一种用于通过超临界溶剂热合成制造表面改性的金属氧化物纳米颗粒的连续流动方法与流程
本发明涉及一种用于通过超临界溶剂热合成制造表面改性的金属氧化物纳米颗粒的连续流动方法,以及用于进行这种方法的装置。本发明的方法可用于制造对于制备纳米复合材料容易可用的复杂纳米颗粒如杂化有机-无机纳米颗粒,这些纳米复合材料可以进而用于各种领域中,如光学、陶瓷、催化、微电子学、燃料电池技术、制药学或化妆品中。
背景技术:
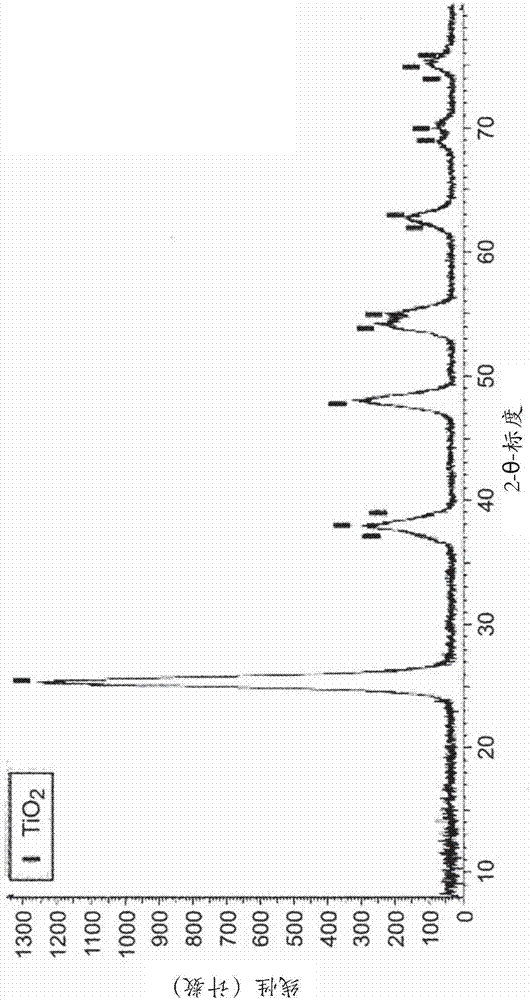
:具有窄粒度分布的精细纳米颗粒可以用各种方法生产,如固态反应、共沉淀、溶胶-凝胶法、水热合成和溶剂热合成、等离子体化学气相沉积或这些方法的组合。在纳米技术中,水热加工具有超过其他常规方法的优势,因为水热加工是简单、有成本效益、节能、无污染的(由于反应在封闭系统中进行),允许成核的较好控制、较高的分散、较高的反应速度和较好的形状控制。溶剂热合成非常类似于水热合成,唯一的区别是用于促进合成期间的前体的相互作用的溶剂不是水性的。超临界水热法是水热技术的延伸。在常规水热与超临界水热技术之间的区别是在温和条件下进行水热技术,而超临界水热技术处理在仅仅接近或高于临界温度的温度下的反应。在超临界条件下,促进了水热反应中无机化合物的成核和晶体生长。其结果是,可以实现无机纳米颗粒如金属氧化物纳米颗粒的快速合成。当与常规水热条件下获得的金属氧化物相比,在超临界水热条件下,可以合成纳米尺寸的金属氧化物颗粒并且纳米颗粒的结晶度高得多,其中形成了块状单晶。纳米复合材料可以通过以下方式形成:将矿物纳米颗粒与聚合物熔体直接混合、接着是挤出(熔融配混),或者将矿物纳米颗粒与聚合物溶液直接混合、接着是溶剂蒸发(膜流延)、或者将矿物纳米颗粒与单体溶液直接混合、接着是聚合(原位聚合)。然而,矿物纳米颗粒具有自发聚集的趋势,是因为它们的大表面积对比体积比和高的表面能。因此,如果在纳米颗粒与单体/聚合物基质之间存在弱的界面相互作用,则可能困难的是获得均匀的分散。为了克服这个问题,可以通过吸附表面活性剂或通过在颗粒表面上接枝足够的官能团以获得稳定的分散来改性纳米颗粒的表面。金属氧化物纳米颗粒的表面改性可以通过在间歇反应器中使用超临界水热合成来进行。例如,mousavand等人(2007)报道了超临界水中的表面改性的tio2纳米颗粒的一锅合成。超临界水热合成在表面改性剂(己醛)存在下进行,将该表面改性剂与金属盐一起加入到间歇反应器中。超临界水热合成导致将己醛化学结合到纳米颗粒的表面上。这种原位表面改性意味着比当己醛与tio2胶体溶液反应(后表面改性)时更有效的己醛在tio2纳米颗粒上的固定。然而,在间歇反应器中在水热合成期间的原位表面改性具有数个缺点。首先,它不允许控制纳米颗粒的尺寸分布。此外,由于反应动力学和位阻,难以控制接枝到纳米颗粒的表面上的表面改性剂的密度。当将两种或更多种表面改性剂加入到间歇反应器中时,这更是如此。在那种情况下,接枝到纳米颗粒的表面上的表面改性剂的相对量还是难以控制的。此外,间歇反应器在体积上是受限制的,并且因此在间歇反应器中产生的纳米颗粒的体积是受限制的。现有技术披露了在处于连续模式的超临界水热合成期间的原位表面改性,其中将表面改性剂和金属前体在相同的注入点处引入到连续室内。然而,这种方法不允许控制纳米颗粒的尺寸分布或控制纳米颗粒官能化的方式。因此,存在开发一种用于制备表面改性的金属氧化物纳米颗粒的方法的需要,尤其当将一种或数种表面改性剂接枝到纳米颗粒的表面上时,该方法可以允许控制纳米颗粒的尺寸分布以及控制纳米颗粒官能化的方式。技术实现要素:本发明涉及一种用于制备表面改性的金属氧化物纳米颗粒如杂化有机-无机纳米颗粒的方法。本发明的方法通过使用超临界溶剂热合成和原位表面改性在一个步骤中进行。本发明的方法是在多次注入连续流动加热室中进行的连续流动方法。根据本发明,通过在该连续流动室内流动的反应介质中的超临界溶剂热合成形成表面改性的金属氧化物纳米颗粒。将起始材料(即金属氧化物前体和表面改性剂)优选地作为在溶剂中的溶液中的加压的流引入到该连续流动加热室内。该连续流动室优选地是管式反应器。本发明的方法在高于室温的温度下并且在大于大气压的压力p下进行。根据本发明,该加热的连续流动室包括两个区域:-水解区域,其中该反应介质(水性的或非水性的)不处于超临界状态并且条件是使得可以引发金属氧化物纳米颗粒的成核和生长;-超临界区域,其中该反应介质处于超临界状态并且可以进行金属氧化物纳米颗粒的超临界溶剂热合成。