耐蚀镁合金及其制备方法
1.本发明涉及镁合金技术领域,特别涉及一种耐蚀镁合金及其制备方法。
背景技术:
2.镁合金作为最轻的结构材料,具有极高的比强度和比刚度,在工业生产的轻量化中有着重要应用,例如,在3c产品、汽车以及船舶制造业,使用镁合金制造的3c产品、汽车或船舶的壳体不仅能提高产品服役寿命,还能实现轻量化的目标。传统的镁合金耐腐蚀性能较差,在正常使用时通常外表面会涂覆耐蚀涂层,例如油漆等。但是无论是3c产品、汽车还是船舶,在服役的过程中,其表面的耐蚀涂层不可避免地会受到损伤,一旦镁合金直接与电解质溶液接触,会在合金表面生成一层氧化膜,而镁的氧化物比较疏松,腐蚀介质会通过氧化镁之间的缝隙对镁合金进行持续的腐蚀,导致镁合金的推广应用受到巨大的阻碍。
3.现有技术中有通过微合金化来提升镁合金耐蚀性的方案。例如cn114540683a公开了一种微合金化的耐腐蚀低成本镁合金及其制备方法,通过在镁合金中添加al、mn、zn以及re等元素配合严格控制的高速挤压变形工艺,来控制镁合金中第二相的种类、尺寸、数量和分布,以提升镁合金的耐蚀性。这种通过微合金化调控微观结构来提升耐蚀性的方法对制备工艺要求较高,很难在生产过程中实现精确控制。
技术实现要素:
4.鉴于现有技术中存在的上述技术问题,本发明的第一方面,提供了一种镁合金,该镁合金包含2.0-6.0wt%的稀土元素,0.1-1.0wt%的成核元素,可选的1.0-2.0wt%的用于调控铸造性能的调控元素,以及可选的0.1-0.5wt%的用于去除镁合金中杂质的净化元素,余量为mg及不可避免的杂质;其中,成核元素的氢氧化物在常温下的溶度积常数ksp≤1
×
10-32
,并且小于稀土元素的氢氧化物的溶度积常数;以及稀土元素的氧化物的pbr值为1.0-2.0。
5.镁合金在接触到电解质溶液后,作为合金组分的镁、稀土元素以及成核元素都会受到电解质溶液不同程度的腐蚀。由于其中成核元素的氢氧化物在常温下的溶度积很小,因此能迅速地在镁合金与电解质溶液接触的表面生成成核元素的氢氧化物,该成核元素的氢氧化物能够作为晶核,使致密的稀土元素氧化物在其表面包覆,以对腐蚀产生的氧化镁之间的缝隙进行有效填补,使镁合金表面形成致密的氧化膜,阻挡电解质溶液对镁合金的进一步腐蚀,以提升镁合金的耐腐蚀性。此外,稀土元素的氧化物的pbr值为1.0-2.0,可使腐蚀后镁合金表面形成致密的氧化膜。
6.优选地,上述镁合金中,稀土元素的含量为3.0-5.0wt%。
7.优选地,上述镁合金中,成核元素的含量为0.3-0.8wt%。
8.优选地,上述镁合金还含有1.0-2.0wt%的调控元素。
9.优选地,上述镁合金还含有0.1-0.5wt%的净化元素。
10.优选地,上述镁合金的化学组成满足下述条件中的一个以上:
11.稀土元素为选自la、nd、gd、y中的一种以上,优选为la和/或nd;
12.成核元素为选自al、ce、zr、sc中的一种以上,优选为al和/或ce;
13.调控元素为zn;
14.净化元素为mn。
15.优选地,本发明的镁合金中杂质元素的含量为:fe≤0.05wt%,cu≤0.03wt%,ni≤0.01wt%。
16.fe、ni、cu等杂质元素容易在晶界偏聚,形成分布于基体中的、具有活跃阴极特性的杂质元素相,例如al3fe相,这些杂质元素相会促进镁合金表面微电池的形成,引起电偶腐蚀。因此,本发明通过控制fe、ni、cu等杂质元素在镁合金中的含量在尽量低的范围,能够提高镁合金的耐蚀性。
