调色剂和调色剂的制造方法与流程
1.本公开涉及一种适用于诸如电子照相法、静电记录法、调色剂喷射法等图像形成方法的调色剂以及该调色剂的制造方法。
背景技术:
2.近年来,打印机和复印机已经实现了高水平的图像品质和耐久性,同时,特别是对于打印机而言,小型化和无废弃物操作已被广泛要求。
3.打印机必须能适应例如大量的人通过网络使用的网络打印机和在soho中使用的个人打印机等各种业务情况。此外,在办公室或soho环境中,强烈期望消除与诸如废调色剂更换等废弃物的产生相关的维护。因此,对于节省空间的打印机,即小型化和无废弃物操作的打印机,仍然存在强烈的需求。
4.定影装置和处理盒的小型化主要对打印机的小型化是有效的。尤其是,处理盒占据了打印机的大部分体积,处理盒的小型化会极大有助于打印机的小型化。
5.显影装置和清洁装置的小型化对于打印机的小型化是有效的。关于显影装置的小型化,有二组分和单组分电子照相显影系统,但适合于小型化的是单组分显影系统。这是因为没有使用如载体等构件。
6.关于清洁装置,完全不具有清洁装置的无清洁器系统是极其适用于处理盒的小型化的。在许多打印机中,在转印步骤中残留的静电潜像承载构件(感光构件)上的调色剂(转印残留调色剂)被清洁刮板等刮掉并收集在清洁容器中,成为废调色剂。而在无清洁器系统中,没有清洁刮板或清洁容器,转印残留调色剂被再次收集在显影装置中并有助于显影,因此可以显著减小处理盒的尺寸,不会产生废调色剂,并且为向无废弃物操作的过渡做出重大贡献。
7.在这样的背景下,无论有无清洁装置,转印残渣少的调色剂都非常重要。通常,获得具有小的转印残渣的调色剂的方法可以举例为降低与感光构件的粘附力的调色剂颗粒的球形化,但是由于球形化也降低了调色剂颗粒之间的粘附力,所以很可能出现转印粉尘。为了解决这个问题,日本专利申请公开no.2007-241310提出了一种通过控制调色剂的表面形状而同时实现转印率的改善和转印粉尘的减少的调色剂。
技术实现要素:
8.虽然根据日本专利申请公开no.2007-241310,可以获得同时实现转印率的改善和转印粉尘的减少的调色剂,但这对于近年来要求长寿命的打印机而言不能说是充分的。此外,例如在日本专利申请公开no.2007-241310中提出的不规则形状的调色剂在显影期间当与刮板或辊摩擦时容易局部受到过度的力,并且在耐久性方面不能说是充分的。
9.本公开要解决的问题是提供一种具有优异耐久性的调色剂,其可以以高水平实现有助于打印机的小型化和向无废弃物操作的过渡的转印率的改善以及转印粉尘的减少,并且这些转印性能长期保持,并且还提供了一种调色剂的制造方法。
10.本公开涉及一种调色剂,其包括含有粘结剂树脂和硼酸的调色剂颗粒,其特征在于,
11.所述调色剂的平均圆形度为0.95以上,并且
12.所述调色剂的形状因子sf1为105至125。
13.本公开还涉及一种调色剂的制造方法,其特征在于,
14.所述调色剂的制造方法依下述顺序具有以下步骤(1)至(3):
15.(1)制备包含所述粘结剂树脂的粘结剂树脂微粒分散液的分散步骤,
16.(2)使所述粘结剂树脂微粒分散液中包含的粘结剂树脂微粒聚集以形成聚集体的聚集步骤,和
17.(3)将所述聚集体加热和熔合的熔合步骤,并且
18.在所述聚集步骤和所述熔合步骤中的至少一个步骤中添加硼酸源。
19.本公开提供一种具有优异耐久性的调色剂,其以高水平实现了转印率的改善和转印粉尘的减少,并且这些转印性能长期保持。
20.本发明的进一步特征将从以下对示例性实施方案的描述中变得显而易见。
具体实施方式
21.在本公开中,表示数值范围的“从xx至yy”或“xx至yy”的表达是指包括作为端点的下限和上限的数值范围,除非另有说明。此外,当以逐步方式描述数值范围时,每个数值范围的上限和下限可以适当组合。
22.在本公开中,“(甲基)丙烯酸”是指“丙烯酸”和/或“甲基丙烯酸”。
23.下面将更详细地描述本公开的调色剂。
24.作为为解决现有技术的上述问题而进行的努力研究的结果,本发明人发现上述问题可以通过包括含有粘结剂树脂的调色剂颗粒的调色剂来解决,其中将调色剂控制为特定形状,并且调色剂颗粒包括硼酸。
25.本公开涉及一种调色剂,其包括含有粘结剂树脂和硼酸的调色剂颗粒,其特征在于,
26.所述调色剂的平均圆形度为0.95以上,并且
27.所述调色剂的形状因子sf1为105至125。
28.首先,通过将调色剂的平均圆形度设为0.95以上、将形状因子sf1设为105至125,可以获得具有高转印率的调色剂。另一方面,具有在上述范围的形状的调色剂产生转印粉尘。然而,硼酸存在于具有在上述范围的形状的调色剂颗粒中使得可以以高水平改善转印率并减少转印粉尘。这可能是因为源自于硼酸的硼具有准金属性质(metalloid property),因此在调色剂颗粒之间或在调色剂颗粒内部进行适当的电荷转移,抑制了显影的调色剂颗粒之间的静电排斥,并且在聚集状态下进行转印。
29.为了获得调色剂形状,优选使用化学调色剂制造方法在水系介质中获得调色剂颗粒,例如乳化聚集法或悬浮聚合法。具体地,上述调色剂形状可以通过制造过程中的球形化步骤、冷却步骤和退火步骤获得。
30.球形化步骤例举为例如在90℃以上、优选92℃以上、优选95℃以下进行热处理的热处理步骤。
31.冷却步骤例举为以0.1℃/秒以上、优选0.5℃秒以上、更优选2℃/秒以上、还更优选4℃/秒以上的冷却速度进行的冷却处理。
32.退火步骤例举为热处理步骤,其中在调色剂的玻璃化转变温度(tg)最大的温度下在5小时内进行热处理。通过在适当条件下进行的这些步骤,可以容易地实现本公开的调色剂形状。特别地,为了获得通过截面抛光仪截面法(cp截面法)形成的调色剂截面的形状因子sf1为105至125,以及调色剂的下述接触面积比率(d/s)为14%以下,优选经受冷却步骤和退火步骤。通过经受这些步骤,可以抑制在调色剂表面形成凹痕,并且易于将截面的形状因子sf1减小到125以下并将接触面积比率(d/s)减小到14%以下。
33.调色剂的平均圆形度为0.95以上,优选为0.97以上,更优选为0.98以上。此外,平均圆形度优选为0.99以下。调色剂的形状因子sf1为105至125,优选为108至120。
34.当调色剂的平均圆形度和形状因子sf1在上述范围内时,调色剂的形状是接近真球形的球形,从而抑制了调色剂对感光构件的粘附并且改善了转印率。
35.通过截面抛光仪截面法(cp截面法)形成的调色剂截面的形状因子sf1的值优选为105至125,更优选为108至120。截面的形状因子sf1可以通过改变上述制造条件来控制。当截面的形状因子sf1在上述范围内时,调色剂表面很少有凹凸,并且局部外力不太可能施加到凸部(peaks)和凹部(valleys),从而大大改善了调色剂的耐久性,并且可长期保持转印率的改善和转印粉尘的减少等转印性能。
36.当在用22-μm开口的筛子筛分10秒的同时调色剂从上方10cm的位置落下并铺在水平玻璃平板上,并且将50个调色剂与玻璃平板之间的接触面的总面积定义为接触面积d,将50个调色剂的总投影面积定义为s时,接触面积比率(d/s)优选为3至14%,更优选为5至8%。可以通过改变上述制造条件来控制接触面积比率(d/s)。当接触面积比率(d/s)在上述范围时,调色剂表面很少有凹凸,局部外力不太可能施加至凸部和凹部,从而调色剂耐久性大大改善,并且可以长期保持如转印率的改善和转印粉尘的减少等转印性能。
37.优选地,调色剂还包含外部添加剂,所述外部添加剂包含二氧化硅微粒,通过x射线光电子能谱装置测量的调色剂颗粒表面被二氧化硅微粒覆盖的覆盖率为50至80面积%,更优选为60至70面积%。当覆盖率在上述范围时,抑制了在被外部添加剂覆盖的部分和未被覆盖的部分之间产生的局部电荷差,从而可以进一步减少转印粉尘并且可以改善转印性。二氧化硅微粒的覆盖率可以通过调整二氧化硅微粒的添加量、外部添加时间等来控制。
38.调色剂表面上的二氧化硅微粒的分散度评价指数优选为0.10至2.00,更优选为0.20至0.40。通过设定为上述范围,抑制了被外部添加剂覆盖的部分和未被覆盖的部分,因此可以进一步减少转印粉尘,并且可以改善转印性。分散度评价指标可以通过调整二氧化硅微粒的添加量、外部添加时间等来控制。
39.优选在通过使用锗作为atr晶体的atr法对调色剂颗粒的ir分析中检测到硼酸。这意味着硼酸存在于调色剂表面附近。当硼酸存在于调色剂表面附近时,在调色剂颗粒之间或在调色剂颗粒内部进行适当的电荷转移,抑制了在显影的调色剂颗粒之间的静电排斥,并且在聚集状态下进行转印。因此,可以减少转印粉尘并且可以改善耐久性。
