一种相变调温纤维及其制备方法与流程
本发明涉及材料
技术领域:
,特别是涉及一种相变调温纤维及其制备方法。
背景技术:
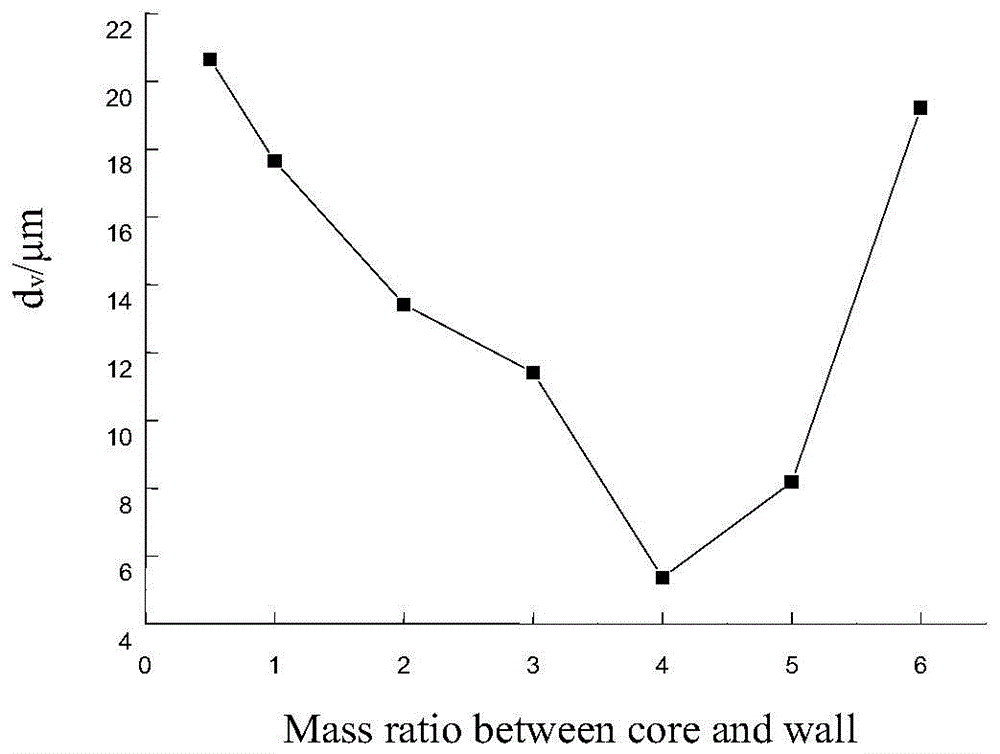
:随着人民生活水平的提高和科学技术的发展,人们对功能性纤维的需求日益加大。研究表明,当人体处于热平衡时,感觉舒适的皮肤平均温度在33.4℃左右,身体任何部位的皮肤温度与皮肤平均温度的差在1.5℃~3.0℃范围内,人体感觉不冷不热,超过±4.5℃范围,人体将有冷暖感。相变材料(phasechangmaterials,简称pcm)是一类在一定温度范围内,依靠自身可逆相变从环境中吸收或向环境释放热量的物质。目前人类已经发现并掌握的天然或合成相变材料约有500种,它们以各种形式存在于自然界中。将相变材料加入到纤维载体中,可以开发出在一定范围内自由调节温度的相变调温纤维。相变调温纤维是一种双向温度调节保温纤维,该纤维可以吸收、储存、重新分配和放出热量,在环境温度较高时具有吸热功能,在环境温度较低时具有放热功能,从根本上扩展了纤维原有性能,属于新型“智能”纤维。相变调温纺织品的研制一般采用微胶囊涂层法,即通过聚合物粘合剂和涂层剂等将相变微胶囊涂敷于织物表面,从而得到相变调温纺织品。triangle公司、bryant和zuckerman等人对这种涂层进行了大量研究。采用微胶囊涂层法制备相变调温纺织品方法简单方便,但织物物理机械性能和透气性能均有所下降。其次是采用微胶囊纺丝法,即将相变微胶囊直接植入纤维内部。由于被永久地封存在纤维内部,因此微胶囊的稳定性好,在随后的纺纱、织造和染色时,可以采用一般工艺,纤维能保持原有的柔软性,但是微胶囊合成粒径、纺丝工艺等控制不当会使纤维强度受到很大的损失。目前,微胶囊复合纺丝生产相变调温纤维的研究主要存在以下问题:(1)由于微胶囊本身具有一定的尺寸和性质,将微胶囊与纺丝液进行共混纺丝时,很容易影响纺丝性能以及复合纤维的机械性能,并造成后续纺织加工性能和应用性能的下降。一般喷丝口的尺寸在数十微米左右,所以,要求纺丝用微胶囊的尺寸必须在1μm左右,而一般微胶囊的尺寸往往在3μm及以上,在纺丝中易堵塞,且其成型往往较差,相变性能差,这将在适合纺丝用微胶囊的合成方面存在一定的难度。(2)在相变材料微胶囊的合成上,由于反复发生相变,使得对微胶囊的包覆性提出了更高的要求。微胶囊壁材的选取直接影响微胶囊纤维的包覆功能,相变微胶囊要求成膜性和密闭性好的壁材,以防在使用过程中发生漏液。技术实现要素:本发明的目的是提供一种相变调温微纤维及其制备方法,以解决上述现有技术存在的问题。为实现上述目的,本发明提供了如下方案:本发明提供一种相变调温纤维,采用溶液复合纺丝法,将相变调温微胶囊加入纺丝液中,进行共混纺丝,得到相变调温纤维;所述相变调温微胶囊包括相变材料芯材及包裹所述相变材料芯材的壁材;所述相变材料芯材为石蜡,所述壁材为甲苯-2,4-二异氰酸酯和己二胺;所述石蜡包括液体石蜡和固体石蜡,所述液体石蜡和所述固体石蜡的质量比为1-3:1。进一步地,所述相变材料芯材与所述壁材的质量比为1-6:1-2;所述相变调温微胶囊的平均粒径为1μm,相变温度34-39℃。