关于流动方向,该超临界区域是在该水解区域的下游。将该表面改性剂引入到该加热的连续流动室允许通过将该表面改性剂接枝到这些纳米颗粒的表面上来对这些纳米颗粒进行表面改性。在本发明的第一方面,在该溶剂热合成已经开始之后,即在所希望的纳米颗粒的成核和生长已经被引发之后,因此在位于该连续流动室的水解区域或超临界区域中的点p2处将该表面改性剂注入到该加热室内,前提是p2是在该金属氧化物前体的注入点(p1)的下游。与现有技术的方法相比,不在该连续流动室的相同注入点处而在不同的注入点处将该金属盐和该表面改性剂引入到该连续流动加热室内。因此该金属氧化物前体的注入点p1和该表面改性剂的注入点p2关于流动方向通过一定距离分开。诸位发明人已经注意到,在该连续反应室内该表面改性剂的注入导致停止或减少该纳米颗粒的生长,因此通过具有所述距离,氧化物纳米颗粒的成核和生长可以在引入该表面改性剂之前开始。进一步地,通过调节在该金属盐的注入点与该表面改性剂的注入点之间的距离,可以控制这些金属氧化物纳米颗粒的合成的反应时间,因此控制纳米颗粒生长的持续时间和条件、以及因此所获得的表面改性的纳米颗粒的尺寸。另一方面,在该纳米颗粒与该表面改性剂之间的反应时间和注入的表面改性剂的量是决定接枝在该纳米颗粒表面上的表面改性剂的量的因素。在本发明的第二方面,为了表面改性所希望的纳米颗粒的目的,在该方法期间,可以将一种或数种表面改性剂引入到该加热室内。通过在不同的注入点处引入两种或更多种表面改性剂,因此可能的是接枝不同类型的表面改性剂,从而导致多官能化的纳米颗粒。此外,引入各种表面改性剂的顺序使能够控制不同表面改性剂接枝到这些纳米颗粒上的方式以及接枝到这些纳米颗粒上的各种表面改性剂的相对量。当表面改性剂比另一种表面改性剂反应性更小时,这是特别有利的。同时引入不同表面改性剂可导致仅仅一种表面改性剂(即最具反应性的表面改性剂)的实际接枝。相比之下,最具反应性的表面改性剂的延迟引入将足够的时间给予较不反应性的表面改性剂以接枝到该纳米颗粒上。此外,在一种或数种表面改性剂的情况下,可以将第一表面改性剂引入在该室内并接枝到这些纳米颗粒的表面上,并且然后可以引入第二改性剂,该第二改性剂进而与该第一改性剂竞争接枝在这些纳米颗粒的剩余自由表面上。因此,通过调节在这两种表面改性剂之间的化学计量,可以控制接枝到该纳米颗粒的表面上的每种表面改性剂的相对量,当将这些表面改性剂同时引入在室内时,这不是那么容易的。在本发明的第三方面,本发明的方法允许通过调节流动的类型,即通过选择湍流或层流,或通过调节流动的速度来控制这些纳米颗粒的尺寸分布。具体实施方式本发明的第一目的是提供一种用于通过在连续流动室内流动的反应介质(水性的或非水性的)中的超临界溶剂热合成制造表面改性的金属氧化物纳米颗粒的连续流动方法,所述连续流动室包含两个区域:-水解区域,其中该反应介质不处于超临界状态并且条件是使得可以引发金属氧化物纳米颗粒的成核和生长;以及-超临界区域,其中该反应介质处于超临界状态并且可以进行金属氧化物纳米颗粒的超临界溶剂热合成,所述方法包括在位于该水解区域或该超临界区域中的点p1处将金属氧化物前体的流引入到该连续流动室内,并且在位于该水解区域或该超临界区域中的点p2处将表面改性剂的流引入到该连续流动室内,其中关于流动方向,p2位于p1的下游。在一个实施例中,该反应介质是水性反应介质并且该溶剂热合成是水热合成。如在本说明书中使用的反应介质(水性的或非水性的)被定义为由该金属氧化物前体流的引入和该表面改性剂流的引入产生的该加热室内的总流。因此,该反应介质的组成不必沿着该连续流动室是均匀的,并且在其中该表面改性剂的流和/或该金属氧化物前体的流包含不同于流动通过该室的另一种溶剂的溶剂情况下,可以取决于该室内的位置而变化。在本发明的含义中,如果在该介质中使用的溶剂含有10mol%的水或更多,则反应介质被认为是水性反应介质。在一个实施例中,本发明中使用的水性反应介质是水或水与一种或多种醇(例如甲醇、乙醇、异丙醇或丁醇)的混合物。当该水性反应介质是水和醇的混合物时,水/醇(如乙醇或异丙醇)的摩尔比可以是从1:5至5:2、特别地从1:4至2:1、特别地从2:3到1:1、特别地大约4:5。可替代地,虽然优选的实施例与在水热条件下使用水性反应介质的本发明的方法和机器有关,但是本发明的方法和机器可以应用于非水性反应介质(即其中该溶剂含有小于10%或甚至没有水的介质)的溶剂热反应,前提是该溶剂使在溶剂热条件下该金属氧化物前体的水解反应成为可能。