一种水相中金纳米簇的富集方法与流程
[0001]
本发明涉及一种水相中金纳米簇的富集方法,属于纳米材料技术领域。
背景技术:
[0002]
金纳米簇是由数个到几十个金原子组成的核以及包裹于其外部的一价金-配体形成的络合物(au(i)-ligand)组成的壳堆积而成的纳米结构。通常金纳米簇核心尺寸小于2nm,由于金纳米簇的尺寸与于费米波长相当,因而表现出离散的分子状电子结构和独特反应性质,由于这些特殊的性质,金纳米簇在光学治疗以及催化等领域得到了广泛的应用。
[0003]
但是,光学治疗以及催化等领域通常需要高浓度的金纳米簇,而化学合成法获得金纳米簇的浓度通常达不到要求,因而,需要对合成的金纳米簇进行富集。已经报道的富集方法有超速离心法、超滤法、免疫捕获法、色谱法以及溶剂沉淀法等。然而,目前的这些方法存在一些缺点,比如超速离心法富集不完全,而超滤法、免疫捕获法、色谱法和溶剂沉淀法有产率低、耗时、费用高等缺点;另外,色谱法和溶剂沉淀法有可能使金纳米簇变性。因此,发展一种高效、方便的金纳米簇富集技术是一个亟待解决的技术问题。
技术实现要素:
[0004]
本发明解决的技术问题在于提供一种水相中金纳米簇的高效、方便的富集方法。发明人实验发现,由氧化型谷胱甘肽和氯金酸反应生成的金纳米簇溶液,与富集溶剂(乙腈与改性剂的混合物)以一定的比例混合后产生油状液滴,该液滴与富集溶剂不相混溶,而且密度比富集溶剂大,更重要的是,原来水相溶液中的金纳米簇几乎全部富集于油状液滴。有鉴于此,本专利提供了一种水相中金纳米簇的富集方法,其特征在于,包括以下步骤:
[0005]
(1)调节待富集金纳米簇溶液的ph值为2~8;
[0006]
所述待富集金纳米簇溶液由氧化型谷胱甘肽和氯金酸反应制得,反应物中氯金酸的起始浓度为1~20mm,氧化型谷胱甘肽与氯金酸的起始摩尔浓度之比为2:1~5:1;
[0007]
(2)将待富集金纳米簇溶液加入富集溶液中,摇匀;所述富集溶液由乙腈和改性剂构成;所述改性剂为氯仿、四氯化碳、四氢呋喃、乙酸乙酯、环己烷和正己烷中一种或多种;
[0008]
(3)相分离;
[0009]
(4)收集金纳米簇富集相。
[0010]
优选的,所述步骤(1)中用200mm naoh或200mm hcl调节待富集金纳米簇溶液的ph值。
[0011]
优选的,所述步骤(2)中待富集金纳米簇溶液与乙腈溶液的体积比为1:(4~7),所述改性剂与乙腈的体积比为(0~1):4。
[0012]
所述步骤(3)中的相分离通过离心或静置的方法实现。
[0013]
通过离心方法实现相分离时,优选的离心速度为500~2000转/分钟,优选的离心时间为5~20分钟。
[0014]
通过静置的方法实现相分离时,优选的静置时间为2~4小时。
[0015]
所述步骤(4)中金纳米簇富集相为液态。
[0016]
本申请提供的金纳米簇富集方法保留了原纳米簇内核组成、形貌、聚集方式和光学特性,富集过程的回收率大于99%,通过离心的方式可在20分钟之内完成分离;通过静置的方式可以实现大规模的纳米簇富集操作。相比于超速离心法、超滤法、免疫捕获法、色谱法等富集方法,本方法操作简便、快速,可以大规模地富集;相比于溶剂沉淀法,本方法所富集的纳米簇可以直接溶解于水,可以满足光治疗以及均相催化的需求。
附图说明
[0017]
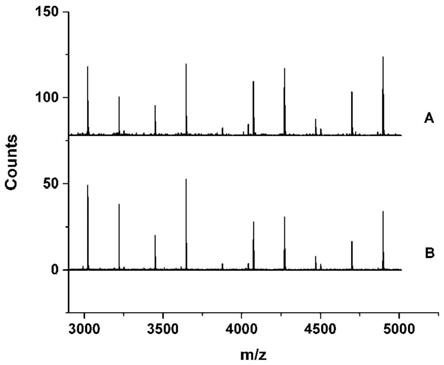
图1为实施例1中水相待富集(a)与富集之后(b)金纳米簇的maldi-tof-ms分析。
[0018]
图2为实施例1中水相金纳米簇的tem图。
[0019]
图3为实施例1中用乙腈浓缩后的金纳米簇的tem图。
[0020]