17.优选地,本发明的镁合金的微观组织包括镁合金基体和分散在所述镁合金基体内的第二相,更优选地,镁合金中第二相的体积分数为5-15%,和/或,第二相的尺寸为10-30微米。
18.镁合金中的第二相通过稀土元素、成核元素和可选的调控元素形成,其可进一步改善镁合金的力学性能和耐蚀性能。第二相的体积分数和尺寸可通过调节镁合金中各组成元素的含量来控制。在本发明的镁合金中,第二相的体积分数为5-15%,优选为5-10%;第二相的尺寸为10-30微米,优选为10-20微米,更优选为15-20微米。
19.本发明人发现,通过将上述体积分数的细小的第二相弥散分布在镁合金基体中,可以使合金的腐蚀失效模式由点蚀转变为均匀腐蚀,从而有助于提升镁合金的力学性能和耐蚀性。相反,粗大的第二相则容易引起电偶腐蚀。
20.优选地,本发明镁合金的屈服强度在90mpa以上,优选为100mpa以上,抗拉强度在150mpa以上,优选为160mpa以上,延伸率为3-10%。本发明的耐蚀镁合金能够满足作为3c产品、汽车和船舶等的外壳使用的标准。
21.优选地,将本发明的镁合金置于质量分数为3.5%的nacl溶液中24hr.后,耐蚀镁合金表面生成氧化膜,该氧化膜的平均厚度在15微米以下,优选为5-15微米,更优选为10-15微米;和/或,本发明的镁合金根据astm-g31-72测得的腐蚀速度在0.5mm/year以下,优选为0.3mm/year以下。
22.本发明的镁合金即使暴露在电解质溶液中更长时间,其表面生成的氧化膜的厚度也基本不随时间变化,能持续保持在15微米以下。氧化膜太薄无法提供足够的保护作用,太厚则容易应力开裂。本发明的镁合金表面生成的氧化膜厚度的最大值和最小值的差在10微米以内,优选为在5微米以内,显示出良好的均匀性。
23.本发明的镁合金优选可以满足根据astm-g31-72测得的腐蚀速度在0.5mm/year以下,表现出优异的耐蚀性。
24.本发明的第二方面,提供了一种制备上述镁合金的方法,该方法包括依次进行的熔炼和浇铸步骤。
25.具体地,熔炼步骤依次按如下进行:
26.第一阶段,将纯镁熔化后在690-700℃加入成核元素,熔化后搅拌均匀,将熔液静置10-20分钟;
27.第二阶段:将第一阶段中获得的熔液升温至720-730℃后,加入稀土元素,熔化后
2010《金属材料拉伸实验第1部分:室温试验方法》,采用lfv-100hh万能材料试验机进行测试。
51.本发明中模拟体液降解速率依据astm-g31-72《金属的实验室浸渍腐蚀试验》,将待测金属试样置于质量分数为3.5%的nacl溶液中采用析氢法进行测试。
52.本领域公知,稀土元素包括:镧(la)、铈(ce)、镨(pr)、钕(nd)、钷(pm)、钐(sm)、铕(eu)、钆(gd)、铽(tb)、镝(dy)、钬(ho)、铒(er)、铥(tm)、镱(yb)、镥(lu)、钇(y)和钪(sc)。
53.在本发明的镁合金中,各化学元素的设计原理具体如下所述:
54.稀土元素:本发明的稀土元素的腐蚀产物的pbr值均为1.0以上,即稀土元素的氧化物的pbr值为1.0以上,以使得膜层更加致密。pbr,即pilling-bedworth ratio,是金属与氧结合在金属表面生成的氧化物膜中的每个金属离子体积与金属中的每个金属原子体积之比,表示氧化膜的致密程度。优选地,本发明的稀土元素的氧化物的pbr值为1.0-2.0。若pbr值大于2.0,则其氧化物膜在生长过程中容易出现应力破损,从而使氧化物膜丧失保护性。本发明中的稀土元素优选为la和/或nd,其中,la的氧化物的pbr值为1.8,nd的氧化物的pbr值为1.3,均具有较好的保护性。