因此,在以下说明书中,除非明确引用含水量,否则术语“反应介质”不限于水性反应介质,并且在使用术语“水热”或“水性反应介质”的情况下,这些方法和机器可以分别加上必要的变更适配于溶剂热方法和非水性反应介质,前提是该溶剂使该金属氧化物前体在溶剂热条件下的水解反应成为可能。优选地,在该连续流动加热室内的反应介质是具有大约4:5的摩尔比的水与醇的混合物,优选地具有大约4:5摩尔比的异丙醇或乙醇与水的混合物。确实,使用这种混合物允许在超临界条件下在低于当使用唯一水作为该反应介质时所要求的温度的温度下形成金属氧化物纳米颗粒。该金属氧化物前体的流和该表面改性剂的流在高于大气压的压力p下被加压,以便实现允许在该连续流动加热室内的超临界水热合成的条件。典型地,该流可以通过使用泵来加压。在一个实施例中,压力p是从10mpa至30mpa、特别地从15mpa至25mpa、更特别地大约22mpa。在一个实施例中,以沿着流动方向的增加的温度梯度加热该连续流动室,该增加的温度梯度范围是从至少th(引发金属氧化物纳米颗粒的成核和生长的水解温度)和tc(该连续流动加热室内的反应介质处于超临界状态的温度)。因此,该加热的连续流动室包括至少两个区域:-水解区域,其中该室的温度是从th至tc;-超临界区,其中该室的温度高于tc。在该水解区域内,在亚临界条件下的同时,该反应介质的温度高于该水解温度,这允许引发金属氧化物颗粒的成核和生长。然后,这些金属氧化物颗粒经过该超临界区域。在这些超临界条件下,该反应介质的解离增强,这增加了该金属盐的水解并导致完全结晶的纳米尺寸金属氧化物颗粒的形成。th取决于该反应介质的组成并根据所希望的纳米颗粒尺寸来确定。在一个实施例中,确定th以便获得纳米颗粒的最低和最窄的尺寸分布。典型地,th是在两个以下条件之间最下游的温度:该温度达到足以允许纳米颗粒的成核的温度,并且将该纳米颗粒前体引入在该连续流动室中。在一个实施例中,th是至少100℃、特别地从130℃至250℃、更特别地从150℃至200℃。tc是该连续流动加热室内的反应介质是在超临界状态下的温度。tc取决于该反应介质的组成,并且可以基于该反应介质的相图来确定。在一个实施例中,tc是至少240℃、特别地从280℃至400℃、更特别地从300℃至380℃。在该水热合成期间在该连续流动加热室内的表面改性剂的引入导致将表面改性剂接枝到这些金属氧化物纳米颗粒的表面上,从而导致形成表面改性的金属氧化物纳米颗粒。如先前所解释的,将该表面改性剂的流在注入点p2处引入在该连续流动加热室内,该注入点p2不同于p1并且是在p1的下游,其中p1是该金属氧化物前体的注入点。因此,与现有技术的方法相比,不在该金属氧化物前体的相同注入点处将该表面改性剂引入到该连续流动加热室内。以这种方式,在这些颗粒的成核和生长已经开始之后引入该表面改性剂,这允许控制这些金属氧化物纳米颗粒的晶体安排、尺寸和尺寸分布。相反地,如果该金属盐和该表面改性剂在相同的注入点处注入,则可能导致形成具有较低结晶度的较小的金属氧化物纳米颗粒以及这些纳米颗粒的较高尺寸分散。对于给定的相对量的表面改性剂和金属氧化物前体,在p1与p2之间的距离决定了在这些金属氧化物纳米颗粒的表面处接枝的表面改性剂的量以及所得表面改性的金属氧化物纳米颗粒的尺寸。特别地,在p1与p2之间的距离决定了通过表面改性剂的存在而不受干扰的颗粒生长和成核的持续时间,并且它决定了与该表面改性剂的接枝同时发生的颗粒生长和成核的持续时间。它还可能影响超临界生长和成核的持续时间,没有表面改性剂和超临界同时接枝和生长。因此,在p1与p2之间的距离以及金属氧化物前体和表面改性剂的各自量决定了这些金属氧化物纳米颗粒的平均粒度、尺寸分散以及在表面处接枝的表面改性剂的量。在一个实施例中,p1和p2两者位于该水解区域中。在另一个实施例中,p1位于该水解区域中并且p2位于该超临界区域中。根据本发明的方法,在p1与p2之间的距离越小,这些表面改性的金属氧化物纳米颗粒的尺寸就越小。因此,可以通过调节在p1与p2之间的距离来控制这些表面改性的金属氧化物纳米颗粒的尺寸。此外,在p1与p2之间的距离越小,在这些金属氧化物纳米颗粒的表面处接枝的表面改性剂的量越大。因此,可以通过调节在p1与p2之间的距离来控制在这些表面改性的金属氧化物纳米颗粒的表面处接枝的表面改性剂的量。