图4为实施例1中待富集纳米簇的荧光发射光谱图(激发波长430nm)。
[0021]
图5为实施例1中富集之后的油相中金纳米簇的荧光发射光谱图(激发波长430nm)。
[0022]
图6为实施例2中气相色谱
–
热导检测法分析油状液滴中的易挥发成分。图中1~3对应的峰分别为水,乙腈,正丁醇。
[0023]
图7为实施例2中氧化型谷胱甘肽(a)、油状液滴(b)以及上层溶液(c)的毛细管电泳分析谱图。图中峰1为氧化型谷胱甘肽,标准溶液中氧化型谷胱甘肽的浓度为0.05mm。
[0024]
图8为实施例6中本发明的方法(1)和乙腈沉淀法(2)对纳米簇的富集效果图。
[0025]
图9为实施例6中本发明的方法(1)和乙腈沉淀法(2)富集的纳米簇的重新溶解。
具体实施方式
[0026]
本专利提供了一种水相中金纳米簇的富集方法,包括以下步骤:
[0027]
(1)将氧化型谷胱甘肽和氯金酸混合,氯金酸的起始浓度为1~20mm,氧化型谷胱甘肽与氯金酸的起始摩尔浓度之比为2:1~5:1;反应后得金纳米簇溶液,用200mm naoh或200mm hcl调节待富集金纳米簇溶液的ph至2~8;
[0028]
(2)将待富集金纳米簇溶液与富集溶液以1:(4~7)的体积比混合后摇匀;富集溶液由乙腈与改性剂组成;改性剂为氯仿、四氯化碳、四氢呋喃、乙酸乙酯、环己烷和正己烷中一种或多种;改性剂与乙腈的体积比为(0~1):4;
[0029]
(3)通过离心或静置的方式进行相分离;通过离心方法实现相分离时,离心速度为500~2000转/分钟,离心时间为5~20分钟;通过静置的方法实现相分离时,优选的静置时间为2~4小时;
[0030]
(4)收集金纳米簇富集相;金纳米簇富集相为液态,与富集溶液不互溶。
[0031]
为进一步理解本发明,下面结合实施例对本发明提供的金纳米簇的富集方法进行详细说明,本发明的保护范围不受以下实施例的限制。
[0032]
下属实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径购得。
[0033]
按照本发明,氧化型谷胱甘肽和氯金酸为原料合成的待富集金纳米簇溶液可以按照现有技术中公开的方法制备,在本说明书中称为金纳米簇溶液。
[0034]
实施例1
[0035]
本实施例对富集前后金纳米簇的组成、形貌和荧光性质进行比较研究。
[0036]
1.1金纳米簇的制备
[0037]
常温剧烈搅拌下,将新鲜配制的5ml 20mm氯金酸(haucl
4
)和5ml 100mm氧化型谷胱甘肽(gssg)的水溶液与90ml三次蒸馏水在圆底烧瓶中混合(此条件下haucl
4
和gssg的起始浓度分别为1mm和5mm),静置两分钟后,将混合物在温和搅拌下(500转/分钟)置于80℃的水浴中恒温加热24小时。待溶液自然冷却至室温后,以2000转/分的转速离心15分钟除去沉淀物,将上清液储存于4℃冰箱中备用。
[0038]
1.2水相中金纳米簇的富集
[0039]
将1.1中制备的待富集金纳米簇溶液用200mm hcl调至ph=2,取1ml金纳米簇溶液,将其加入到盛有4ml乙腈的离心管中,摇匀,静置5分钟后,2000转/分离心5分钟,即可在离心管底部发现淡黄色的油状液滴,体积约25μl。
[0040]
1.3富集前后金纳米簇的质谱表征
[0041]
金纳米簇是一种具有类似分子的、以金原子为主体的几何结构,其结构中的金原子数影响金纳米簇的光学性质和催化性能。而质谱是解析金纳米簇中原子个数信息的重要工具。基质辅助激光解析电离飞行时间质谱(maldi-tof-ms)谱图(图1)可以看出,富集前后金纳米簇质谱主要离子峰的m/z相同,而且峰的强度相近。表1的质谱分析结果显示,gssg与haucl
4