40.在调色剂颗粒的荧光x射线测量中,硼的强度优选为0.1至0.6kcps,更优选为0.2至0.6kcps。当硼的强度在上述范围时,调色剂颗粒中含有适量的硼酸,适量的电荷在调色剂颗粒之间或在调色剂颗粒内部转移,抑制了显影的调色剂颗粒之间的静电排斥,并且在
聚集状态下进行转印。因此,可以减少转印粉尘并且可以改善耐久性。
41.硼酸可以通过使用硼酸源作为内部添加剂或絮凝剂而包含在调色剂颗粒中。特别地,通过添加硼酸源作为絮凝剂,可以将硼酸引入调色剂颗粒表面附近。
42.下面将更详细地描述构成调色剂的各组分和调色剂的制造方法。
43.粘结剂树脂
44.调色剂颗粒包含粘结剂树脂。粘结剂树脂的量相对于调色剂颗粒中的树脂组分的总量优选为50质量%以上。
45.粘结剂树脂没有特别限制,其实例包括苯乙烯丙烯酸系树脂、环氧树脂、聚酯树脂、聚氨酯树脂、聚酰胺树脂、纤维素树脂、聚醚树脂、它们的混合树脂和复合树脂等。苯乙烯丙烯酸系树脂和聚酯树脂是优选的,因为它们便宜、容易获得并且能够实现优异的低温定影性。
46.聚酯树脂可以通过从多元羧酸、多元醇、羟基羧酸等中选择并组合适当的组分,使用如酯交换法、缩聚法等以往公知的方法进行合成而获得。
47.多元羧酸是在一分子中含有两个以上的羧基的化合物。其中,优选使用二羧酸,其为一分子中含有两个羧基的化合物。
48.二羧酸的实例包括草酸、琥珀酸、戊二酸、马来酸、己二酸、β-甲基己二酸、壬二酸、癸二酸、壬烷二甲酸、癸烷二甲酸、十一烷二甲酸、十二烷二甲酸、富马酸、柠康酸、二甘醇酸、环己烷-3,5-二烯-1,2-羧酸、六氢对苯二甲酸、丙二酸、庚二酸、辛二酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、四氯邻苯二甲酸、氯邻苯二甲酸、硝基邻苯二甲酸、对-羧基苯乙酸、对苯二乙酸、间苯二乙酸、邻苯二乙酸、二苯基乙酸、二苯基-p,p'-二甲酸、萘-1,4-二甲酸、萘-1,5-二甲酸、萘-2,6-二甲酸、蒽二甲酸、环己烷二甲酸等。
49.除了二羧酸之外的多元羧酸的实例包括偏苯三酸、均苯三酸、均苯四酸、萘三甲酸、萘四甲酸、芘三甲酸、芘四甲酸、衣康酸、戊烯二酸、正十二烷基琥珀酸、正十二烯基琥珀酸、异十二烷基琥珀酸、异十二烯基琥珀酸、正辛基琥珀酸、正辛烯基琥珀酸等。这些可以单独使用或以两种以上组合使用。
50.多元醇是在一分子中含有两个以上的羟基的化合物。其中,优选使用二醇,其为一分子中含有两个羟基的化合物。
51.具体实例包括乙二醇、二甘醇、三甘醇、1,2-丙二醇、1,3-丙二醇、1,4-丁二醇、1,5-戊二醇、1,6-己二醇、1,7-庚二醇、1,8-辛二醇、1,9-壬二醇、1,10-癸二醇、1,11-十一烷二醇、1,12-十二烷二醇、1,13-十三烷二醇、1,14-十四烷二醇、1,18-十八烷二醇、1,14-二十烷癸二醇(1,14-eicosanedecanediol)、二甘醇、三甘醇、二丙二醇、聚乙二醇、聚丙二醇、聚四亚甲基醚二醇、1,4-环己二醇、1,4-环己烷二甲醇、1,4-丁烯二醇、新戊二醇、1,4-环己二醇、聚丁二醇、氢化双酚a、双酚a、双酚f、双酚s、上述双酚的氧化烯(环氧乙烷、环氧丙烷、环氧丁烷等)加合物等。
52.其中,优选碳原子数为2至12的亚烷基二醇和双酚的氧化烯加合物,特别优选双酚的氧化烯加合物及其与碳原子数为2至12的亚烷基二醇的组合。
53.三元以上的多元醇的实例包括甘油、三羟甲基乙烷、三羟甲基丙烷、季戊四醇、六羟甲基三聚氰胺、六羟乙基三聚氰胺、四羟甲基苯并胍胺、四羟乙基苯并胍胺、山梨糖醇、三苯酚pa、苯酚酚醛清漆、甲酚酚醛清漆、上述三元以上的多酚的氧化烯加合物等。这些可以
单独使用或以两种以上组合使用。
54.苯乙烯丙烯酸系树脂的实例包括由以下聚合性单体组成的均聚物、通过组合这些中的两种以上而获得的共聚物、或它们的混合物。
55.苯乙烯系单体,例如苯乙烯、α-甲基苯乙烯、β-甲基苯乙烯、邻甲基苯乙烯、间甲基苯乙烯、对甲基苯乙烯、2,4-二甲基苯乙烯、对正丁基苯乙烯、对叔丁基苯乙烯、对正己基苯乙烯、对正辛基苯乙烯、对正壬基苯乙烯、对正癸基苯乙烯、对正十二烷基苯乙烯、对甲氧基苯乙烯和对苯基苯乙烯;
56.(甲基)丙烯酸系单体,例如(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸正丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸正丁酯、(甲基)丙烯酸异丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸正戊酯、(甲基)丙烯酸正己酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸正辛酯、(甲基)丙烯酸正壬酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸苄酯、磷酸二甲酯(甲基)丙烯酸乙酯、磷酸二乙酯(甲基)丙烯酸乙酯、磷酸二丁酯(甲基)丙烯酸乙酯和(甲基)丙烯酸2-苯甲酰氧基乙酯、(甲基)丙烯腈、(甲基)丙烯酸2-羟乙酯、(甲基)丙烯酸和马来酸;
57.乙烯基醚单体,例如乙烯基甲基醚和乙烯基异丁基醚;
58.乙烯基酮单体,例如乙烯基甲基酮、乙烯基乙基酮、乙烯基异丙烯基酮等;和
59.聚烯烃类,例如乙烯、丙烯、丁二烯等。
60.苯乙烯丙烯酸系树脂可以根据需要使用多官能聚合性单体。多官能聚合性单体的实例包括二甘醇二(甲基)丙烯酸酯、三甘醇二(甲基)丙烯酸酯、四甘醇二(甲基)丙烯酸酯、聚乙二醇二(甲基)丙烯酸酯、1,6-己二醇二(甲基)丙烯酸酯、新戊二醇二(甲基)丙烯酸酯、三丙二醇二(甲基)丙烯酸酯、聚丙二醇二(甲基)丙烯酸酯、2,2'-双(4-((甲基)丙烯酰氧基二乙氧基)苯基)丙烷、三羟甲基丙烷三(甲基)丙烯酸酯、四羟甲基甲烷四(甲基)丙烯酸酯、二乙烯基苯、二乙烯基萘、二乙烯基醚等。
61.另外,为了控制聚合度,还可以进一步添加公知的链转移剂和公知的阻聚剂。
62.用于获得苯乙烯丙烯酸系树脂的聚合引发剂的实例包括有机过氧化物类引发剂和偶氮类聚合引发剂。
63.有机过氧化物类引发剂的实例包括过氧化苯甲酰、过氧化月桂酰、过氧化二-α-枯基、2,5-二甲基-2,5-双(过氧化苯甲酰)己烷、双(4-叔丁基环己基)过氧化二碳酸酯、1,1-双(叔丁基过氧化)环十二烷、叔丁基过氧化马来酸、双(叔丁基过氧化)间苯二甲酸酯、过氧化甲乙酮、叔丁基过氧化-2-乙基己酸酯、二异丙基过氧化碳酸酯、异丙苯过氧化氢、过氧化2,4-二氯苯甲酰、过氧化新戊酸叔丁酯等。
64.偶氮类聚合引发剂的实例包括2,2'-偶氮二-(2,4-二甲基戊腈)、2,2'-偶氮二异丁腈、1,1'-偶氮二(环己烷-1-甲腈)、2,2'-偶氮二-4-甲氧基-2,4-二甲基戊腈、偶氮二甲基丁腈、2,2'-偶氮二-(异丁酸甲酯)等。
65.另外,作为聚合引发剂,也可以使用组合了氧化性物质和还原性物质的氧化还原类引发剂。
66.氧化性物质的实例包括过氧化氢、过硫酸盐(钠盐、钾盐和铵盐)的无机过氧化物和四价铈盐的氧化性金属盐。
67.还原性物质的实例包括还原性金属盐(二价铁盐、一价铜盐和三价铬盐),氨,低级
胺(碳原子数为1至6的胺,例如甲胺和乙胺),胺化合物例如羟胺,还原性硫化合物例如硫代硫酸钠、连二亚硫酸钠、亚硫酸氢钠、亚硫酸钠、甲醛合次硫酸氢钠,低级醇(碳原子数为1至6),抗坏血酸或其盐,低级醛(碳原子数为1至6)。
68.聚合引发剂参考10小时半衰期温度选择,单独或组合使用。聚合引发剂的添加量根据所期望的聚合度而改变,但是相对于聚合性单体100.0质量份,通常为0.5至20.0质量份。
69.脱模剂
70.公知的蜡可以用作调色剂的脱模剂。
71.具体实例包括以固体石蜡、微晶蜡、矿脂为代表的石油类蜡及其衍生物、褐煤蜡及其衍生物、通过费-托法获得的烃蜡及其衍生物、以聚乙烯为代表的聚烯烃蜡及其衍生物、以巴西棕榈蜡和小烛树蜡为代表的天然蜡及其衍生物,衍生物包括氧化物、与乙烯基单体的嵌段共聚物、接枝改性物。
72.其他实例包括醇例如高级脂肪醇,脂肪酸例如硬脂酸、棕榈酸等或其酰胺、酯和酮,硬化蓖麻油及其衍生物,植物蜡和动物蜡。这些可以单独或组合使用。
73.其中,优选使用聚烯烃、通过费托法生产的烃蜡或石油类蜡,因为显影性能和转印性趋于得到改善。在不影响调色剂特性的范围内,可以将抗氧化剂添加到这些蜡中。