本发明的多次注入方法还允许在该纳米颗粒上接枝不同类型的表面改性剂,从而导致多官能化的纳米颗粒。关于流动方向,这些不同表面改性剂的引入的顺序使能够控制这些不同表面改性剂接枝到如以上提及的这些纳米颗粒上的方式。此外,这些纳米颗粒的尺寸分布可以通过调节流动的类型进行控制,即通过选择湍流或层流,更湍流导致这些颗粒的更窄的尺寸分布,通过使该室内部的速度曲线均匀化,或通过调节该流动的速率,该流动速率影响时间周期(在其期间该混合物在该室内部),因此影响可用于该颗粒的生长的时间。增加的流动的速度或流动速率引起颗粒尺寸减少。在一个实施例中,该连续流动加热室中的流动是具有高于3000、特别地从3000至8000的雷诺数的湍流。金属盐可以用作这些金属氧化物纳米颗粒的前体。在一个实施例中,该金属盐可溶于水性反应介质中。例如,它可以是无机酸盐,如cu、ba、ca、zn、al、y、si、sn、zr、ti、sb、v、cr、mn、fe、co或ni的硝酸盐、氯化物、硫酸盐、氧基盐酸盐、磷酸盐、硼酸盐、亚硫酸盐、氟化物或含氧酸盐,或者有机酸盐,如cu、ba、ca、zn、al、y、si、sn、zr、ti、sb、v、cr、mn、fe、co或ni的醇盐、甲酸盐、乙酸盐、柠檬酸盐、草酸盐或乳酸盐。还可以使用这些金属盐的混合物。在另一个实施例中,该前体不可溶于水性反应介质中。在那种情况下,使用使在溶剂热条件下该金属氧化物纳米颗粒的前体的水解成为可能的非水性反应介质。这样一对前体/非水性溶剂是本领域技术人员众所周知的。优选地,该金属盐是钛(iv)或锆的盐,如异丙醇钛(iv)、丙醇钛(iv)、乙酸锆、异丙醇锆、丙醇锆或乙酰丙酮锆。该金属氧化物前体在该反应介质中的浓度不受限制,只要它溶解于该反应介质中。该金属氧化物前体在该反应介质中的浓度可以是从0.0001mol/l至1mol/l、特别地从0.001mol/l至0.1mol/l、更特别地从0.01mol/l至0.1mol/l。该浓度可以根据这些纳米颗粒的所希望的尺寸在实验上进行调节:该浓度越低,这些纳米颗粒越小。用于本发明中的表面改性剂是能够与待处理的纳米颗粒的表面强烈相互作用的任何化合物。在一个实施例中,它是能够与纳米颗粒表面共价结合的任何化合物。可替代地,该表面改性剂可以通过化学吸附或物理吸附接枝到这些纳米颗粒的表面上。该表面改性剂必须可溶于该反应介质中。在一个实施例中,该表面改性剂是有机配体,从而导致杂化有机-无机纳米颗粒(官能化的纳米颗粒)。在一个具体的实施例中,该有机配体含有酸基团,如羧酸基团、膦酸基团或磺酸基团,硅烷基团,胺基团或硫醇基团。在更具体的实施例中,该有机配体含有羧酸基团、膦酸基团,或者它可以是醛。它可以例如是己酸、辛基膦酸、苯基膦酸或亚磷酸。注入到该连续流动加热室内的表面改性剂的量取决于所希望的纳米颗粒的官能化的速率而调节。典型地,表面改性剂/金属氧化物前体在该反应介质中的摩尔比是从0.05至10、特别地从0.1至1、更特别地从0.15至0.2。为了进行连续方法,优选地将该金属氧化物前体和该表面改性剂两者作为在与该反应介质易混合的溶剂中的溶液中的流引入到该加热室内。此外,优选的是,该金属氧化物前体和表面改性剂可溶于该反应介质中,否则它可能引起管线、泵和过滤器堵塞问题。优选地,该反应介质是水性反应介质。该金属氧化物前体的流的溶剂和该表面改性剂的流的溶剂可以是相同或不同的。每种流的组成和流动速率可以取决于该加热室内的所希望的反应介质的组成并且取决于所希望的金属氧化物前体和表面改性剂的相对量来调节。优选地,该金属氧化物前体的流的溶剂和该表面改性剂的流的溶剂是水或水与一种或多种醇(例如甲醇、乙醇、异丙醇或丁醇)的混合物。典型地,该金属氧化物前体和表面改性剂两者分别从具有给定浓度的金属氧化物前体和表面改性剂的储备溶液注入到该连续流动室内。这些流可以与没有金属氧化物前体和表面改性剂两者的流组合,以便在该反应介质中获得所希望浓度的金属氧化物和表面改性剂。根据本发明的方法的主要实施例,在该连续流动加热室的超临界区域的末端处回收表面改性的金属氧化物纳米颗粒的流。在一个实施例中,该表面改性的金属氧化物纳米颗粒的流在低于th的温度下通过使用冷却装置如冷凝器淬灭,这允许以液体悬浮液的形式回收这些表面改性的金属氧化物纳米颗粒。在通过过滤器过滤该悬浮液之后或在蒸发该悬浮液的溶剂之后,可以以干燥形式回收这些表面改性的金属氧化物纳米颗粒。