反应产生的是金纳米簇的混合物,这一点与还原型谷胱甘肽(gsh)与haucl
4
反应合成进纳米簇的情况类似(chemelectrochem,2020,7,1092-1096)。而且,本专利中有些金纳米簇与gsh合成产生的产物相同,比如,[au
15
s
5
+4cl]-,[au
17
s
6
+4cl]-和[au
22
s
7
+4cl]-等(the journal of physical chemistry letters,2010,1,2903
–
2910)。以上结果说明,富集前后金纳米簇结构中原子的成分没有变化。
[0042]
表1质谱分析荷质比与对应的金纳米簇
[0043][0044]
1.4富集前后金纳米簇的透射电子显微镜表征
[0045]
金纳米簇的结构和固态聚集状态影响其性能,包括催化性能和光学性能等。将水相金纳米簇和富集后处于油状液滴中的金纳米簇分别做透射电子显微镜(tem)实验,结果
显示,经过富集之后的金纳米簇无论尺寸和形貌与水相金纳米簇没有明显差别(图2、图3)。说明富集过程没有引起金纳米簇结构的变化。
[0046]
1.5富集前后金纳米簇的荧光强度对比
[0047]
金纳米簇的荧光性质反应了纳米簇的内核金原子晶体结构、外层au(i)-gssg络合物的分布状态(包括au(i)与au(0)的相对含量以及外层au(i)-gssg络合物包裹的刚度等)的信息,同时也间接反映金纳米簇作为一个整体在溶液中的聚集特性。
[0048]
由于富集后的纳米簇油状液滴中含有乙腈,而乙腈的存在会影响金纳米簇的荧光性质。有鉴于此,实验中采用离心超滤的方法去除液体中的小分子。具体操作如下:
[0049]
取1ml步骤1.1中获得的金纳米簇水溶液,放入超滤离心管(截止分子量3kda,millipore)中,500转/分钟离心15分钟后,再向超滤管中加1ml三次蒸馏水,摇匀,再次以500转/分钟离心15分钟,小心将截留的纳米簇溶解于2ml三次蒸馏水中待测。
[0050]
对于步骤1.2得到的富集后的油状液滴,将其溶解于1ml三次蒸馏水,放入超滤离心管(截止分子量3kda,millipore)中,500转/分钟离心15分钟后,再向超滤管中加1ml三次蒸馏水,摇匀,再次以500转/分钟离心15分钟,小心将截留的纳米簇溶解于2ml三次蒸馏水中待测。
[0051]
荧光测量的激发波长为424nm,在发射波长为606nm处测量荧光发射强度。测得水相纳米簇的发射强度为4.7656
×
10
5
(图4),富集后的油状液滴经过去除乙腈后分散于相同体积的三次蒸馏水中的荧光发射强度为4.8574
×
10
5
(图5)。富集前后的金纳米簇的荧光特性没有明显的改变,说明两种金纳米簇在水中的聚集行为相似。
[0052]
以上的实验结果说明,将水相金纳米簇加入乙腈中,形成了新的一相,该相沉于底部,与上层的乙腈/水混合相不相混溶,可以实现水相中金纳米簇的富集,富集过程没有改变金纳米簇内核的金原子排布以及其外层包裹的au(i)-gssg性质,没有改变固相纳米簇的聚集行为特性;而且,富集后的纳米簇可以直接溶解于三次蒸馏水,溶解于水之后的金纳米簇聚集特性与待富集纳米簇溶液相近。
[0053]
实施例2
[0054]
2.1金纳米簇的制备
[0055]
常温剧烈搅拌下,将新鲜配制的40ml 50mm haucl
4
和40ml 100mm gssg水溶液与20ml三次蒸馏水在圆底烧瓶中混合(此条件下haucl
4
和gssg的起始浓度分别为20mm和40mm),静置两分钟后,将混合物在温和搅拌下(500转/分钟)置于80℃的水浴中恒温加热24小时。待溶液自然冷却至室温后,以2000转/分的转速离心15分钟除去沉淀物,将上清液储存于4℃冰箱中备用。