55.此外,稀土元素加入到镁合金中可以提高合金的力学性能,在本发明中主要用于在镁合金表面形成比氧化镁更致密的保护膜,提高镁合金的耐腐蚀性。镁合金中稀土元素添加量大于6.0wt%会导致强烈的微电偶腐蚀效应;添加量小于2.0wt%,其形成的氧化物过少不能起到填补氧化镁缝隙的作用,获得的镁合金也无法达到本发明要求的力学性能。优选地,本发明中稀土元素的添加量为3.0-5.0wt%,可以获得更优的力学性能和耐腐蚀性能的平衡。
56.成核元素:是指在溶液中形成的氢氧化物沉淀的溶度积常数ksp较小的元素。在本发明中,添加的成核元素的氢氧化物的ksp应当小于稀土元素的氢氧化物的ksp,这样成核元素的氢氧化物能够更快沉积在镁合金表面,为稀土元素氧化物提供附着位点,其添加不宜过多,但添加量不足又不能为稀土元素氧化物提供充足的生成位点,综合考虑,在本发明中,将成核元素的添加量控制在0.1-1.0wt%,优选0.3-0.8wt%。
57.在本发明中,成核元素优选为al或ce中的至少一种。常温下,氢氧化铝的溶度积常数为1
×
10-33
,氢氧化铈的溶度积常数为2
×
10-48
,均在1
×
10-32
以下。因此,含al和/或ce的耐蚀镁合金,能在其与电解质溶液接触的表面迅速生成al和/或ce的氢氧化物作为晶核,帮助致密的稀土元素氧化物在镁合金表面生成。
58.用于调控铸造性能的调控元素:调控元素可以增加铸造过程中熔体的流动性。将调控元素的添加量控制在1.0-2.0wt%。在本发明中,调控元素优选为zn,zn的添加量过少起不到增加熔体流动性的作用,添加量过多会影响合金的耐蚀性。
59.净化元素:净化元素是指在熔炼过程中与原材料中的杂质fe,ni,cu等反应沉淀至坩埚底从而降低镁合金铸锭中的杂质含量的元素,由于该类元素一般都具有强烈的钝化倾向,综合考虑,在本发明中,将净化元素的添加量控制在0.1-0.5wt%。
60.在本发明中,净化元素为mn。在熔炼过程中,mn会与合金中fe,ni,cu等杂质元素结合生成沉淀,净化熔体,有助于提高镁合金的品质和耐腐蚀能力。
61.本发明的耐蚀镁合金及其制备方法具有如下的优点以及有益效果:
62.本发明的耐蚀镁合金通过对镁合金中的元素组成进行优化设计,使得微合金化后
的合金相间电位差较小,从而有效减小电偶腐蚀;同时在服役环境下容易在合金表面沉淀生成具有保护性的腐蚀产物膜层,从而提高了镁合金的耐腐蚀性能。
63.下面结合实施例对本发明的技术方案作进一步详细的说明。应明确,以下实施例仅用于对本发明的具体实施方式的描述,并不用于对本发明的保护范围构成任何限制。
64.实施例
65.实施例1-6的镁合金通过以下步骤制得:
66.1)熔炼:根据表1所示的配方,对镁合金进行熔炼。
67.具体地,
68.实施例1-4的镁合金的熔炼过程按如下依次进行:
69.①
第一阶段:在保护气氛中,将纯镁熔化后在690-700℃加入成核元素,熔化后搅拌均匀,静置10-20分钟。
70.②
第二阶段:在保护气氛中,将第一阶段获得的熔液升温至720-730℃后加入稀土元素以及净化元素,熔化后搅拌均匀,静置10-30分钟。净化元素与稀土元素的添加顺序并无限制,可以是同时添加,也可以是先后添加,只要最终所加入的元素在熔体中均匀分布即可。
71.③