本发明的方法可用于制造表面改性的金属氧化物纳米颗粒,这些纳米颗粒选自tio2、zro2、zno、batio3、nimoo3、niwo3、al2o3、ga2o3、in2o3、sio2、geo2、v2o5、ceo2、coo、α-fe2o3、γ-fe2o3、nio、co3o4、mn3o4、γ-mno2、cu2o、cofe2o4、znfe2o4、znal2o4、fe2coo4、bazro3、bafe12o19、limno2o4、licoo2或la2o3。作为非限制的实例,tio2或zro2颗粒可以用羧酸或用膦酸官能化;batio3颗粒可以用硅烷基团(-si(or)3)或胺(-nh2)官能化;tio2或zno颗粒可以用硫醇基团(-sh)或磺酸(-so2oh)官能化;nimoo3或niwo3颗粒可以用羧酸官能化。在以下实例中,该接枝通过在该表面改性剂的两个或三个氧原子与该微晶的氧化物之间的共价键合进行操作,因此具有两个或三个氧原子的表面改性剂是优选的。但是还可以使用包含至少一个具有两个或三个氧原子的部分的羧酸(膦酸、硝酸、砷酸...等)的酸衍生物。这些纳米颗粒的尺寸范围典型地是从1nm至50nm的直径,特别地从3nm至20nm,例如在5nm与10nm之间。根据本发明的方法,可以通过调节在该金属盐的注入点p1与该表面改性剂的注入点p2之间的距离来控制这些表面改性的金属氧化物纳米颗粒的尺寸。取决于该金属氧化物前体的类型并且取决于该表面改性剂的类型,如单环和/或四方结构,可以用本发明的方法制备官能化纳米颗粒的不同结晶结构。本发明的另一个目的是如先前描述的用于进行本发明的方法的装置。参照图6,本发明的装置包括用加热器(2a,2b)加热的连续流动室(1),该加热器以沿着流动方向的增加的温度梯度加热连续流动室(1)。该温度梯度在该连续流动加热室中限定了至少两个区域:-水解区域(h),其中该反应介质不处于超临界状态并且条件是使得可以引发金属氧化物纳米颗粒的成核和生长,以及-超临界区域(sc),其中该反应介质处于超临界状态并且可以进行金属氧化物纳米颗粒的超临界溶剂热合成。此外,连续流动室(1)具有:-入口(3),该入口用于在注入点p1处将该金属氧化物前体的流引入到该连续流动室(1)内,-一个或数个入口(4a,4b),该一个或数个入口用于在不同于p1并且在p1下游的注入点p2处将该表面改性剂的流引入到在该水解区域内或在该超临界区域内的所述连续流动加热室(1)内。该装置还可以包括:-用于回收在该超临界区域中产生的表面改性的金属氧化物纳米颗粒的流的出口(5),-连接到出口(5)上的冷却装置(6),接着是用于回收这些呈干燥形式的表面改性的金属氧化物纳米颗粒的过滤装置(7)以及用于回收该反应介质的溶剂的接收器(13),或者接着是用于回收这些呈悬浮液形式的表面改性的金属氧化物纳米颗粒的容器。连续流动室(10)优选地是管式反应器。在一个实施例中,该连续流动加热室由数个串联连接的模块组成。将每个模块用加热器(例如加热筒)彼此独立地加热,该加热器以大致均匀的方式加热该模块。该水解区域可以被一个或数个模块覆盖。该超临界区域可以被一个或数个模块覆盖。具有数个覆盖该水解区域或该超临界区域的模块的优点是可以在两个模块之间注入该金属氧化物前体或该表面改性剂。以这种方式,这些模块不必配备有注入入口,这允许使用现有技术的常规的连续流动反应器。因此,用于将该表面改性剂的加压的流引入到所述连续流动加热室(1)内的入口(4a,4b)可以位于这些模块之间。在一个实施例中,连续流动室(10)在流动方向上包括两个用于在亚临界条件下进行水热合成的水解模块以及两个用于在临界条件下进行水热合成的超临界模块。可替代地,该连续流动室可以由单个模块组成,该单个模块通过数个加热器加热,以便获得在该连续流动室内限定水解区域和超临界区域的温度梯度。该金属氧化物前体的流和该表面改性剂的流可以分别从储备溶液(10)和(11)注入到该连续流动室内。可以将该金属氧化物前体的流在进入室(1)之前用预热器(10)预热。连续流动室(1)优选地是由不锈钢或incorek制成的。其尺寸取决于所希望的雷诺数和停留时间进行调节。停留时间是将该纳米颗粒生长至所希望的尺寸所需要的反应时间。典型地,该管的长度可以是从1m至50m、特别地从3m至25m、更特别地从10m至15m。内径可以是从0.5mm至100mm、特别地从1mm至10mm、更特别地从1.5mm至5mm。该金属氧化物前体的流可以通过使用泵(8)、特别是高压泵来加压。当这些泵必须在已经加压的系统中注入液体时,它们可能需要是高压泵。