本发明还提供一种上述的相变调温纤维的制备方法,具体步骤如下:(1)采用纤维素浆粕经浸渍、压榨、粉碎、老成、黄化后制得纤维素黄酸脂;(2)向纤维素黄酸脂中加入相变调温微胶囊混合搅拌,并经过滤、熟成,制得纺丝胶;(3)将制得的纺丝胶经喷头喷入到凝固浴成型,并经过牵伸、精练、烘干等后处理制得相变调温纤维。进一步地,步骤(1)中,所述浸渍的碱质量分数20%,浸渍温度40℃,浸渍时间30min,浴比1:40;所述压榨倍数:2.8;所述粉碎度150g/l;所述老成的温度25℃,时间72h;所述黄化的时间2h,温度25℃,二硫化碳用量125%。进一步地,步骤(2)中,所述纺丝胶中调温微胶囊相对纤维素有效含量10%,含碱质量比7.5%。进一步地,步骤(3)中,所述凝固浴含有:硫酸20g/l、硫酸锌15g/l、硫酸钠330g/l;凝固温度48℃。进一步地,所述相变调温微胶囊的制备方法,包括以下步骤:将乙二胺加入到蒸馏水中,得到乙二胺溶液,在所述乙二胺溶液中加入十二烷基硫酸钠,加热,得到乙二胺和十二烷基硫酸钠水溶液;石蜡加热至融化,将所述乙二胺和十二烷基硫酸钠水溶液置于高剪切条件下,迅速加入所述融化的石蜡和甲苯-2,4-二异腈酸酯,加入乳化液,乳化,乳化完毕得到稳定分散的乳化液;将所述乳化液在搅拌回流条件下,超声震荡反应1-4h,反应完毕,抽滤,洗涤,干燥,得到所述相变调温微胶囊。进一步地,所述乳化液为苯乙烯-顺丁烯二酸酐共聚物的水解物,所述乳化液浓度为2.5-20g/l,ph为7.4-13.4;所述高剪切条件以乳化机实现,转速12000-28000r/min,乳化时间0.5-4h。进一步地,所述搅拌回流条件的搅拌转速为400-900r/min,温度50-90℃。本发明公开了以下技术效果:本发明以相变材料石蜡为芯材,甲苯-2,4二异氰酸酯(tdi)和己二胺为壁材,合成共聚物苯乙烯-顺丁烯二酸酐的水解物(smh)为乳化剂,采用原位聚合技术,将相变材料包覆在微胶囊内部,制成相变调温微胶囊。通过正交试验研究实验参数对合成微胶囊的影响,最终确定微胶囊合成工艺为:超声波时间2h,芯材与壁材的质量比为4:1,smh的浓度为5g/l,反应时间为2h,反应温度为70℃。该工艺技术指标为:调温微胶囊平均粒径在1μm左右,相变温度34-39℃。采用本发明的相变调温微胶囊制备的相变调温纤维的工艺技术指标为:粘胶基微胶囊相变调温纤维平均断裂强度18.97cn/tex,平均断裂伸长率在13.1%。通过相变调温纤维纺丝工艺的优化,保证了微胶囊相变介质在纺丝液中的分散均匀性,稳定了相变调温效果及良好的耐洗涤性。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1为微胶囊芯材与壁材的质量比对体均粒径影响示意图;图2为超声波时间对体均粒径的影响示意图;图3为微胶囊体均粒径分布示意图;图4为微胶囊数均粒径分布示意图;图5为乳化剂smh的浓度(g/l)对体均粒径影响示意图;图6为乳化剂smh的ph对体均粒径影响示意图;图7为乳化转速(r/m)对体均粒径影响示意图;图8为乳化时间(h)对体均粒径影响示意图;图9为反应转速(r/min)对体均粒径影响示意图;图10为反应温度对体均粒径影响示意图;图11为相变调温微胶囊的扫描电镜图;图12为芯材和微胶囊a、b升温过程dsc图;图13为芯材和微胶囊a、b降温过程dsc图;图14为芯材和微胶囊a、b和壳材的tg图;图15为相变调温纤维的升温曲线;图16为相变调温纤维的降温曲线。