74.此外,从相对于粘结剂树脂的相分离或其结晶温度的观点来看,可以优选例举山萮酸山萮醇酯、癸二酸二山萮醇酯等高级脂肪酸酯。
75.相对于粘结剂树脂100.0质量份,脱模剂的量优选为1.0至30.0质量份。
76.脱模剂的熔点优选为30至120℃,更优选为60至100℃。通过使用熔点为30至120℃的脱模剂,有效地发挥脱模效果,并确保更宽的定影区域。
77.增塑剂
78.优选将结晶性增塑剂用于本发明的调色剂以改善迅速熔融性。增塑剂没有特别限制,可以使用如下所述的适用于调色剂的公知增塑剂。
79.一元醇和脂肪族羧酸的酯,例如山萮酸山萮醇酯、硬脂酸硬脂酯和棕榈酸棕榈醇酯,或一元羧酸和脂肪族醇的酯;二元醇和脂肪族羧酸的酯,例如乙二醇二硬脂酸酯、二山萮醇癸二酸酯和己二醇二山萮酸酯,或二元羧酸和脂肪族醇的酯;三元醇和脂肪族羧酸的酯,例如甘油三山萮酸酯,或三元羧酸和脂肪醇的酯;四元醇和脂肪族羧酸的酯,例如季戊四醇四硬脂酸酯和季戊四醇四棕榈酸酯,或四元羧酸和脂肪族醇的酯;六元醇和脂肪族羧酸的酯,例如二季戊四醇六硬脂酸酯和二季戊四醇六棕榈酸酯,或六元羧酸和脂肪族醇的酯;多元醇与脂肪族羧酸的酯,例如聚甘油山萮酸酯,或多元羧酸和脂肪族醇的酯;以及天然酯蜡,例如巴西棕榈蜡和米蜡。这些可以单独或组合使用。
80.着色剂
81.调色剂颗粒可以包括着色剂。公知的颜料和染料可以用作着色剂。从优异耐候性的观点来看,颜料优选作为着色剂。
82.青色类着色剂的实例包括铜酞菁化合物及其衍生物、蒽醌化合物、碱性染料色淀化合物等。
83.具体实例包括c.i.颜料蓝1、7、15、15:1、15:2、15:3、15:4、60、62和66。
84.品红色着色剂的实例包括缩合偶氮化合物、二酮吡咯并吡咯化合物、蒽醌化合物、
喹吖啶酮化合物、碱性染料色淀化合物、萘酚化合物、苯并咪唑酮化合物、硫靛化合物、苝化合物等。
85.具体实例包括c.i.颜料红2、3、5、6、7、23、48:2、48:3、48:4、57:1、81:1、122、144、146、150、166、169、177、184、185、202、206、220、221和254,以及c.i.颜料紫19。
86.黄色着色剂的实例包括缩合偶氮化合物、异吲哚啉酮化合物、蒽醌化合物、偶氮金属配合物、次甲基化合物和烯丙基酰胺化合物。
87.具体实例包括c.i.颜料黄12、13、14、15、17、62、74、83、93、94、95、97、109、110、111、120、127、128、129、147、151、154、155、168、174、175、176、180、181、185、191和194。
88.黑色着色剂的实例包括使用上述黄色着色剂、品红色着色剂和青色着色剂调色为黑色的那些,以及炭黑。
89.这些着色剂可以单独使用或作为混合物使用,并且它们可以以固溶体的形式使用。
90.优选以相对于粘结剂树脂100.0质量份为1.0至20.0质量份的量使用着色剂。
91.电荷控制剂和电荷控制树脂
92.调色剂颗粒可以包括电荷控制剂或电荷控制树脂。
93.作为电荷控制剂,可以使用公知的那些,特别优选摩擦带电速度快、能够稳定地维持一定的摩擦带电量的电荷控制剂。此外,当通过悬浮聚合法制造调色剂颗粒时,特别优选具有低聚合抑制性并且在水系介质中基本上不提供可溶材料的电荷控制剂。
94.控制调色剂为带负电的电荷控制剂的实例包括单偶氮金属化合物,乙酰丙酮金属化合物,芳族羟基羧酸、芳族二羧酸、羟基羧酸和二羧酸的金属化合物,芳族羟基羧酸、芳族单羧酸和多羧酸及其金属盐、酸酐和酯,苯酚衍生物如双酚,脲衍生物,含金属的水杨酸类化合物,含金属的萘甲酸类化合物,硼化合物,季铵盐,杯芳烃,和电荷控制树脂等。
95.电荷控制树脂的实例包括具有磺酸基、磺酸碱或磺酸酯基的聚合物或共聚物。具有磺酸基、磺酸碱或磺酸酯基的聚合物的特别优选实例包括以2质量%以上、更优选5质量%以上的共聚比例包含含磺酸基的丙烯酰胺类单体或含磺酸基的甲基丙烯酰胺类单体的聚合物。
96.电荷控制树脂的玻璃化转变温度(tg)优选为35至90℃,峰值分子量(mp)为10,000至30,000,重均分子量(mw)为25,000至50,000。当使用这种电荷控制时,可以赋予良好的摩擦带电特性,而不影响调色剂颗粒所需的热特性。此外,在电荷控制树脂包含磺酸基的情况下,例如,电荷控制树脂本身在聚合性单体组合物中的分散性和着色剂等的分散性得到改善,并且着色强度、透明性和摩擦带电特性可以进一步改善。
97.这些电荷控制剂或电荷控制树脂可以单独或以其两种以上的组合使用。
98.电荷控制剂或电荷控制树脂的添加量相对于100.0质量份的粘结剂树脂优选为0.01质量份至20.00质量份,更优选为0.5至10.0质量份。
99.调色剂的制造方法
100.调色剂的制造方法没有特别限制,可以使用如粉碎法、悬浮聚合法、溶解悬浮法、乳化聚集法、和分散聚合法等公知的方法。这里,优选通过下示的方法来制造调色剂。即,调色剂优选通过乳化聚集法来制造。
101.调色剂的制造方法优选包括按下述顺序进行的以下步骤(1)至(3):
102.(1)分散步骤,制备包含粘结剂树脂的粘结剂树脂微粒分散液,
103.(2)聚集步骤,将粘结剂树脂微粒分散液中包含的粘结剂树脂微粒聚集以形成聚集体,和
104.(3)熔合步骤,将聚集体加热并熔合,并且
105.在聚集步骤和熔合步骤的至少一个中添加硼酸源。
106.此外,优选在熔合步骤期间或之后按以下顺序进行以下步骤(4)至(6):
107.(4)球形化步骤,通过进一步升高温度加热聚集体,
108.(5)冷却步骤,以0.1℃/秒以上的冷却速度冷却聚集体,和
109.(6)退火步骤,将聚集体在等于或高于粘结剂树脂的结晶温度或玻璃化转变温度的温度下加热并保持。
110.调色剂优选通过乳化聚集法制造,因为可以控制调色剂的形状并且硼酸很可能均匀分散在调色剂表面附近。现在将描述乳化聚集法的细节。
111.乳化聚集法
112.在乳化聚集法下,预先制备包含调色剂颗粒的构成材料并且与目标粒径相比充分小的微粒的水系分散液,将这些微粒在水系介质中聚集直至达到调色剂颗粒的粒径,并且通过加热等使树脂熔合,来制造调色剂颗粒。
113.即,在乳化聚集法中,通过进行以下步骤来制造调色剂颗粒:分散步骤,制造包含调色剂颗粒的构成材料的微粒分散液;聚集步骤,聚集包含调色剂颗粒的构成材料的微粒以控制粒径直到达到调色剂颗粒的粒径;熔合步骤,使所得到的聚集颗粒中所含的树脂进行熔合;球形化步骤,通过进一步加热等熔融以控制调色剂的表面形状;之后的冷却步骤;金属除去步骤,筛选获得的调色剂并除去过量的多价金属离子;过滤/洗涤步骤,用离子交换水等洗涤;以及从洗涤后的调色剂颗粒中除去水并干燥的步骤。
114.树脂微粒分散液的制备步骤(分散步骤)
115.树脂微粒分散液可以通过公知的方法制备,但不限于这些方法。公知的方法的实例包括例如,乳液聚合法、自乳化法、向通过溶解于有机溶剂中获得的树脂溶液中添加水系介质以使树脂乳化的转相乳化法、或在不使用有机溶剂的水系介质中通过高温处理对树脂强制乳化的强制乳化法等。
116.具体而言,将粘结剂树脂溶解在能够溶解粘结剂树脂的有机溶剂中,添加表面活性剂或碱性化合物。此时,在粘结剂树脂是具有熔点的结晶性树脂的场合下,可以通过加热到熔点或高于熔点来进行熔化。接着,一边用均化器等搅拌一边缓慢添加水系介质,以使树脂微粒析出。然后,通过加热或降低压力除去溶剂,以制备树脂微粒的水系分散液。作为用于溶解树脂的有机溶剂,可以使用能够溶解树脂的任意有机溶剂,但从抑制生成粗颗粒的观点来看,优选使用如甲苯等与水形成均相的有机溶剂。
117.在上述乳化时使用的表面活性剂没有特别限定,其实例包括阴离子表面活性剂,例如硫酸酯和盐、磺酸盐、羧酸盐、磷酸酯和皂等;阳离子表面活性剂,例如胺盐和季铵盐等;和非离子表面活性剂,例如聚乙二醇型、烷基酚环氧乙烷加合物和多元醇型等。表面活性剂可以单独或以其两种以上的组合使用。
118.分散步骤中使用的碱性化合物的实例包括无机碱如氢氧化钠和氢氧化钾等,以及有机碱如氨、三乙胺、三甲胺、二甲氨基乙醇和二乙氨基乙醇等。碱性化合物可以单独或以
其两种以上的组合使用。
119.另外,树脂微粒的水系分散液中的粘结剂树脂微粒的基于体积分布的50%粒径(d50)优选为0.05至1.0μm,更优选为0.05至0.4μm。通过将基于体积分布的50%粒径(d50)调节在上述范围内,变得容易获得调色剂颗粒的合适的体积平均粒径为3至10μm的调色剂颗粒。