可以通过使用背压调节器(9)来控制室(1)的压力。例如,该溶液可以通过注入流体的标准泵和背压调节器的组合作用来加压,该背压调节器允许该流体仅当达到压力阈值时通过。可以通过将具有热电偶探头的热电偶插入在室(1)内部或者通过将充足的控制提供给这些加热器并且监测加热器参数来监测室(1)的温度。本发明现在将在以下实例中进一步描述。提供这些实例以说明本发明,并且决不应该被视为限制本发明。图1示出了如在实例1中使用的根据本发明的连续流动反应器系统的示意图。图2表示在超临界条件下在水和乙醇的混合物中通过超临界水热合成获得的tio2纳米颗粒的xrd图案。图3表示在超临界条件下在水和乙醇的混合物中通过超临界水热合成获得的tio2纳米颗粒的hr-tem显微照片。图4表示在超临界条件下在水和乙醇的混合物中通过超临界水热合成获得的tio2纳米颗粒的粒度分布。图5表示作为该表面改性剂的注入点的函数的用本发明的方法制备的表面改性tio2的粒度。图6示出了根据本发明的连续流动反应器系统的示意图。实例1:tio2纳米颗粒的官能化图1示出了该连续流动反应器系统的示意图。roh=乙醇hpp=高压泵p=压力计v=阀vr=调节阀,还被称为背压调节器f=过滤器c=冷凝器该系统包括四个串联连接的模块r1至r4。r1和r2是用于在亚临界条件下进行该水热合成的水解模块。r3和r4是用于在超临界条件下进行该水热合成的超临界模块。该表面改性剂的注入点被定位在反应器r1之前、不同模块(r1-r2、r2-r3、r3-r4)之间并且在反应器r4之后。在以下条件下,用水和乙醇(水/乙醇摩尔比=0.8)的混合物进行tio2纳米颗粒的超临界水热合成:·钛前体:在水性溶液中的ti(o-ic3h7)4,具有水/乙醇摩尔比为8,在储备溶液中的浓度=4.10-2mol.l-1,·在r1-r4内的压力p=22mpa,·在r1-r4内的总流量q=11.6g.min-1·流动的类型:湍流(re=3287),·r1和r2:ο具有12m的总长度、由两个模块(每个具有6m的长度)组成的不锈钢的管式反应器,ο150℃的温度,·r3和r4:ο具有12m的总长度、由两个模块(每个具有6m的长度)组成的不锈钢的管式反应器,ο380℃的温度。在该合成后,将tio2纳米颗粒(裸露或官能化的)作为水和乙醇中的溶液回收。将它们离心并用乙醇洗涤5次以去除未反应的表面改性剂。图2表示所获得的tio2纳米颗粒的x射线衍射(xrd)图案,而没有向该反应系统中加入表面改性剂。它可以归因于对应于体心四方晶格的icdd-pdf卡片00-021-1272(锐钛矿相,空间群i41/amd,)。从应用于25.326°处的峰(101)和48.1°处的峰(200)的德拜-谢乐方程式,估算微晶的平均尺寸是7.3nm。图3表示tio2纳米颗粒的hr-tem显微照片(高分辨率透射电子显微镜)。它示出了它们的单晶态。因此,可以认为这些微晶的平均尺寸等于颗粒的平均尺寸。粒度和粒度分布的估算是从在tem显微照片上观察的约200个纳米颗粒的计数来进行的。聚集颗粒范围是从6nm至15nm。最大群体具有大约10nm的尺寸,并且平均尺寸与用xrd估计的平均尺寸相关。图4表示tio2纳米颗粒的粒度分布。进行与以上相同的实验,但是其中在该水热合成期间在该系统的不同注入点处加入表面改性剂:-在r1与r2之间,-在r2与r3之间,-在r3与r4之间,或-在r4之后。注入的表面改性剂是己酸(ha)或辛基膦酸(opa)。每秒注入的ti原子与每秒注入的己酸分子的接枝头的摩尔比(ti/ha比率)是6(ha6)或12(ha12)。调节注入的表面改性剂的量以便具有6(opa6)或12(opa12)的每秒注入的ti原子与每秒注入的膦酸分子的接枝头的摩尔比(ti/opa)。该表面改性剂是在与该钛前体的溶剂相同的组成和相同的水/醇摩尔比的水-乙醇混合物中的溶液中。官能化试剂与tio2纳米颗粒的相互作用通过评估其对计算的微晶尺寸(被认为是粒度)的影响而证实。表1给出了取决于注入点并且取决于该前体中每个ti原子所注入的表面改性剂的摩尔比的微晶的平均尺寸(通过德拜-谢乐方程式计算的)。