/benlangmead/bowtie-majref。
23、s3-2.基于s3-1的序列构建人源参考基因组index;
24、s3-3.将s2的fastq序列与s3-2的序列进行比较,输出未比对上的fastq格式的序列,并给出比对统计结果。
25、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
26、步骤s3-2和s3-3的过程通过生信分析软件bowtie2来实现。
27、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
28、所述步骤s4中比对分析借助生物信息分析软件kraken2实现。
29、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
30、所述数据库的构建方法如下所示:
31、s0-1.确定构建数据库用的数据来源;
32、s0-2.确定检测物种目录:确定寄生虫的检测范围内的所有全基因组序列以及对应的taxonomyid;
33、s0-3.将s0-2的全基因组序列以及对应的taxonomyid,基于k-mer的算法,构建物种的基因组哈希索引。
34、进一步地,本发明提供的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征还在于:
35、所述数据库的更新方法:针对每个新上传的物种,基于其对应的在ncbi taxonomy数据库中对应唯一的物种分类编号,通过该唯一编号,获得物种准确的物种分类信息后,借助生信数据分析软件bioawk将基因组fasta序列与物种taxonomyid进行合并,并整合到已有数据库,实现数据库referencehashindex的更新。
36、另外,本发明还提供了一种载体,其特征在于:包括基于上述方法构建的数据库,以及运行基于全基因组测序的寄生虫虫种鉴定分析方法中所列的步骤的程序或启动模块。
37、另外,本发明还提供了一种基于全基因组测序的寄生虫虫种鉴定分析设备,其特征在于:装载并运行上述载体。
技术特征:
1.一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于,包含如下步骤:
2.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
3.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
4.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
5.如权利要求4所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
6.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
7.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
8.如权利要求1所述的一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于:
9.一种载体,其特征在于:包括如权利要求7或8方法构建的数据库,以及运行如权利要求1-6任一所述的基于全基因组测序的寄生虫虫种鉴定分析方法中所列的步骤的程序或启动模块。
10.一种基于全基因组测序的寄生虫虫种鉴定分析设备,其特征在于:装载并运行如权利要求9所述的载体。
技术总结
本发明提供了一种基于全基因组测序的寄生虫虫种鉴定分析方法,其特征在于,包含如下步骤:S1.获取原始数据;S2.对原始数据进行数据质控和数据统计后,输出fastq序列,所述数据质控,通过排除数据中的低质量碱基列,获得优化后的序列;S3.去除宿主人源基因组序列;S4.将经S3处理后的测序数据与数据库进行比对分析,从而输出基于k‑mer的所有基因组的最低共同祖先LCA,对每条基因组序列进行分类,并按照门纲目科属和种给出对应的统计信息。本发明可以在短时间内获知寄生虫虫种的相关序列,从而进行相关虫种的鉴定,提供了一种快速精准的寄生虫虫种鉴定技术。
技术研发人员:丰俊,陈敏,庄源,陈洪友,潘淼,张曦,袁政安,吴寰宇,徐桢
受保护的技术使用者:上海市疾病预防控制中心
技术研发日:
技术公布日:2024/3/12
- 还没有人留言评论。精彩留言会获得点赞!