120.使用动态光散射型粒度分布计nanotrac upa-ex150(nikkiso co.,ltd.制)测量基于体积分布的50%粒径(d50)。
121.着色剂微粒分散液
122.根据需要,可以使用着色剂微粒分散液。着色剂微粒分散液可以通过下面的公知方法制备,但不限于这些方法。因而,着色剂微粒分散液可以通过用如已知的搅拌机、乳化机、和分散机等混合机混合着色剂、水系介质和分散剂来制备。作为这里使用的分散剂,可以使用如表面活性剂和高分子分散剂等公知的分散剂。
123.无论分散剂是表面活性剂还是高分子分散剂,都可以在后述的洗涤步骤中除去,但从洗涤效率的观点来看,优选表面活性剂。
124.表面活性剂的实例包括阴离子表面活性剂,例如硫酸酯和盐、磺酸盐、磷酸酯和皂等;阳离子表面活性剂,例如胺盐和季铵盐;和非离子表面活性剂,例如聚乙二醇、烷基酚环氧乙烷加合物和多元醇。其中,优选非离子表面活性剂或阴离子表面活性剂。此外,可以组合使用非离子表面活性剂和阴离子表面活性剂。表面活性剂可以单独或以其两种以上的组合使用。水系介质中的表面活性剂的浓度优选为0.5至5质量%。
125.着色剂微粒分散液中的着色剂微粒的含量没有特别限制,但相对于着色剂微粒分散液的总质量,优选为1至30质量%。
126.另外,关于着色剂的水系分散液中的着色剂微粒的分散粒径,从最终得到的调色剂中的着色剂的分散性的观点来看,基于体积分布的50%粒径(d50)优选为0.5μm以下。另外,出于相同的理由,优选的是,基于体积分布的90%粒径(d90)优选为2μm以下。分散在水系介质中的着色剂微粒的分散粒径通过动态光散射型粒度分布计(nanotrack upa-ex150:nikkiso co.,ltd.制)测量。
127.用于在水系介质中分散着色剂的如已知的搅拌机、乳化机、和分散机等混合机的实例包括超声波均质机、喷射磨机、加压均质机、胶体磨、球磨机、砂磨机和油漆搅拌机。这些可以单独或组合使用。
128.脱模剂(脂肪族烃化合物)微粒分散液
129.根据需要,可以使用脱模剂微粒分散液。脱模剂微粒分散液可以通过以下公知的方法来制备,但不限于这些方法。
130.脱模剂微粒分散液可以通过将脱模剂添加到包括表面活性剂的水系介质中,加热至脱模剂的熔点或高于脱模剂的熔点,用具有强剪切能力的均质机(例如,"clearmix w motion",m technique co.,ltd.制)或压力排放式分散机(例如,"gaulin homogenizer",gaulin co.,ltd.制)分散成颗粒状,然后冷却至低于脱模剂熔点的温度来制造。
131.另外,脱模剂的水系分散液中的脱模剂微粒分散液的分散粒径以基于体积分布的50%粒径(d50)计优选为0.03至1.0μm,更优选为0.1至0.5μm。另外,优选不存在1μm以上的粗颗粒。
132.当脱模剂微粒分散液的分散粒径在上述范围内时,脱模剂可以以微细分散状态存在于调色剂中,使在定影时的外迁移(outmigration)效果最大化,并且可以获得良好的脱模性。通过分散在水系介质中获得的脱模剂微粒分散液的分散粒径可以用动态光散射型粒度分布计(nanotrack upa-ex150:nikkiso co.,ltd.制)来测量。
133.混合步骤
134.在混合步骤中,混合树脂微粒分散液以及根据需要的脱模剂微粒分散液和着色剂微粒分散液中的至少一种来制备混合液。这可以使用如均质机和混合器等公知的混合设备来进行。
135.形成聚集体颗粒的步骤(聚集步骤)
136.在聚集步骤中,使混合步骤中制备的混合液中所含的微粒聚集以形成具有目标粒径的聚集体。此时,通过添加和混合絮凝剂并根据需要适当地施加热和/或机械力中的至少一者,使树脂微粒以及根据需要的脱模剂微粒和着色剂微粒中的至少一种聚集形成聚集体。
137.絮凝剂的实例包括有机絮凝剂,例如季盐型阳离子表面活性剂、聚乙烯亚胺等;无机金属盐,例如硫酸钠、硝酸钠、氯化钠、氯化钙、硝酸钙等;无机铵盐,例如硫酸铵、氯化铵、硝酸铵等;以及无机絮凝剂,例如二价以上的金属配合物等。此外,也可以添加酸以降低ph和实现软聚集,例如可以使用硫酸、硝酸等。
138.絮凝剂可以以干粉形式或作为通过溶解在水系介质中获得的水溶液添加,但优选以水溶液的形式添加絮凝剂以实现均匀的聚集。另外,优选在等于或低于混合液中所含的树脂的玻璃化转变温度或熔点的温度下添加并混合絮凝剂。通过在这些温度条件下混合,相对均匀地进行聚集。向混合液中混合絮凝剂可以使用如均质机和混合器等公知的混合设备来进行。聚集步骤是在水系介质中形成调色剂颗粒大小的聚集体的步骤。在聚集步骤中生成的聚集体的体积平均粒径优选为3至10μm。体积平均粒径可以通过利用coulter方法的粒度分布分析仪(coulter multisizer iii:beckman coulter,inc.制)来测量。
139.获得包含调色剂颗粒的分散液的步骤(熔合步骤)
140.在熔合步骤中,首先在与聚集步骤中相同的搅拌下,停止在聚集步骤中获得的含有聚集体的分散液中的聚集。通过添加能够调节ph的如碱或螯合化合物等聚集停止剂、或如氯化钠等无机盐化合物来停止聚集。
141.在分散液中的聚集颗粒的分散状态由于聚集停止剂的作用而稳定后,进行加热到粘结剂树脂的玻璃化转变温度或熔点或者高于粘结剂树脂的玻璃化转变温度或熔点,以使聚集颗粒熔合并调节粒径为所需值。调色剂颗粒的体积基准的50%粒径(d50)优选为3至10μm。
142.获得所需调色剂表面形状的步骤(球形化步骤)
143.优选在熔合步骤期间或之后经历球形化步骤,其中进一步升高并保持温度直到调色剂颗粒具有所需的圆形度或表面形状。特定的球形化步骤的温度例如为90℃以上,优选为92℃以上,更优选为95℃以下。球形化步骤中的加热时间的实例包括3小时以上、5小时以上和8小时以上的加热时间。通过该步骤,来自硼酸的氢键可能在调色剂颗粒中形成。
144.冷却步骤
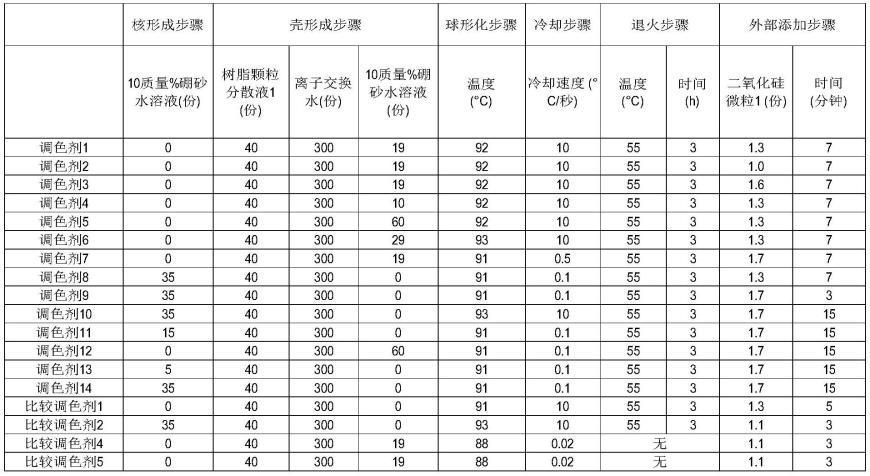
145.在球形化步骤之后,优选经历控制冷却速度以将含有获得的调色剂颗粒的分散液
的温度降低至低于粘结剂树脂的结晶温度和/或玻璃化转变温度的温度的冷却步骤。通过经历冷却步骤,抑制了由于如调色剂颗粒中的材料的膨胀或收缩等体积变化而在调色剂颗粒表面上形成凹凸,从而变得易于将截面的形状因子sf1控制为105至125,或将调色剂的接触面积比率(d/s)控制为14%以下。此外,通过增加冷却速度,可以进一步抑制体积变化,从而可以抑制调色剂颗粒表面的凹痕的生成,可以保持在球形化步骤中获得的所需圆形度或表面形状,调色剂的形状因子sf1和调色剂截面的形状因子sf1可以设为125以下,并且调色剂的接触面积比率(d/s)可以设为14%以下。具体的冷却速度为0.1℃/秒以上,优选为0.5℃/秒以上,更优选为2℃/秒以上,还更优选为4℃/秒以上。
146.退火步骤
147.冷却步骤后,优选在等于或高于粘结剂树脂的结晶温度或玻璃化转变温度的温度下经历加热和保持的退火步骤,并且当含有脱模剂时,温度为等于或低于脱模剂的结晶温度。通过经历退火步骤,可以进一步抑制体积变化,从而能够抑制调色剂颗粒表面上的凹痕的生成。因此,可以保持通过冷却步骤获得的所需圆形度或表面形状,调色剂的形状因子sf1和调色剂截面的形状因子sf1可以设为125以下,并且调色剂的接触面积比率(d/s)可以控制为14%以下。具体的退火温度为45至75℃,优选为50至70℃,更优选为55至65℃。退火步骤中的热处理时间例如为5小时以下,优选为2至3小时。
148.后处理步骤
149.在调色剂的制造方法中,可以进一步进行如洗涤步骤、固液分离步骤、干燥步骤等后处理步骤,通过进行后处理步骤,可以得到在干燥状态下的调色剂颗粒。
150.外部添加步骤
151.获得的调色剂颗粒可以原样地用作调色剂。
152.在外部添加步骤中,根据需要,将无机微粒外部添加到干燥步骤中获得的调色剂颗粒中。具体而言,优选通过在干燥状态下施加剪切力来添加并混合如二氧化硅等无机微粒或如乙烯基树脂、聚酯树脂和有机硅树脂等树脂微粒。二氧化硅微粒优选以相对于100质量份调色剂颗粒为1.2至1.8质量份的量混合2至16分钟的时间,更优选以1.3至1.7质量份的量混合6至10分钟的时间。当以上述范围内的添加量和混合时间添加并混合二氧化硅微粒时,通过x射线光电子能谱仪测量的二氧化硅微粒的调色剂颗粒表面的覆盖率易于控制在50至80面积%的范围内,调色剂表面上的二氧化硅微粒的分散度评价指数易于控制在0.10至2.00的范围内。