[0056]
2.2水相中金纳米簇的富集
[0057]
将2.1中制备的待富集金纳米簇溶液用200mm naoh调至ph=8,取2ml金纳米簇溶液,将其加入到盛有14ml乙腈的离心管中,摇匀,静置。静置过程中发现上层乙腈/水相中有淡黄色的乳状物,随着时间的推移渐渐下移,4小时后上层的乙腈/水相变澄清,在离心管底部发现淡黄色的油状液滴,体积为110μl。
[0058]
2.3油状液滴中金纳米簇的含量及富集回收率的测定
[0059]
样品处理:
[0060]
①
待富集金纳米簇溶液的处理方法:取2ml步骤2.1中获得的金纳米簇溶液,加入
haucl
4
和40ml 100mm gssg的水溶液与20ml三次蒸馏水在圆底烧瓶中混合(此条件下haucl
4
和gssg的起始浓度分别为20mm和40mm),将混合物在温和搅拌下(500转/分钟)反应2小时。用200mm naoh将溶液调至ph=8,取2ml溶液,将其加入到盛有14ml乙腈的离心管中,摇匀,静置。静置过程中可以发现上层乙腈/水相中有白色乳状物,但经过2小时后,在离心管底部形成沉淀。由于常温下,haucl
4
与过量的gssg反应生成的是gsosg,上述实验说明,gsosg不是形成油状液滴的重要成分。
[0074]
结合以上实验可以推断,反应中生成的gso
3
h是形成油状液滴的主要动力,推测其与乙腈之间能形成弱强度的氢键,而金单质是一种缺电子的元素,能与水分子之间发生弱相互作用,上述物质混合后形成了与乙腈/水相不相混溶、流动性好、稳定性高的新相。
[0075]
实施例3
[0076]
本实施例探究待富集纳米簇水相溶液ph值对富集效果的影响。
[0077]
金纳米簇的合成同2.1,富集之前用200mm hcl和200mm naoh调节纳米簇溶液ph值。取2ml金纳米簇溶液,将其加入到盛有14ml乙腈的离心管中,摇匀,静置4小时后测量离心管底部淡黄色的油状液滴的体积,见表2。结果显示,在水相ph2~8范围内,油状液滴的体积为97~115μl,说明在较宽的水相溶液ph值范围内都能实现有效富集。
[0078]
需要特别指出的是,由于gssg分子结构上既有羧基也有氨基,因此本身是两性化合物。由于制备金纳米簇的反应中gssg过量,因此金纳米簇水溶液含有gssg,溶液本身就有一定的缓冲能力,其溶液的ph值为2.2。实验发现,将没有调节ph值的金纳米簇溶液直接加入乙腈中,也能形成油状液滴。
[0079]
表2待富集纳米簇水相溶液ph值对富集效果的影响
[0080][0081]
实施例4
[0082]
本实施例探究金纳米簇水溶液与乙腈体积比对形成油状液滴能力的影响。
[0083]
金纳米簇的合成参见2.1,用200mm naoh将溶液调至ph=5,参照表3的条件将v1体积的金纳米簇水溶液加入到v2体积的乙腈中,摇匀后静置4小时,观察离心管底部是(以√表示)否(以
×
表示)形成油状液滴。
[0084]
表3金纳米簇水溶液体积(v1)与乙腈体积(v2)对形成油状液滴的影响
[0085][0086]
由该结果可以看出,水相:乙腈相的体积比例在1:4至1:8之间可以形成油状液滴。
[0087]
实施例5
[0088]
本实施例考察在乙腈中加入改性剂对富集的影响。
[0089]
所述改性剂为三氯甲烷、四氯化碳、四氢呋喃、乙酸乙酯、环己烷和正己烷,分别向乙腈中加入不同体积的改性剂,使乙腈与改性剂的体积之比在1:1~20:1之间。由于改性剂的化学结构不同,极性差异较大,因而与乙腈混溶能力不一样。实验发现,对于环己烷和正己烷,当乙腈与改性剂的体积之比在1:1~5:1之间时,不能混溶,用“n.a.”表示(表4)。而对于三氯甲烷、四氯化碳、四氢呋喃、乙酸乙酯,乙腈与改性剂的体积之比在1:1~20:1之间都可以混溶。