第三阶段:在保护气氛中,将上述第二阶段获得的熔液升温至750-760℃,加入调控元素,熔化后搅拌均匀,静置15-30分钟。随后降温至720-730℃,再在保护气氛中加入精炼剂进行精炼搅拌,静置30分钟以去除熔液中的杂质。
72.稀土元素、成核元素、净化元素以及调控元素的加入形式分别为相应的mg中间合金。例如,当稀土元素为la时,其添加形式为mg-la中间合金。
73.实施例5-6的镁合金的熔炼过程按如下依次进行:
74.①
第一阶段:在保护气氛中,将纯镁熔化后在690-700℃加入成核元素,熔化后搅拌均匀,静置10-20分钟。
75.②
第二阶段:在保护气氛中,将第一阶段获得的熔液升温至720-730℃后加入稀土元素,熔化后搅拌均匀,静置10-30分钟。
76.③
在保护气氛中加入精炼剂进行精炼搅拌,静置30分钟以去除熔液中的杂质。
77.上述熔炼过程中采用的精炼剂是jdmj系列精炼剂,例如是jdmj-1精炼剂。
78.2)浇铸:在保护气氛中,将熔炼后的熔液导入预先准备的模具中进行砂型铸造,模具温度在190-200℃之间,冷却速度为10-30℃/s,获得耐蚀镁合金。
79.对比例1-4按照与上述实施例1-4相同的工艺制备得到,不同之处在于,对比例1-4的镁合金中成核元素或稀土元素的含量不在本发明限定的范围内。
80.对比例5不含成核元素,因此采用了不同的熔炼工艺,具体地:在保护气氛中,直接将纯镁熔液升温至720-730℃加入稀土元素,熔化后搅拌均匀,静置10-30分钟,再在保护气氛中加入精炼剂进行精炼搅拌,静置30分钟以去除熔液中的杂质。浇铸工艺与实施例采用的工艺条件相同。
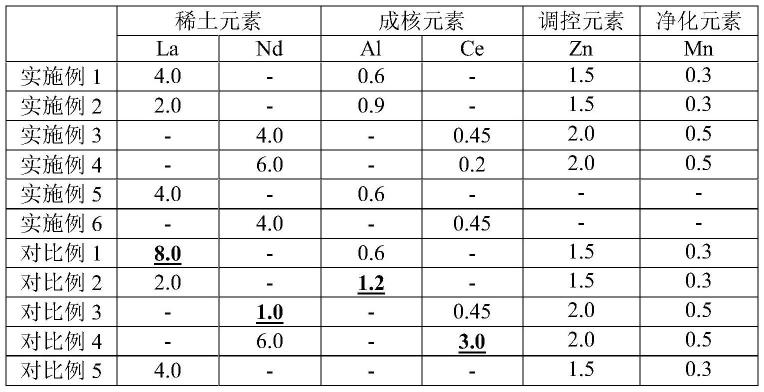
81.表1示出实施例1-6和对比例1-5中镁合金的化学组分,单位为wt%,余量为mg及不可避免的杂质。
82.表1
[0083][0084]
实施例1-6以及对比例1-5的镁合金中fe≤0.05wt.%,cu≤0.03wt.%,ni≤0.01wt.%。
[0085]
表2示出制备实施例1-6和对比例1-5中的镁合金采用的具体工艺参数。
[0086]
表2
[0087][0088][0089]
图1和图3分别为实施例1和对比例1的镁合金一截面的电镜照片。由图1可知,实施例1的镁合金的微观组织包括作为连续相的镁合金基体,以及弥散分布在镁合金基体内的第二相,该第二相的尺寸在10-30微米左右;而与图1相比,图3中镁合金中第二相含量很大,导致微电偶腐蚀加剧,影响耐蚀性。
[0090]
图2是实施例5的镁合金一截面的电镜照片。与实施例1类似,实施例5的镁合金中第二相弥散分布于基体中,且第二相的尺寸也在10-30微米左右。
[0091]
图4和图5分别为实施例1和对比例1的镁合金在电解质溶液中浸泡后一截面的电镜照片。具体地,浸泡条件为:常温下镁合金样品在质量分数为3.5%的nacl溶液中浸泡24小时。
[0092]
图4中镁合金的界面腐蚀产物(即氧化膜)的厚度明显要大于对比例1,且该氧化膜实现了对镁合金表面的全面包覆,说明实施例1的镁合金在试验条件下能在镁合金表面形成完整的氧化膜,以实现对镁合金芯部的保护。且图4中氧化膜的厚度为5-15微米,形貌显
示该膜层极为致密,基本不存在缺陷。在镁合金和电解质溶液界面生成的氧化膜的状态与界面处镁合金中成膜元素和成核元素的分布,以及电解质溶液的溶质分布有关,因此在不同位点生成的氧化膜厚度略有差异。
[0093]
图5中镁合金表面的氧化膜不连续,主要是因为合金中la含量过高,其和mg的电位差相对较大,使得在腐蚀初期有大量氢气泡在反应界面产生,阻碍了有效的氧化膜在镁合金表面生成。