153.调色剂的制造方法优选具有壳形成步骤,该壳形成步骤通过聚集步骤形成聚集颗粒(核颗粒),然后进一步添加包含壳用树脂的树脂微粒以引起聚集并形成壳。即,优选调色剂颗粒具有包含粘结剂树脂的核颗粒和在核颗粒表面的壳。作为壳用树脂,可以使用与粘结剂树脂相同的树脂,或者可以使用其它树脂。相对于100质量份的核颗粒中所含的粘结剂树脂,壳的树脂的添加量优选为1至10质量份,更优选为2至7质量份。
154.在该情况下,优选调色剂的制造方法具有以下步骤:
155.(1)分散步骤,制备包含粘结剂树脂的粘结剂树脂微粒分散液,
156.(2-1)使粘结剂树脂微粒分散液中所含的粘结剂树脂微粒聚集以形成聚集体的聚集步骤,
157.(2-2)通过向包含聚集体的粘结剂树脂微粒分散液进一步添加包含壳用树脂的树
脂微粒并且使包含壳用树脂的树脂微粒聚集而形成具有壳的聚集体的壳形成步骤,和
158.(3)熔合步骤,将聚集体加热并熔合。
159.即,上述聚集步骤(2)(使粘结剂树脂微粒分散液中所含的粘合剂树脂微粒聚集以形成聚集体的聚集步骤)优选包括以下步骤(2-1)和(2-2)。
160.(2-1)使粘结剂树脂微粒分散液中所含的粘结剂树脂微粒聚集以形成聚集体的聚集步骤,和
161.(2-2)通过向包含聚集体的粘结剂树脂微粒分散液进一步添加包含壳用树脂的树脂微粒并且使包含壳用树脂的树脂微粒聚集而形成具有壳的聚集体的壳形成步骤。
162.此外,更优选在熔合步骤期间或之后按以下顺序进行以下步骤(4)至(6):
163.(4)球形化步骤,通过进一步升高温度加热聚集体,
164.(5)冷却步骤,以0.1℃/秒以上的冷却速度冷却聚集体,和
165.(6)退火步骤,将聚集体在等于或高于粘结剂树脂的结晶温度或玻璃化转变温度的温度下加热并保持。
166.为了促进在调色剂颗粒表面附近包含硼酸,优选将硼酸源与包含壳用树脂的树脂微粒一起在壳形成步骤中添加到包含聚集体的分散液中。
167.硼酸源可以为硼酸或在调色剂制造期间通过ph控制等可以变成硼酸的化合物。例如,可以提及选自由硼酸、硼砂、有机硼酸、硼酸盐、硼酸酯等组成的组中的至少一种。例如,可以添加硼酸源并且可以进行控制以在聚集体中包含硼酸。优选地,在聚集步骤中将ph控制为酸性条件,然后进行壳形成步骤。
168.硼酸可以以未取代的状态存在于聚集体中。硼酸源优选为选自由硼酸和硼砂组成的组中的至少一种。当在水系介质中制造调色剂时,从反应性和制造稳定性的观点来看,优选添加硼酸盐作为硼酸源。具体而言,硼酸源更优选包括选自由四硼酸钠、硼砂、硼酸铵等组成的组中的至少一种,更优选为硼砂。
169.硼砂由四硼酸钠na2b4o7的十水合物并且在酸性水溶液中变成硼酸。因此,在水系介质的酸性环境中使用时优选硼砂。作为添加方法,可以添加干粉或通过溶解在水系介质中获得的水溶液,但为了诱发均匀聚集,优选以水溶液的形式添加。水溶液的浓度可以根据调色剂中的浓度适当地改变,例如为1至20质量%。为了变成硼酸,优选在添加之前、期间或之后将ph设为酸性条件。例如,可以进行控制为1.5至5.0,优选为2.0至4.0。
170.接下来,将描述根据本公开的用于测量各物性的方法。
171.调色剂或调色剂颗粒的重均粒径(d4)和数均粒径(d1)的测量
172.调色剂或调色剂颗粒的重均粒径(d4)和数均粒径(d1)以下述方式来计算。基于具有100μm口管的孔电阻法的精确粒度分布测量设备(coulter counter multisizer 3(注册商标),beckman coulter,inc.制)以及用于设定测量条件和测量数据的分析的测量设备的专用软件(beckman coulter multisizer 3version 3.51,beckman coulter,inc.制)用于测量中。使用25,000个有效测量通道进行测量,然后分析并计算测量数据。
173.可以使用通过将特级氯化钠以其浓度为约1质量%溶解于离子交换水中而获得的溶液,例如"isoton ii"(beckman coulter制)作为用于测量中的电解质水溶液。
174.在进行测量和分析之前,将专用软件以如下方式设定。在专用软件中的“标准操作方法(som)更改”界面中,将控制模式的总计数设定为50,000个颗粒;将测量的次数设定为1
次;将kd值设定为通过使用“标准颗粒10.0μm”(beckman coulter)而获得的值。通过按下“阈值/噪声水平测量按键”,自动设定阈值和噪声水平。另外,将电流设定为1,600μa;将增益设定为2;将电解质溶液设定为isoton ii;并且勾选“测量后冲洗口管”选项。在专用软件的“脉冲向粒径的转换设定”界面中,将元件间隔(bin interval)设定为对数粒径;将粒径元件(particle diameter bin)设定为256个粒径元件;并且粒径范围设定为2至60μm。
175.具体的测量方法如下。
176.1.将200ml电解质水溶液放入到multisizer 3专用的250ml圆底玻璃烧杯中,并且将该烧杯置于样品台上,并且搅拌棒以24转/秒的速度进行逆时针旋转。通过进行专用软件的“口管冲洗”功能,除去口管中的污染物和气泡。
177.2.将约30ml的电解质水溶液放入到100ml平底玻璃烧杯中。向烧杯中添加约0.3ml通过用离子交换水约三质量倍稀释“contaminon n”(用于清洁精密测量仪器的中性洗涤剂的10质量%水溶液,ph为7并且包括非离子表面活性剂、阴离子表面活性剂和有机助洗剂,购自wako pure chemical industries,ltd.制)而获得的稀释液作为分散剂。
178.3.准备超声波分散机(ultrasonic dispersion system tetora 150,nikkaki bios co.,ltd.制),其电力输出为120w,其中封装有2个振荡频率为50khz的振荡器,使得它们的相位错开180
°
。将规定量的离子交换水放入超声波分散机的水槽中,并且将约2ml的contaminon n添加至该水槽中。
179.4.将上述步骤(2)中提及的烧杯置于超声波分散机上的烧杯固定孔中,并且启动超声波分散机。调整烧杯的高度使得烧杯内的电解质水溶液的液面的共振状态最大化。
180.5.在用超声波照射根据上述部分(4)中提及的烧杯内的电解质水溶液的同时,将约10mg调色剂一次一小点添加到电解质水溶液中并分散于其中。继续超声波分散处理另外60秒。当进行超声波分散时,将水浴的温度适当地调整为10℃至40℃的温度。
181.6.借助于移液管,将分散有调色剂的上述部分(5)中提及的电解质水溶液滴入到置于样品台上的上述部分(1)中提及的圆底烧杯中,并且调节测量浓度至约5%。进行测量直到测量的颗粒数达到50,000个。
182.7.通过使用附带的专用软件分析测量数据,计算重均粒径(d4)和数均粒径(d1)。当专用软件设定为图表/体积%时,在“分析/体积基准统计值(算术平均)”界面上的“平均直径”为重均粒径(d4)。当专用软件设定为图表/个数%时,在“分析/数值基准统计值(算术平均)”界面上的“平均直径”为数均粒径(d1)。
183.调色剂的平均圆形度的测量方法
184.在校准操作的测量和分析条件下,使用“fpia-3000”流动颗粒图像分析仪(sysmex corporation)测量调色剂的平均圆形度。
185.在向20ml离子交换水中添加适量的表面活性剂(烷基苯磺酸盐)作为分散剂后,添加0.02g测量样品,并且通过使用振荡频率为50khz和电输出为150w的台式超声波清洗机分散机(商品名:vs-150,velvo-clear co.,ltd.制)进行分散处理2分钟,以制备测量用分散液。此时,适当进行冷却,从而使分散液的温度变为10至40℃。
186.配备有标准物镜(10x)的流动式颗粒图像分析仪用于测量中,并且使用颗粒鞘“pse-900a”(sysmex corporation制)作为鞘液。将根据上述程序制备的分散液引入流动式颗粒图像分析仪,在hpf测量模式下以总计数模式测量3000个调色剂颗粒,将在颗粒分析时
的二值化阈值设置为85%,分析颗粒的直径限制为1.98至19.92μm的圆当量直径,并且获得调色剂(颗粒)的平均圆形度。
187.在测量中,在测量开始之前,使用标准胶乳颗粒(例如,用离子交换水稀释的5100a(商品名),duke scientific corporation制)进行自动焦点调节。之后,优选从测量开始每2小时进行焦点调节。
188.调色剂的形状因子sf1的测量方法