表1样品观察的参数微晶的尺寸(nm±10%)ti002用dibup的非原位的官能化8ti003用bis2p的非原位的官能化7.1ti004用opa的非原位的官能化7.7ti005用3op的非原位的官能化7.5ti009在r4之后ha6的注入8.1ti010在r4之后ha12的注入7.7ti011在r4之后opa6的注入7.9ti012在r3与r4之间ha6的注入7.7ti013在r3与r4之间ha12的注入7.8ti014在r3与r4之间opa6的注入7.5ti015在r2与r3之间ha6的注入7.6ti016在r2与r3之间ha12的注入8.2ti017在r2与r3之间opa6的注入7ti018在r1与r2之间ha6的注入6.7ti019在r1与r2之间ha12的注入6.7ti020在r1与r2之间opa6的注入5.4管线ti002至ti005对应于以下实验,其中首先合成作为裸露纳米颗粒的tio2纳米颗粒并且在回收以溶液形式的纳米颗粒之后第二次进行官能化,如通过使用单词“非原位”表达的。图5表示根据该表面改性剂的注入点的微晶尺寸。清楚地示出,在该合成方法中当更早注入该表面改性剂时,微晶尺寸减小,尤其用辛基膦酸时。表面改性剂的注入越早,微晶越小。这是在这些tio2纳米颗粒与该表面改性剂之间的相互作用的证据,并且该接枝的表面改性剂阻碍这些纳米颗粒的生长。这还表明了辛基膦酸似乎具有比己酸更大的与tio2微晶的相互作用,因为它对微晶尺寸的影响更强。此外,至少对于己酸,每12个ti原子1个己酸分子的接枝头的比率(12的ti/ha比率)似乎不足以在没有水解步骤的情况下具有对微晶的有效接枝,因为如果在该水解步骤之后注入己酸ha12,则纳米颗粒的尺寸不变。此外,那些结果示出了,定位该注入点使得在该方法的水解步骤期间进行该注入确保了对微晶尺寸的更大影响。对于通过在r2与r3之间注入该表面改性剂、用辛基膦酸官能化的tio2进行的ftir(傅里叶变换红外光谱学)分析示出了对应于烷基链的三个带[2960cm-1:νas(-ch2-ch3),2925cm-1:νas(-ch2-),2850cm-1:νs(-ch2-)],这是在tio2纳米颗粒的表面处存在官能化试剂的证据。此外,在1100-1000cm-1:νs(-p-o3)处的带是充分可见的,经由p-o官能团评估在这些纳米颗粒的表面处的该改性剂的接枝。从这个注入点可以得出结论,tio2纳米颗粒用辛基膦酸进行官能化。对于通过在r1与r2之间注入该表面改性剂、用辛基膦酸官能化的tio2进行的相同的fitr分析示出了,在1460cm-1:δsc(-ch2-)处的亚烷基带的存在,以及比当该注入点是在r2与r3之间时更强的在1100-1000cm-1:νs(-p-o3)处用辛基膦酸接枝的证据。在通过在r4之后注入该表面改性剂用辛基膦酸官能化的tio2上进行的tga-ms(使用质谱仪的热重量分析仪)分析示出了比裸露纳米颗粒更高的质量损失:7.5%比2.9%。此外,通过该损失输出的气体通过tga-ms进行分析并且发现归因于与该辛基膦酸的辛基部分相对应的有机碎片。因此,虽然ftir不能准确指出官能化的量,但是tga-ms确认了通过注入opa改性剂获得的tio2颗粒被辛基膦酸或其衍生物之一官能化,甚至当该注入点位于该超临界通道之后(紧邻在r4之后)。对于通过在r3与r4之间注入该表面改性剂、用辛基膦酸官能化的tio2的相同分析示出了10%的质量损失,其中7.1%可归因于具有对应于该辛基膦酸的辛基部分的信号的有机部分。对于通过在r1与r2之间注入该表面改性剂、用辛基膦酸官能化的tio2的相同分析示出了20%的质量损失,其中仅仅2.9对应于裸露颗粒,因此17.1%可归因于具有对应于所使用的膦酸的辛基链的信号的有机部分。可以得出结论,本发明的连续多次注入方法允许在一个步骤中将膦酸分子原位接枝在tio2微晶上。因此,尤其通过使用超临界水/乙醇系统,小且非常良好结晶的tio2纳米颗粒是容易获得的。该表面改性剂关于流动方向的注入点的位置具有对接枝表面改性剂的量以及所得纳米颗粒的尺寸的影响。早期注入允许更高的官能化和微晶尺寸(并且最可能地还有粒度)的减少。然而,诸位本发明人已经发现,重要的是在注入该官能化表面改性剂之前,使纳米颗粒的成核发生,否则tio2微晶的形成被废物污染。确实,在那些情况下,所得产物具有非常复杂且不良分辨的xrd图案。这意味着该材料的一部分似乎是无定形的。进一步地,所产生的种类不是通过例如opa链但可能地ti-ox-py材料的颗粒官能化的纯tio2颗粒。这是由于p与金属的高反应性(高于o与金属),并且加入p改性剂太早防止tio2形成。实例2:zro2纳米颗粒的官能化使用与实例1中使用的系统相同的系统、用相同操作条件制备zro2微晶。反应物:-zr前体:乙酰丙酮锆、乙酸锆、丙醇锆或异丙醇锆。