具体实施方式现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见得的。本申请说明书和实施例仅是示例性的。关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。本发明提供一种相变调温纤维的制备方法,采用溶液复合纺丝法,将相变微胶囊加入纺丝液中,进行共混纺丝,制备相变调温粘胶纤维。选用优质棉、木浆粕等纤维素浆粕为原料,经过浸渍、压榨、粉碎、老成、黄化、溶解、混合、过滤、熟成、脱泡、纺丝、牵伸、精练、烘干等步骤,制备工序与普通的粘胶纤维一致,具体如下:(1)采用纤维素浆粕经浸渍、压榨、粉碎、老成、黄化后制得纤维素黄酸脂。工艺参数为:碱质量分数20%,浸渍温度40℃,浸渍时间30min,浴比1:40,压榨倍数:2.8,粉碎度150g/l;低温老成温度25℃,时间72h;黄化时间2h,温度25℃,二硫化碳用量125%(基数为纤维素浆粕的质量(g))。(2)向纤维素黄酸脂中加入调温微胶囊混合搅拌,并经过滤、熟成,制得调温微胶囊相对纤维素有效含量约10%,含碱质量比约7.5%的纺丝胶;(3)将制得的纺丝胶经喷头喷入到温度48℃,含硫酸20g/l、硫酸锌15g/l、硫酸钠330g/l,的凝固浴成型,并经过牵伸、精练、烘干等后处理制得相变调温纤维。作为本发明的进一步改进,所述相变调温微胶囊,包括相变材料芯材及包裹所述相变材料芯材的壁材;所述相变材料芯材为石蜡,所述壁材为甲苯-2,4二异氰酸酯和己二胺。作为本发明的进一步改进,所述石蜡包括液体石蜡和固体石蜡,所述液体石蜡和所述固体石蜡的质量比为1-3:1。作为本发明的进一步改进,所述甲苯-2,4二异氰酸酯和己二胺的质量比为1-5:1-2。相变调温微胶囊芯材的选择:将液体石蜡和固体石蜡进行一定质量比例的混合,加热混合均匀后冷却放置。以此方法得到一系列不同质量比的混合石蜡,以备选择。测量不同质量比例混合石蜡的熔程范围、吸热焓、凝固范围和放热焓,结果见表1。综合熔程、峰值以及吸热焓等因素,固体石蜡与液体石蜡比例为3:1时吸热焓、放热焓最大,故选择3:1。表1作为本发明的进一步改进,所述相变材料芯材与所述壁材的质量比为1-6:1-2。芯材与壁材的质量比对相变微胶囊体均粒径影响:芯材与壁材质量比分别为1:2、1:1、2:1、3:1、4:1、5:1和6:1进行实验,结果见图1。当芯材与壁材质量比逐渐增大时,体均粒径急剧减小,从21μm左右减小到5μm左右,芯材的比例再增加时,体均粒径又增加。可以得出结论,此单因素对体均粒径影响显著。当加入的石蜡较少时,体系中存在过量的四乙烯五胺和异氰酸酯,另外,异氰酸酯与水反应生成氨基,这不影响链段的继续增长,这样只形成了小部分的微胶囊,绝大部分为聚脲颗粒,故体积较大;当石蜡比例逐渐上升后,开始大量生成微胶囊,故体均粒径减小;当加入的石蜡增加到过量时,壁材的用量不足以在每个小分散的石蜡颗粒上形成包覆,因此石蜡会出现团聚的情况,体均粒径又会增加。