[0090]
金纳米簇的合成参见2.1,用200mm naoh将溶液调至ph=5,取2ml金纳米簇溶液,将其加入到盛有10ml乙腈的离心管中,摇匀后静置4小时。
[0091]
表4改性剂对纳米簇富集能力的影响
[0092]
[0093][0094]
上述改性剂加入乙腈后,除四氢呋喃没有引起水相金纳米簇溶液富集行为改变外,其它改性剂加入乙腈之后都引起了富集行为的变化。在体积比为20:1的乙腈/三氯甲烷的混合液(富集溶剂)中,当水相金纳米簇溶液与富集溶剂的体积比为1:10时也能形成油状液滴,拓宽了待富集相的体积范围;而对于体积比为8:1的乙腈/乙酸乙酯混合液,水相金纳米簇溶液与富集溶剂的体积比为1:(3~10)的范围内都能形成油状液滴;用乙腈/环己烷和乙腈/正己烷为富集溶剂时(乙腈/环己烷和乙腈/正己烷的体积比都为8:1),形成油状液滴的时间缩短到1.5小时。
[0095]
实施例6
[0096]
本实施例研究金纳米簇的浓缩和再溶解,和文献报道的方法相对照,并探究了用于富集的乙腈有机相的再生。
[0097]
6.1金纳米簇的浓缩
[0098]
金纳米簇的合成同1.1。
[0099]
6.1.1本发明的方法
[0100]
将50ml金纳米簇溶液加入到盛有300ml乙腈/乙酸乙酯(乙腈与乙酸乙酯的体积比为10:1)的圆底烧瓶中,摇匀,静置2小时后发现瓶底有黄色液体,将其吸出,测量其体积为985μl。该油状液体有很好的流动性(见图8-1)。
[0101]
6.1.2对比例的方法
[0102]
采用文献(journal of the american chemical society,2016,138,390
–
401)报道的溶剂沉淀法对纳米簇进行富集。取15ml金纳米簇溶液,加入5ml乙腈,混匀5分钟后以5000转/分钟的速度离心后得深黄色沉淀(见图8-2)。
[0103]
6.2浓缩纳米簇再溶解
[0104]
6.2.1本发明富集的纳米簇再溶解
[0105]
将油状液体取出,加入2ml三次蒸馏水中,轻轻摇动,油状液体溶解,形成淡黄色透明液体(图9-1)。
[0106]
6.2.2对比例富集的纳米簇再溶解
[0107]
将图8-2中的上清液倒掉,留下离心管底深黄色沉淀,向离心管中加入2ml三次蒸馏水,摇动,沉淀不溶解,形成黄色悬浊液(图9-2)。
[0108]
以上实验结果说明,本发明方法富集的纳米簇可以直接溶解在水中,适合于后期应用于作为光治疗载体、均相催化等应用场景;而现有的用有机溶剂方法富集的纳米簇形成的沉淀无法直接溶解在水中,说明此时金纳米簇已经变性,限制了其应用。
[0109]
6.3富集溶剂的再生
[0110]
6.1.1中将油状溶液与上层液相分离后,将上层液相在60℃下旋蒸回收,得液体287ml,气相色谱分析表明其中乙腈、乙酸乙酯和水的体积比为1:0.092:0.0079。将此液体应用于富集水相中金纳米簇的实验,重复6.1.1的步骤,得到的油状液体的体积为1007μl,与之前的985μl非常接近。
[0111]
本说明书通过以上实施例说明本申请提供的金纳米簇富集方法得到的产品保留了原纳米簇内核组成、形貌、在水溶液中聚集方式和光学特性,富集过程的回收率大于99%,通过离心的方式可在20分钟之内完成分离,而通过静置的方式可以实现大规模的纳米簇富集操作。相比于超速离心法、超滤法、免疫捕获法、色谱法等富集方法,本方法操作简便、快速;相比于溶剂沉淀法,本方法所富集的纳米簇可以溶解于水,可以满足直接应用于光学治疗以及均相催化的需求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1