189.使用fe-sem(商品名:s-4700,hitachi,ltd.制)在设置为2000倍的观察放大率下观察调色剂。
190.使用olympus corporation制的图像分析软件(商品名:analysis pro)分析调色剂图像。获得调色剂的绝对最大长度r、周长l和截面积s。由获得的调色剂的周长,圆当量直径r通过圆当量直径r=l/π获得,该值在使用coulter计数器通过上述方法获得的重均粒径d4的
±
10%的范围内的对象取为对应的颗粒。
191.随机选取对应的颗粒共50个,其截面的绝对最大长度的平均值取为rave,截面积的平均值取为save,调色剂的形状因子sf1(截面)的值由下式获得。
192.形状因子sf1=(rave2×
π)/(save
×
4)
×
100
193.调色剂截面的形状因子sf1的测量方法
194.使用jeol ltd.制的截面抛光机(商品名:sm-09010)通过cp截面法测量调色剂的形状因子。然后,如下所述制备调色剂的截面。
195.将碳双面压敏粘合片部分粘贴在硅晶片片上,固定mo网(直径:3mm/厚度:30μm),大约一层调色剂(大约一个调色剂颗粒的厚度)附着在其上。在其上气相沉积铂之后,使用截面抛光机在4kv的加速电压和3小时的处理时间的条件下形成调色剂的截面。
196.使用fe-sem(商品名:s-4700;hitachi,ltd.制)在设置为2000倍的观察放大率下观察由此形成的调色剂截面。
197.使用olympus corporation制的图像分析软件(商品名:analysis pro)分析调色剂截面的图像。获得调色剂截面的绝对最大长度r、周长l和截面积s。由获得的调色剂的周长,圆当量直径r通过圆当量直径r=l/π获得,该值在使用coulter计数器通过上述方法获得的重均粒径d4的
±
10%的范围内的对象取为对应的颗粒。
198.随机选取对应的颗粒共50个,其截面的绝对最大长度的平均值取为rave,截面积的平均值取为save,调色剂截面的形状因子sf1的值由下式获得。
199.截面的形状因子sf1=(rave2×
π)/(save
×
4)
×
100
200.调色剂的接触面积比率(d/s)的测量方法
201.准备无色透明的载玻片(厚度约2mm),并在其上准备22μm的开口。将调色剂铺在筛网上,通过施加振动10秒从10cm的高度筛分,将少量调色剂均匀地铺在载玻片上。随后,在激光显微镜(kh-3000,hirox co.,ltd.制)中将放大倍率增加到100倍,从载玻片侧拍摄调色剂图像,并将调色剂图像取入图像分析装置。通过在分析软件(image-pro plus 4.5,media cybernetics,inc.制)中拍摄的图像上随机采样50个颗粒来进行图像分析。在图像分析中,假设调色剂和玻璃表面接触的区域的面积为d,并且整个调色剂的投影面积为s,则d/s是调色剂的接触面积比率的值。
202.通过使用锗(ge)作为atr晶体(atr crystal)的atr方法对调色剂(颗粒)的ir分析
203.使用调色剂的atr-ir分析通过以下方法进行。通过下文描述的方法从调色剂中除去外部添加剂而获得的调色剂颗粒也可以用作样品。
204.ir分析通过使用配备有通用atr测量附件(universal atr sampling accessory)的傅里叶变换红外光谱分析仪(spectrum one:perkinelmer co.制)的atr法进行。具体的测量程序如下。
205.红外光的入射角(λ=5μm)设置为45
°
。作为atr晶体,使用ge atr晶体(折射率=4.0)。其它条件如下。
206.范围
207.起点:4000cm-1
208.终点:650cm-1
(ge atr晶体)
209.持续时间
210.扫描数:16
211.分辨率:4.00cm-1
212.高等(advanced):带co2/h2o校正
213.(1)ge atr晶体(折射率=4.0)附接到设备上。
214.(2)扫描类型设置为背景,单位设置为egy,测量背景。
215.(3)扫描类型设置为样品,单位设置为a。
216.(4)在atr晶体上称量0.01g调色剂或调色剂颗粒的总和。
217.(5)用压力臂对样品加压(测力计为90)。
218.(6)测量样品。
219.检测1380cm-1
的吸收光谱。当在1380cm-1
附近检测到吸收峰时,确定检测到对应于硼酸的峰。
220.调色剂颗粒的荧光x射线测量中的硼强度的测量
221.通过校准曲线法测量调色剂颗粒的荧光x射线测量中的硼强度。具体而言,将铝环(内径40mm、外径43mm、高度5mm)设置在半自动minipress机(specac limited制)的样品成型模具上。将约3g的调色剂颗粒放入其中并在15t的压制压力下压制成型1分钟以制备测量用丸粒。使用成型为约3mm厚度和约40mm直径的丸粒。
222.通过使用波长色散型荧光x射线分析仪“axios”(panalytical co.制)以及用于设定测量条件和分析测量数据而设置的专用软件“superq ver.4.0f”(panalytical co.制)在以下条件下进行测量。rh用作x射线管的阳极,测量气氛为真空,测量直径(准直器掩模直径)为27mm,测量时间为10秒。在硼的情况下,使用比例计数器(pc)进行检测。
223.在上述条件下进行测量,基于获得的x射线的峰值位置识别硼,并且测量作为每单位时间的x射线光子数的计数率(单位:cps)。
224.通过从调色剂中除去外部添加剂获得调色剂颗粒的方法
225.将总共160g蔗糖(kishida chemical co.,ltd.制)添加到100ml的离子交换水中并溶解在水浴中以制备蔗糖浓缩液。总共31g蔗糖浓缩液和6ml的contaminone n(用于清洗精密测量仪器的中性洗涤剂的10质量%水溶液,其包含非离子表面活性剂、阴离子表面活性剂和有机助洗剂,ph为7,wako pure chemical industries,ltd.制)置于离心管(50ml容量)中以制备分散液。向该分散液中添加1.0g调色剂,用抹刀等将调色剂块弄松散。用振荡
器(as-1n,由as one corporation出售)以300spm(每分钟冲程)将离心管振荡20分钟。振荡后,将溶液转移至摆动转子用玻璃管(50ml),通过离心机(h-9r,kokusan co.,ltd.制)以3500rpm进行分离30分钟。
226.通过该操作,调色剂颗粒和外部添加剂分离。目视确认调色剂颗粒和水溶液的充分分离,并用抹刀等收集在最上层分离的调色剂颗粒。用真空过滤器过滤收集的调色剂颗粒,然后用干燥器干燥1小时以上以获得测量用样品。此操作多次进行以确保所需的量。
227.调色剂颗粒表面用二氧化硅微粒覆盖的覆盖率的测量方法
228.调色剂颗粒表面用二氧化硅微粒覆盖的覆盖率通过以下方法确定。首先,通过esca(x射线光电子能谱法)测量来自调色剂颗粒表面上存在的二氧化硅微粒的硅(以下简称为si)的原子量。
229.esca仪器和测量条件如下。
230.使用的设备:quantum 2000,ulvac-phi,inc.制
231.分析方法:窄分析(narrow analysis)
232.测量条件:
233.x射线源:al-kα
234.x射线条件:光束直径100μm、25w、15kv
235.光电子吸收角:45
°
236.通过能量:58.70ev
237.测量范围:
238.在分析方法中,首先,将源自于碳1s轨道的c-c键的峰校正为285ev。然后,通过使用由ulvac-phi,inc.提供的相对灵敏度因子,由源自于硅2p轨道的峰面积(其中在100至105ev检测到峰顶)计算来自二氧化硅的si在调色剂中的构成元素的总量中的比率“a(原子%)”。
239.接下来,通过与上述相同的方法测量相对于调色剂中使用的二氧化硅单体(simple substance)的si量,并且获得来自二氧化硅的si在二氧化硅单体中的构成元素的总量中的比率“b(原子%)”。该比率b(原子%)认为是100%覆盖率的值。
240.此时,二氧化硅覆盖率由下式计算:
241.二氧化硅覆盖率(%)=比率a/比率b
×
100
242.当使用第一外部添加剂和第二外部添加剂这两种外部添加剂时,其中第一外部添加剂和第二外部添加剂都是二氧化硅,因为在外部添加剂单体中的si量与第一和第二外部添加剂二者中的是相同的,因此通过上述方法进行测量。