综合考虑,最佳核壳比为4:1。作为本发明的进一步改进,所述相变调温微胶囊的平均粒径为1μm,相变温度34-39℃。作为本发明的进一步改进,上述的相变调温微胶囊的制备方法,包括以下步骤:将乙二胺加入到蒸馏水中,得到乙二胺溶液,在所述乙二胺溶液中加入十二烷基硫酸钠,加热,得到乙二胺和十二烷基硫酸钠水溶液;石蜡稍许加热至融化,将所述乙二胺和十二烷基硫酸钠水溶液置于高剪切条件下,迅速加入所述融化的石蜡和甲苯-2,4-二异腈酸酯,加入乳化液,乳化,乳化完毕得到稳定分散的乳化液;将所述乳化液在搅拌回流条件下,超声震荡反应,反应完毕,抽滤,洗涤,干燥,得到所述相变调温微胶囊。作为本发明的进一步改进,所述超声震荡的时间为1-4h。超声波时间对相变微胶囊体均粒径的影响:超声波震荡有助于将反应液中大的颗粒团分散成一个个小颗粒,并且将极细小的颗粒聚集成稍大的颗粒团,使微胶囊的粒径分布变窄,体均粒径大大减小。由图2可以看出,随着超声波时间的增加,体均粒径明显减小,当超过2小时后,体均粒径基本保持不变。故选择超声波震荡时间为2h。体均粒径(dv)是按照不同颗粒大小微胶囊所占总体的体积分数计算,而数均粒径(dn)是按照不同颗粒大小微胶囊所占总体的数量分数计算,两者计算方法不同。也就是说,体均粒径的大小是由体积较大的一部分的决定;而数均粒径的大小是由数目较多的一部分的决定。分布指数pdi=dv/dn,pdi值越接近1,说明微胶囊的分散性越好。实验结果可以证明,超声波震荡有助于将反应液中大的颗粒团分散成一个个的小颗粒,并且将极细小的颗粒聚集成稍大的颗粒团,使微胶囊的粒径分布变窄,体均粒径大大减小。通过激光粒度分析仪测试实验制得微胶囊的体均粒径和数均粒径分别为4.08μm和1.04μm,如图3、图4所示,则pdi=4.08/1.04=3.93。作为本发明的进一步改进,所述乳化液为苯乙烯-顺丁烯二酸酐共聚物的水解物,所述乳化液浓度为2.5-20g/l,ph为7.4-13.4。乳化剂的浓度对相变微胶囊粒径的影响:分别选用乳化剂smh(苯乙烯-顺丁烯二酸酐聚合物)浓度为2.5g/l、5g/l、10g/l、15g/l、20g/l,进行单因素实验。如图5所示,当smh浓度由2.5g/l逐渐增大到20g/l后,体均粒径先减小后逐渐增大。这是因为:smh用量较小时,达不到较好的分散乳化效果,石蜡分散的不均匀,所以体均粒径较大;逐渐增加smh的用量时,其分散乳化的效果会逐渐增强,体均粒径就会显著下降;当用量逐渐增加时,多余的smh被包覆于微胶囊中,故粒径有所变大。当smh用量为5g/l时体均粒径达到最小值,为smh用量的最佳值。乳化剂smh水溶液ph对相变微胶囊体均粒径的影响:分别选用smh水溶液的ph为7.4、8.4、9.4、10.4、11.4、12.4、13.4,进行单因素实验。如图6所示:随着smh水溶液ph从7.4增加到13.4,微胶囊体均粒径差值在1.5μm以内,故可以得出结论,smh水溶液ph对体均粒径影响较小。