[0094]
图6和图7分别为实施例1和对比例1的镁合金在电解质溶液中浸泡后表面的电镜照片。具体地,浸泡条件为:常温下在质量分数为3.5%的nacl溶液中浸泡24小时。可以看出,实施例1的镁合金表面腐蚀痕迹远远少于对比例1的镁合金,这也是因为对比例1中镁合金的la元素含量太高,腐蚀初期在反应界面有大量氢气泡产生,影响了随后的膜层沉积。
[0095]
将最终制得的实施例1-6和对比例1-5的镁合金分别取样,并对每个样品的力学性能进行测试,测试结果列于表3。
[0096]
测试方法如下:
[0097]
实施例1-6的镁合金和对比例1-5的镁合金的屈服强度和抗拉强度按照gb/t 228.1-2010《金属材料拉伸实验第1部分:室温试验方法》进行测试。
[0098]
耐蚀性测试:实施例1-6的镁合金和对比例1-5的镁合金的模拟体液降解速率依据astm-g31-72《金属的实验室浸渍腐蚀试验》进行测试(即下文所称的析氢失重测试)。
[0099]
在进行析氢失重测试之前,通过线切割工艺将样品切割成10mm
×
10mm
×
5mm的方块,随后用320#、1200#、3000#粒度砂纸依次对样品的六个表面进行打磨,用酒精清洗并吹干后,可用于析氢失重测试。在测试之前,用游标卡尺多次(例如是3次)测量样品的三维尺寸并记录,采用多次测量结果的平均值计算样品的表面积a。
[0100]
本发明中所用的析氢失重测试装置图8所示。将打磨好的被测试样3通过鱼线系牢,在被测试样3上倒扣一个漏斗4,将被测试样3固定于倒扣的漏斗4内部一起置于1000ml的烧杯中。烧杯中提前加入配好的约700ml的电解质溶液2,电解质溶液2是质量分数为3.5%的nacl溶液。随后将装有溶液的酸式滴定管1与漏斗4连接,烧杯内部溶液液面应没过酸式滴定管1与漏斗4的连接处以形成液封,酸式滴定管1的活塞处涂抹凡士林进行密封处理。记录酸式滴定管1内的初始液面高度处对应的体积刻度值,随后每隔一段时间记录一次数据,滴定管内溶液减少的体积,即为相应时间段内析出的氢气体积。
[0101][0102]vh
为浸泡实验中被测样品的析出氢气的速度,单位可以为ml/cm2/day,通过上式计算,其中,δv为某一时间段t内析出的氢气体积,单位可以是ml,时间t可以以天计,a是被测样品的表面积,单位可以是cm2。相同时间析氢的量越少,说明相应镁合金的耐腐蚀性能越好。腐蚀速度vr与析氢速度vh存在以下关系:
[0103]vr
=2vh[0104]
其中,vr的单位为mm/year。
[0105]
表3
[0106][0107][0108]
由表3可知,本发明的实施例1-6的镁合金表现出良好的力学性能,屈服强度可达90-120mpa,抗拉强度可达150-180mpa,延伸率为3-10%,可以满足常规3c产品的使用需求。特别是实施例1-6的镁合金由于合理的成分控制,腐蚀速度在0.3mm/year以下,在耐腐蚀性能方面明显优于对比例1-5。
[0109]
虽然通过参照本发明的某些优选实施方式,已经对本发明进行了描述,但本领域的普通技术人员应该明白,以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。本领域技术人员可以在形式上和细节上对其作各种改变,包括做出若干简单推演或替换,而不偏离本发明的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1