243.同时,当使用除二氧化硅以外的物质作为第一外部添加剂时,由于外部添加剂单体(simple substance)中的si量在第一和第二外部添加剂之间不同,因此通过以下方法进行测量。
244.测量仅外部添加了第一外部添加剂的调色剂中的si的比率a1,并且类似地,测量仅外部添加了第二外部添加剂的调色剂中的si的比率a2。该情况下的二氧化硅覆盖率使用上述si的比率“b(原子%)”,通过下式计算出。
245.二氧化硅覆盖率(%)=(比率a1/比率b+比率a2/比率b)
×
100
246.调色剂表面上的二氧化硅微粒的分散度评价指数
247.使用扫描电子显微镜“s-4800”计算调色剂表面上的二氧化硅微粒的分散度评价指数。在放大10,000倍的视野中,在1.0kv的加速电压下在同一视野中观察外部添加有二氧化硅微粒的调色剂。使用图像处理软件“imagej”(可获得自https://imagej.nih.gov/ij/)从观察到的图像以下列方式进行计算。
248.进行二值化,从而仅提取二氧化硅微粒。具体而言,对在同一视野内的调色剂表面进行能量色散型x射线光谱(edx)分析以确定颗粒是否为二氧化硅微粒。计算二氧化硅微粒的数量n和所有二氧化硅微粒的重心坐标,并计算距每个二氧化硅微粒最近的二氧化硅微粒的距离dn min。
249.假设图像中的二氧化硅微粒之间的最近距离的平均值为d ave,分散度由下式表示:
[0250][0251]
随机观察的50个调色剂颗粒的分散度通过上述程序来获得,并且其平均值用作分散度评价指数。分散度评价指数越小,分散性越好。
[0252]
实施例
[0253]
下文中,通过实施例更详细地描述本发明。本发明不限于以下实施例。文本中的配方中的“份”是基于质量的,除非另有明确说明。
[0254]
调色剂1的制造例
[0255]
二氧化硅微粒1的制备
[0256]
将总共10.0份聚二甲基硅氧烷(粘度=100mm2/s)喷雾在100份气相二氧化硅(商品名;aerosil380s,bet法比表面积380m2/g,一次颗粒的数均粒径7nm,nippon aerosil co.,ltd.制)上并继续搅拌30分钟。然后,边搅拌边升温至300℃,进行另外搅拌2小时,以制备二氧化硅微粒1。
[0257]
聚酯树脂1的合成
[0258]
·
双酚a环氧乙烷2mol加合物:9mol份
[0259]
·
双酚a环氧丙烷2mol加合物:95mol份
[0260]
·
对苯二甲酸:50mol份
[0261]
·
富马酸:30mol份
[0262]
·
十二碳烯基琥珀酸:25mol份
[0263]
将上述单体放入装备有搅拌器、氮气导入管、温度传感器、精馏柱的烧瓶中,在1小时内升温至195℃,确认反应体系内部均匀搅拌。将总共1.0份二硬脂酸锡添加到100份这些单体中。此外,一边蒸馏出生成的水,一边在5小时内从195℃升温至250℃,在250℃下进行另外2小时的脱水缩合反应。
[0264]
结果,获得玻璃化转变温度为60.2℃、酸值为16.8mg koh/g、羟值为28.2mg koh/g、重均分子量为11,200和数均分子量为4100的聚酯树脂1。
[0265]
聚酯树脂2的合成
[0266]
·
双酚a环氧乙烷2mol加合物:48mol份
[0267]
·
双酚a环氧丙烷2mol加合物:48mol份
[0268]
·
对苯二甲酸:65mol份
[0269]
·
十二碳烯基琥珀酸:30mol份
[0270]
将上述单体放入装备有搅拌器、氮气导入管、温度传感器、精馏柱的烧瓶中,在1小时内升温至195℃,确认反应体系内部均匀搅拌。将总共0.7份二硬脂酸锡添加到100份这些单体中。此外,一边蒸馏出生成的水,一边在5小时内从195℃升温至240℃,在240℃下进行另外2小时的脱水缩合反应。然后,将温度降低至190℃,逐渐添加5mol份偏苯三酸酐,并且在190℃下继续反应1小时。
[0271]
结果,获得玻璃化转变温度为55.2℃、酸值为14.3mg koh/g、羟值为24.1mg koh/g、重均分子量为43,600和数均分子量为6200的聚酯树脂2。
[0272]
树脂颗粒分散液1的制备
[0273]
·
聚酯树脂1:100份
[0274]
·
甲乙酮:50份
[0275]
·
异丙醇:20份
[0276]
将上述甲乙酮和异丙醇放入容器中。然后,逐渐添加聚酯树脂1,搅拌,完全溶解,以获得聚酯树脂1溶液。将包含聚酯树脂1溶液的容器设定为65℃,边搅拌边逐渐滴加10%氨水溶液至总共5份,并以10ml/分钟的速率逐渐滴加230份离子交换水,以诱导转相乳化。此外,用蒸发器通过减压除去溶剂,从而获得聚酯树脂1的树脂颗粒分散液1。树脂颗粒的体积平均粒径为135nm。用离子交换水将树脂颗粒固体成分量调整为20%。
[0277]
树脂颗粒分散液2的制备
[0278]
·
聚酯树脂2:100份
[0279]
·
甲乙酮:50份
[0280]
·
异丙醇:20份
[0281]
将上述甲乙酮和异丙醇放入容器中。然后,逐渐添加聚酯树脂2,搅拌,完全溶解,以获得聚酯树脂2溶液。将包含聚酯树脂2溶液的容器设定为40℃,边搅拌边逐渐滴加10%氨水溶液至总共3.5份,并以10ml/分钟的速率逐渐滴加230份离子交换水,以诱导转相乳化。此外,通过减压除去溶剂,从而获得聚酯树脂2的树脂颗粒分散液2。树脂颗粒的体积平均粒径为155nm。用离子交换水将树脂颗粒固体成分量调整为20%。
[0282]
着色剂颗粒分散液的制备
[0283]
·
铜酞菁(颜料蓝15:3):45份
[0284]
·
离子表面活性剂neogen rk(dks co.,ltd.制):5份
[0285]
·
离子交换水:190份
[0286]
将上述组分用均质机(ultra-turrax,ika制)混合并分散10分钟,然后使用ultimizer(反碰撞湿式粉碎机:sugino machine limited制)在250mpa的压力下分散20分钟,从而获得体积平均粒径为120nm、固体成分量为20%的着色剂颗粒分散液。
[0287]
脱模剂颗粒分散液的制备
[0288]
·
脱模剂(烃蜡,熔点:79℃):15份
[0289]
·
离子表面活性剂neogen rk(dks co.,ltd.制):2份
[0290]
·
离子交换水:240份
[0291]
将上述组分加热至100℃,用ika制的ultra-turrax t50充分分散,然后用压力排
出型gaulin均质机加热至115℃,并且进行分散处理1小时,从而获得体积平均粒径为160nm、固体成分含量为20%的脱模剂颗粒分散液。
[0292]
调色剂颗粒1的制造
[0293]
·
树脂颗粒分散液1:500份
[0294]
·
树脂颗粒分散液2:400份
[0295]
·
着色剂颗粒分散液:50份
[0296]
·
脱模剂颗粒分散液:80份
[0297]
首先,作为核形成步骤,将上述材料放入圆形不锈钢烧瓶中并混合。随后,使用均质机ultra-turrax t50(ika制)以5000r/分钟分散10分钟。添加1.0%硝酸水溶液并将ph调整为3.0后,一边在水浴中使用搅拌叶片进行加热并适当地调整转数,一边进行加热至58℃。
[0298]
适当地使用coulter multisizer iii确认所形成的聚集颗粒的体积平均粒径,当形成尺寸为5.0μm的聚集颗粒(核)时,通过添加以下材料进行壳形成步骤,进一步搅拌1小时,以形成壳。
[0299]
·
树脂颗粒分散液1:40份
[0300]
·
离子交换水:300份
[0301]
·
10.0质量%硼砂水溶液:19份
[0302]
(硼砂:四硼酸钠十水合物,wako pure chemical industries,ltd.制)
[0303]
之后,作为球形化步骤,通过使用5%氢氧化钠水溶液将ph调节至9.0,一边继续搅拌一边进行加热至92℃。
[0304]
当获得所需的表面形状时,停止加热,作为冷却步骤,通过快速加冰以使冷却速度为10℃/秒以上,进行冷却至40℃,另外,作为退火步骤,在55℃下进行3小时退火。
[0305]
之后,进行冷却至25℃,过滤和固液分离,接着用离子交换水洗涤。洗涤结束后,通过使用真空干燥机干燥,获得重均粒径(d4)为6.8μm的调色剂颗粒1。
[0306]
对调色剂颗粒1进行外部添加。使用亨舍尔混合器(mitsui mining co.,ltd.制),将总共100.0份调色剂颗粒1和1.3份二氧化硅微粒1干混7分钟,从而获得调色剂1。表2显示了获得的调色剂1的物理特性。