作为本发明的进一步改进,所述高剪切条件以乳化机实现,转速12000-28000r/min,乳化时间0.5-4h。乳化转速对微胶囊体均粒径的影响:选用5种不同的乳化机转速进行实验,分别为12000r/min、16000r/min、20000r/min、24000r/min、28000r/min,结果见图7。随着乳化转速从12000r/m提高到28000r/m,微胶囊的体均粒径从8.2μm降为5.3μm,并且在转速提高到20000r/m以上后,体均粒径下降趋势平缓,保持在5.5μm左右。由此可以得出结论:乳化转速是影响微胶囊的体均粒径的重要因素,提高乳化转速可以明显降低微胶囊的体均粒径,但达到一定转速时,提升的效果减弱。综合考虑实验条件与实验结果,确定乳化转速为20000r/min。乳化时间对微胶囊体均粒径的影响:分别选用乳化时间为0.5h、1.0h、2.0h、3.0h、4.0h,进行实验,如图8所示。随着乳化时间的增加,体均粒径明显减小,当超过2小时后,体均粒径稍有增大,这可能是由于继续增加反应时间,使包覆在石蜡表面的聚合物膜膜层厚度变大所导致。乳化少于2小时,则未充分反应。确定最佳乳化时间为2小时。作为本发明的进一步改进,所述搅拌回流条件的搅拌转速为400-900r/min,温度50-90℃。反应转速对微胶囊体均粒径的影响:分别选用反应转速为400r/min、500r/min、600r/min、700r/min、800r/min和900r/min,进行实验,如图9所示。当反应转速从400r/min提高到900r/min,体均粒径差值在2μm以内,可见:反应转速对微胶囊体均粒径影响较小。选择反应转速为700r/min即可,速度太大容易引起反应液的飞溅。反应温度对微胶囊体均粒径影响:分别选用反应温度为50℃、60℃、70℃、80℃、90℃,进行实验,如图10所示,当温度从50℃提高至90℃后,体均粒径明显减小,可能是因为随着反应温度的升高加快了单体的扩散速度,迅速吸附于石蜡表面,形成薄膜。另外一方面,由于芯材为具有较大粘度的石蜡,它的粘度会随着温度的升高而减小,这样就会有利于石蜡的分散,从而得到小粒径的微胶囊。由此可以得出结论,反应温度是影响微胶囊体均粒径的主要因素之一。确定最佳反应温度为70℃。根据上述的条件,选择相变调温微胶囊制备过程中有无超声波、芯材与壁材的质量比、smh的浓度、反应时间和反应温度为影响因素进行正交实验,寻找制备相变微胶囊过程中各因素对其体均粒径影响的大小。smh的水溶液ph固定在12,乳化转速固定在20000r/min,反应时转速固定在700r/min。实验条件见表2.由表2可知,“有无超声波”一项中,其极差达到25.375,说明其对于相变微胶囊的体均粒径影响最大;“芯材与壁材的比例”一项中,极差为12.175,说明其对于相变微胶囊的体均粒径也有着较重要的影响;“反应时间”“反应温度”“smh的浓度”的极差分别为5.775、5.475和2.700,相比以上两个因素而言,这三项因素对相变微胶囊的体均粒径影响较小。从4项均值中,可确定出最佳工艺条件:超声波时间2h,芯材与壁材的质量比为4:1,smh的浓度为5g/l,反应时间为2h,反应温度为70℃。