-表面改性剂:己酸、辛基膦酸、苯基膦酸、亚磷酸或sik7709-10(12-十二烷基膦酸)三乙基溴化铵)。-溶剂:水和乙醇或异丙醇。在每种情况下,将注入的表面改性剂的量调节为具有0.16的酸分子/氧化锆摩尔比,这对应于tio2实例中为6的ti/ha或ti/p。在该合成后,将zro2纳米颗粒(裸露或官能化的)作为水和乙醇或异丙醇中的溶液回收。将它们离心并用乙醇洗涤5次以去除未反应的表面改性剂。在tga分析之后在残余物的ftir观察下,可以在zro2微晶上发现对应于p-o-金属结合的峰。此外,在tga分析的1000℃下煅烧期间排放的气体的相关质谱不能检测释放的含磷碎片。这些组合的结果意味着在1000℃下的tga分析之后,膦酸接枝头,即至少磷原子,仍然化学吸附在zro2的表面上,并且在tga分析期间它们不参与样品的质量损失。要注意的是,在tga分析之后,对于用opa官能化的tio2纳米颗粒残余物的ftir分析,观察到相同的峰。结果提供在表2和3中。m=单斜晶系的t=四方的w/e=水/乙醇w/ip=水/异丙醇x意味着在该合成介质中没有分散δ意味着可接受但不是太好的分散pa=亚磷酸ppa=苯基膦酸表2表3这些结果示出了,对于zro2纳米颗粒,这些官能化的纳米颗粒的结构取决于该表面改性剂的性质。确实,不论使用己酸、辛基膦酸、苯基膦酸还是亚磷酸,xrd图案是不同的。用己酸的情况下,维持裸露纳米颗粒的单斜晶系结构,并且用辛基膦酸的情况下获得了zro2的四方结构。用这两种表面改性剂的情况下,获得了良好结晶的材料,而用亚磷酸和苯基膦酸的情况下,最终材料是不良结晶的,并且难以清楚地区分一些相,即使可以猜测对于亚磷酸的单斜晶系相的微晶和四方相的微晶的混合物以及对于苯基膦酸的四方相的微晶的存在。表面改性剂sik7709-10含有两种活性位点:膦酸部分和溴化铵部分。用苯基膦酸和(1-丁基)三乙基溴化铵的混合物进行实验,以模拟两种活性位点,并看看是否在这两种部分之间将存在竞争并且哪一种将发挥优势。将这些表面改性剂一起溶解于0.8摩尔比的水/乙醇溶液中,具有0.16的p/zr与n/zr摩尔比。乙酰丙酮锆是在4.10-2mol.l-1的浓度下。测试两个注入点:在r1与r2之间以及在r2与r3之间,具有10ml.min-1的注入流量。将总压力保持在23mpa下。将r1和r2在200℃下加热,并将r3在380℃下加热。获得的纳米颗粒的ftir分析示出了存在含氮化合物的证据。因此,它意味着膦酸超过溴化铵优先地接枝在zro2的纳米颗粒的表面上。进行类似的测试以便比较膦酸和羧酸的相对反应强度,即在所考虑的这两种表面改性剂之间哪种分子将优先地接枝在该纳米颗粒表面上。证明了,膦酸超过羧酸或溴化物优先地被接枝。因此,具有溴化物结尾或具有羧酸官能团的微晶的官能化可以分别用包含膦酸官能团和溴化物两者的表面改性剂并且用包含羧基官能团的表面改性剂来进行。表面改性剂sik7709-10可以用于接枝具有结尾溴化物官能团的微晶,而不接枝悬挂的膦酸基团,这进而将具有不想要的将颗粒彼此桥联的作用,因此导致纳米颗粒的强聚集。多次注入设置还用于在该室内在距彼此的一定距离处分开注入两种改性剂,即苯基膦酸和亚磷酸。这些注入点分别位于r1与r2之间(对于该第一改性剂)以及r2与r3之间(对于该第二改性剂)。这两种表面改性剂都溶解于水中,各自具有0.08的p/zr摩尔比(如与对于单一改性剂实验的0.16的p/zr比率相反)。所使用的前体是溶解于水中4.10-2mol.l-1浓度下的乙酸锆。单独注入两种不同的改性剂有效地导致纳米颗粒双重接枝。使用苯基膦酸作为第一表面改性剂允许获得具有基本上由单斜晶系晶体组成的单晶结构的双重官能化的微晶,而使用亚磷酸作为第一表面改性剂产生了两种类型的微晶:四方微晶和单斜晶系微晶。因此,可以通过调节这些表面改性剂注入到该反应系统内的相对量和顺序来控制纳米颗粒尺寸、结构和接枝量。由于一些表面改性剂可以超过其他表面改性剂优先地接枝,接枝在这些微晶上的表面改性剂的安排将取决于这些表面改性剂的注入的顺序。以上结果示出了:-如果更早地进行该表面改性剂的注入,尤其在已经经过反应时间的2/3之前,接枝在该微晶上的表面改性剂的量更高,但是粒度更小。-膦酸具有比羧酸和溴化物反应性基团对粒度更大的影响。-该前体的性质可具有对某些材料的结晶结构的影响。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1