[0307]
调色剂2至14的制造例
[0308]
除了改变表1中所示的配方和条件以外,以与调色剂1相同的方式获得调色剂2至14。表2显示了调色剂2至14的物理特性。
[0309]
调色剂15的制造例
[0310]
·
聚酯树脂1:60.0份
[0311]
·
聚酯树脂2:40.0份
[0312]
·
铜酞菁颜料(颜料蓝15:3):6.5份
[0313]
·
脱模剂(烃蜡,熔点79℃):5.0份
[0314]
·
增塑剂(乙二醇二硬脂酸酯):15.0份
[0315]
·
硼酸粉末(wako pure chemical industries,ltd.制):1.5份
[0316]
将上述材料用fm混合器(nippon coke industries co.,ltd.制)预混合,然后用双螺杆混炼挤出机(pcm-30型,ikegai iron works co.,ltd.制)熔融混炼。
[0317]
将获得的混炼物冷却,用锤磨机粗粉碎,之后添加130份乙酸乙酯,进行加热至80℃,用t.k.homomixer(special machinery chemical industry co.,ltd.)以5000rpm的旋转速度进行搅拌1小时,接着冷却至30℃以获得溶液。
[0318]
将总共400份水和5份eleminol mon-7(sanyo chemical industries,ltd.制)放入单独的容器中并设置为30℃。然后,一边用t.k.homomixer(special machinery chemical industry co.,ltd.)以13,000rpm的转速搅拌一边添加100份上述溶液,进行进一步搅拌20分钟以获得浆料。一边轻轻搅拌一边将获得的浆料在30℃、减压下脱溶剂8小时,然后在45℃下熟化4小时,然后通过洗涤、过滤和干燥步骤获得调色剂颗粒15。将调色剂颗粒15以与调色剂1的制造相同的方式进行外部添加以获得调色剂15。表2显示了调色剂15的物理特性。
[0319]
比较调色剂1、2、4和5的制造例
[0320]
除了改变表1中所示的配方和条件以外,以与调色剂1相同的方式获得比较调色剂1、2、4和5。表2显示了比较调色剂1、2、4和5的物理特性。
[0321]
比较调色剂3的制造例
[0322]
·
聚酯树脂1:50份
[0323]
·
聚酯树脂2:40份
[0324]
·
铜酞菁(颜料蓝15:3):8份
[0325]
·
脱模剂(烃蜡,熔点79℃):4份
[0326]
·
硼酸粉末(wako pure chemical industries,ltd.制):1.5份
[0327]
将上述材料用fm混合器(nippon coke industries co.,ltd.制)预混合,然后用双螺杆混炼挤出机(pcm-30型,ikegai iron works co.,ltd.制)熔融混炼。
[0328]
将获得的混炼物冷却,用锤磨机粗粉碎,然后用机械粉碎机(t-250,turbo industries co.,ltd.制)粉碎,将获得的细粉碎粉末用使用coanda效应的多级分级机分级,获得重均粒径(d4)为7.0μm的比较调色剂颗粒3。
[0329]
比较调色剂颗粒3以与调色剂1的制造中的相同的方式进行外部添加,获得比较调色剂3。表2显示了比较调色剂3的物理特性。
[0330]
[表1]
[0331][0332]
[表2]
[0333][0334]
表2中,“c.e.”表示“比较例”,并且“c.t.”表示“比较调色剂”。
[0335]
实施例1
[0336]
对调色剂1进行以下评价。
[0337]
调色剂评价
[0338]
使用改造的市售佳能激光束打印机“lbp7600c”。改造涉及通过改变评价机器主体的齿轮和软件将显影辊的旋转速度设为鼓的圆周速度的1.5倍。
[0339]
在低温低湿环境(15℃/10%rh)下,以下列方式在letter尺寸business4200纸(xerox制,75g/m2)上输出图像。
[0340]
(1)输出三幅实心图像。
[0341]
(2)输出三幅厚度为100μm(静电潜像中的厚度)的线条的间隔1cm的网格图案。
[0342]
(3)输出一幅实心图像,在将实心图像转印后的感光构件上的转印残留调色剂用mylar胶带贴上并剥离。
[0343]
(4)输出4000幅打印百分比为1%的图像。之后,再次输出三幅网格图案。
[0344]
(5)输出三幅实心图像。
[0345]
(6)输出三幅厚度为100μm(静电潜像中的厚度)的线条的间隔1cm的网格图案。
[0346]
(7)输出一幅实心图像,在将实心图像转印后的感光构件上的转印残留调色剂用mylar胶带贴上并剥离。
[0347]
初始转印性
[0348]
将(3)中得到的胶带和未进行粘胶的胶带贴附至letter尺寸business4200纸(75g/m2,xerox corp.制)。基于以下基准评价在各个带表面上的反射率差。c以上的评价对应于可接受的水平。
[0349]
a:小于0.6%。
[0350]
b:0.6%以上且小于1.2%。
[0351]
c:1.2%以上且小于2.0%。
[0352]
d:2.0%以上且小于3.0%。
[0353]
e:3.0%以上。
[0354]
初始转印粉尘
[0355]
使用放大倍率为25倍的放大镜观察(2)中输出的第三幅网格图案图像,并基于以下基准评价转印粉尘(调色剂散落在字符和线条图像周围的现象)。c以上的评价对应于可接受的水平。
[0356]
a:线条非常清晰,几乎没有转移灰尘。
[0357]
b:线条清晰,仅有飞溅的少量调色剂。
[0358]
c:调色剂有些飞溅,但线条相对清晰。
[0359]
d:有许多调色剂飞溅,线条形状模糊。
[0360]
耐久性试验中的转印性
[0361]
将(7)中得到的胶带和未进行粘胶的胶带贴附至letter尺寸business4200纸(75g/m2,xerox corp.制)。基于以下基准评价在各个带表面上的反射率差。c以上的评价对应于可接受的水平。
[0362]
a:小于0.6%。
[0363]
b:0.6%以上且小于1.2%。
[0364]
c:1.2%以上且小于2.0%。
[0365]
d:2.0%以上且小于3.0%。
[0366]
e:3.0%以上。
[0367]
耐久性试验中的转印粉尘
[0368]
使用放大倍率为25倍的放大镜观察(6)中输出的第三幅网格图案图像,并基于以下基准评价转印粉尘(调色剂散落在字符和线条图像周围的现象)。c以上的评价对应于可接受的水平。
[0369]
a:线条非常清晰,几乎没有转移灰尘。
[0370]
b:线条清晰,仅有飞溅的少量调色剂。
[0371]
c:调色剂有些飞溅,但线条相对清晰。
[0372]
d:有许多调色剂飞溅,线条形状模糊。
[0373]
耐久性试验中的显影性能
[0374]
图像浓度通过使用"macbeth densitometer rd918"(macbeth co.,ltd.制)根据装置所提供的使用说明书测量相对于具有图像浓度为0.00的白色背景的图像的相对浓度来测量,并将获得的相对浓度用作图像浓度的值。耐久性试验中的显影性能由浓度降低程度确定。根据以下关于初始浓度与耐久试验后浓度之差的值的基准,评价浓度降低程度,其中将(1)中输出的三幅图像中的每一个的中心浓度的平均值取为初始浓度,将(5)中输出的三幅图像中的每一个的中心浓度的平均值取为耐久性试验后浓度。c以上的评价对应于可接受的水平。
[0375]
a:小于0.05。
[0376]
b:0.05以上且小于0.1。
[0377]
c:0.1以上且小于0.15。
[0378]
d:0.15以上且小于0.20。
[0379]
e:0.20以上。
[0380]
实施例2至15
[0381]
使用调色剂1至15分别进行与实施例1中相同的评价。评价结果示于表3中。
[0382]
比较例1至5
[0383]
使用比较调色剂1至5分别进行与实施例1中相同的评价。评价结果示于表3中。
[0384]
[表3]
[0385][0386]
表3中,“c.e.”表示“比较例”,并且“c.t.”表示“比较调色剂”。
[0387]
虽然已参考示例性的实施方案描述了本发明,但将理解本发明不局限于所公开的示例性的实施方案。所附权利要求的范围应符合最宽泛的解释,以涵盖所有此类变型以及等同的结构和功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1