表2一种上述的相变调温微胶囊的制备方法,包括以下步骤:将乙二胺加入到蒸馏水中,得到乙二胺溶液,在所述乙二胺溶液中加入十二烷基硫酸钠,加热,得到乙二胺和十二烷基硫酸钠水溶液;石蜡稍许加热至融化,将所述乙二胺和十二烷基硫酸钠水溶液置于高剪切条件下,迅速加入所述融化的石蜡和甲苯-2,4-二异腈酸酯,加入乳化液,乳化,乳化完毕得到稳定分散的乳化液;将所述乳化液在搅拌回流条件下,超声震荡反应,反应完毕,抽滤,洗涤,干燥,得到所述相变调温微胶囊a。所述芯材与壁材的质量比为4:1。所述超声震荡的时间为2h。所述甲苯-2,4二异氰酸酯和己二胺的质量比为3:1.5。所述乳化液为苯乙烯-顺丁烯二酸酐共聚物的水解物,所述乳化液浓度为5g/l,ph为12。所述高剪切条件以乳化机实现,转速20000r/min,乳化时间2h。所述搅拌回流条件的搅拌转速为700r/min,温度70℃。试验制备的相变调温微胶囊a的性能检验1.扫描电子显微镜分析用jsm-5600lv扫描电子显微镜观测相变微胶囊的表观形态,结果如图11所示。由图11可观察出,微胶囊的粒径较小,粒径分布较窄,大约在1-5μm的范围内。微胶囊整体分散程度较好,颗粒之间基本没有相互粘附的情况。微胶囊表面基本没有破裂,这可能是因为壁材在界面反应充分,能够全部包覆乳化后的相变材料微球。2.调温微胶囊的吸放热性能使用差示扫描量热仪(dsc法)分别测量芯材(固体石蜡与液体石蜡质量比例为3:1混合石蜡)和微胶囊熔融范围、吸热焓、凝固范围和放热焓,具体见表3,其中微胶囊b制备过程中未采用超声波震荡。表3其在升温和降温过程中的dsc如图12和图13,从dsc图中可以看出,大部分的热焓还是集中在30-37℃之间,适合应用于人体温度的调节。3.调温微胶囊的耐热性能使用热重分析仪分别对芯材、微胶囊a、微胶囊b和壳材(聚脲)进行热重分析,裂解温度列于表4,其裂解过程如图14。表4表4表明,芯材、微胶囊a、b和壳材的裂解温度分别为130℃、200℃、200℃和260℃,说明在130-200℃之间,壳材对相变材料石蜡有保护作用;同时也说明相变材料石蜡已经较好的包覆在壳材中,并且有着较好的耐热性能。4.调温微胶囊的耐酸性能称取1g调温微胶囊,加水定溶250ml,放入平底烧瓶中,加入2g硫酸,升温观测,结果如表5,由观测结果可知微胶囊比较耐酸。微胶囊的耐酸性使其在粘胶纺丝过程中酸浴工序及后处理的酸洗工序中不会存在太大问题。表5操作程序试液现象加酸1min微胶囊颗粒悬浮,水透明40℃,30min微胶囊颗粒悬浮,水透明60℃,30min微胶囊颗粒悬浮,水透明80℃,30min少量石蜡析出,微胶囊颗粒悬浮,水透明100℃,30min少量石蜡析出,絮状物出现,水半透明5.调温微胶囊的耐碱性能称取1g调温微胶囊,加水定溶250ml,放入平底烧瓶中,加入2.5g氢氧化钠,升温观测,结果如表6,由观测结果可知微胶囊比较耐碱。微胶囊的耐碱性使其在脱硫工序中不会存在太大问题。表6本发明所制备相变调温纤维性能测试a.调温性能测试外界环境温度升高时的测试:把控温微胶囊纤维和普通粘胶纤维放置恒温恒湿环境中(温度20℃,湿度60%)平衡24h,然后分别放在40℃的保温板上,每间隔2-3s,用红外测温仪测定纤维表面温度,记录并绘制升温曲线,见图15。外界环境温度降低时的测试:把控温微胶囊纤维和普通粘胶纤维放置于42℃烘箱中加热保温2h,然后迅速取出,自然冷却至环境温度(温度20℃,湿度60%),此过程中每间隔2-3s,用红外测温仪测定纤维表面温度,记录并绘制降温曲线,见图16。从图15可以看出:在30℃时,微胶囊调温纤维开始吸收热量,自身升温速率逐渐减慢,35℃时达到最小值,最后缓缓到达37℃;从图16可以看出:在34℃时,微胶囊调温纤维开始释放自身热量,自身降温速率下降,30℃时达到最小值,最后到达20℃。而普通粘胶纤维,无论是升温速度还是降温速度,均比调温纤维相对较快。实验表明:粘胶基微胶囊相变调温纤维的温控范围约在20℃~37℃之间,与其采用的相变微胶囊的吸热和放热温度区间基本一致。b.调温性能特征指标的选择分别采用差示扫描量热法、差热分析法,对相变调温纤维的调温保温效果进行了检测研究,结果见表7、8。表7表8分析上述两表,说明:对于微胶囊及相变调温纤维,两种方法测试结果基本一致,但差热分析(dta)曲线更能准确找到相转变点。因此,最终选择比较直观的差热分析法(dta)测定结果——“相转变温度”,作为相变调温纤维的特征指标。c.调温微胶囊纤维强力测试对于微胶囊有效含量不同的相变调温粘胶纤维试验样品,根据国家标准gb/t14344-2008《化学纤维长丝拉伸性能试验方法》测试纤维断裂强度和断裂伸长率。见表9:表9从测试结果看,微胶囊添加量低于12%时,纤维断裂强度大于18.97cn/tex,断裂伸长率大于13%。对于普通粘胶纤维,产品标准gb/t13758-2008《粘胶长丝》规定:一等品干断裂强度不低于1.75cn/dtex,干断裂伸长率(16.0~25.0)%。可见,加入微胶囊的相变调温纤维断裂伸长率有所下降。d.调温微胶囊粒径的影响微胶囊粒径3μm以上,纺丝困难,成形较差,不适合用于后续加工。分别采用粒径为2μm和1μm的相变微胶囊,添加量12%,试纺粘胶基微胶囊相变调温纤维,其物理性能测试结果如表10。从表中可以看出:微胶囊粒径增大,相变纤维断裂伸长率降低,断裂强力变异系数明显增大。即可纺性能明显降低。表10不同粒径微胶囊制备的粘胶基微胶囊相变调温纤维比较另外,选用纤维素材料常用的两类染料——直接染料、活性染料,分别对微胶囊相变调温粘胶纤维和普通粘胶纤维进行染色,利用分光光度计测定染色过程中染料上染程度及最终上染率;根据染后样品耐皂洗色牢度、耐摩擦色牢度等指标评价固色程度。从而分别研究染料浓度、染浴温度对微胶囊相变调温粘胶纤维和普通粘胶纤维染色性能的影响。实验结果表明:微胶囊相变调温粘胶纤维与普通粘胶纤维的的染色性能基本一致。分析原因,可能是由于纺织用相变材料调温范围通常在40℃以下,对于40℃起染的染色工艺,相变材料基本不起作用。此外,微胶囊相变材料是以物理形式添加到纤维纺丝液中,未发生分子结构的变化,因而染色性能基本不受影响。以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1