用于三氧化硫转化和氢气生产过程的催化剂组合物的制作方法
本文所述的主题一般涉及用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,所述催化剂组合物包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料。本主题还涉及用于制备用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物的方法。
背景技术:
存在可用于通过裂解水来产生作为产物的氢气和作为副产物的氧气的许多热化学方法。有许多这样的热化学循环在过去几十年中作为可行路线已经通过实验进行了分析。在这些循环中,在us4,089,940中公开的最初由generalatomic提出的硫-碘热化学循环由于其更高的效率是最有前途的循环。硫-碘(si)循环在一系列化学反应中产生氢,所述一系列化学反应的设计方式使得每个反应的起始材料是另一反应的产物。在这个循环中,热能通过数个高温化学反应进入。一定量的热通过放热低温反应排出。该反应的输入为水和高温热,并且其释放低温热、氢气和氧气。在该循环中没有产生流出液,并且除水之外的所有试剂都被再循环和再利用。整个循环包括以下三个反应,如下所示:
so2(g)+2h2o(l)+i2(l)→h2so4(aq)+2hi(aq)(25℃-120℃)(1)
2hi(g)→h2(g)+i2(g)(400-700℃)(2)
h2so4(g)→h2o(g)+so2(g)+0.5o2(g)(>800℃)(3)
反应(1)被称为本生(bunsen)反应,一种放热气体(so2)吸收反应,其在25℃至120℃的温度范围内自发进行并产生两种酸:hi和h2so4。hi分解(2)是轻微吸热的反应,释放氢气并在400℃至700℃的温度范围内发生。h2so4分解(3)以产生so2是两步反应。第一步包括h2so4的热分解(h2so4→so3+h2o),第二步是so3催化分解(so3→so2+1/2o2)以产生so2和氧气。较低的so3分压和高温有利于分解反应。如果so3的分解平衡压力较高,则必须升高温度以增加实际过程的分解速率。然而,催化剂通过降低反应的活化能势垒而在改善解离效率方面起到主要作用。
us2,406,930公开了可以在非常高的温度下使硫酸热分解以得到二氧化硫和氧气。us3,888,730公开了可以在低得多的温度下使硫酸分解,条件是使硫酸蒸气与钒催化剂接触。us4,089,940公开了通过使用铂催化剂可以进一步降低分解温度。us4,314,982公开了负载在各种载体(如硫酸钡、氧化锆、二氧化钛、二氧化硅、硅酸锆及其混合物)上的有效的铂催化剂。铂负载型催化剂在分解反应的低温区域(即,至多700℃)中是稳定且有效的。在超过和高于750℃的温度下,使用负载在上述载体上的铜氧化物和铁氧化物作为催化剂。酸的整个催化分解在作为具有负载型铂催化剂的低温床和具有较便宜的铁氧化物或铜氧化物负载形式的高温床的一系列床中发生。在这些床中实现的停留时间分别为1.0秒±50%和0.5秒±50%。用于多级过程的催化剂的组合能够在不超过7秒的总停留时间内实现等于对于最佳温度的平衡值的至少约95%的分解以产生so2。
ko100860538公开了负载或没有负载在氧化铝和二氧化钛上的铜-铁二元氧化物催化剂,其中铜与铁之比为0.5至2,催化剂与载体之比为1:1。所述催化剂可以长时间承受高温并且可以保持更高的活性,空速高至100ml/g催化剂.小时至500,000ml/g催化剂.小时,优选高至500ml/g催化剂.小时至100,000ml/g催化剂.小时。
还发表了一系列探索数种具有高活性和稳定性以实现硫酸分解的催化剂的研究论文。在1977年dokiya等人[1]测试了用于在大气压下在1073k至1133k的范围内进行硫酸分解的一系列氧化物催化剂(tio2、v2o5、cr2o3、mno2、fe2o3、coo4、nio、cuo、zno、al2o3和sio2)。其中,经烧结的fe2o3表现出良好的催化剂活性,然而,该催化剂在高温下随着时间发生活性、表面积和抗碎强度的损失。这些观察结果基于4小时的实验测试。在1982年norman等人[2]总结了在各种载体上的不同的活性材料。他们所使用的活性金属/金属氧化物为pt、fe2o3、cuo、cr2o3,并且载体为各种组合的al2o3、tio2、zro2和baso4。他们得出结论,铬和钒的氧化物是挥发性的并且它们在反应器的后期充当重整催化剂。锰、钴和镍由于过量的硫酸化而表现出具有较低的活性。铂和铁(iii)氧化物被认为是良好的活性材料,并且二氧化钛被认为是用于贵金属催化剂的载体。他们表明,具有二氧化钛载体的铂在较低的温度下充当良好的催化剂,fe2o3和cr2o3在较高温度下是有前途的。在1982年ishikawa等人[3]测试了以1%至5%(w/w)的负载水平负载在氧化铝基体上的pt、fe2o3、cuo,并且活性按pt>fe2o3>v2o5>cuo的顺序降低。在他们的实验中,负载在多孔氧化铝上的活性材料表现出为非多孔氧化铝四倍高的活性,但是非多孔氧化铝表现出更好的稳定性。在1989年tagawa等人[4]对铁、铬、铝、铜、锌、钴、镍和镁的各种廉价金属氧化物进行了更系统的研究。从他们的实验中发现,所有催化剂在高于850℃时表现出相似的转化率。当在低于850℃时操作时,铁(iii)氧化物最初表现出高转化率,并且由于硫酸盐物质的形成而随时间降低。发现活性的顺序为pt>cr2o3>fe2o3>cuo>ceo2>nio>al2o3。
在2006年barbarossa等人[5]在500℃至1100℃的温度范围内在7秒的停留时间下用ag-pd金属间合金和负载在石英棉上的铁氧化物进行了实验。两种催化剂最初都具有高活性,并且在16小时的时间之后,铁(iii)氧化物的活性保持恒定,而ag-pd的活性损失归因于在催化剂的表面上形成pdo薄膜。在2006年kim等人[6]报道了通过共沉淀法制备的负载在al或ti上的fe-催化剂的活性。fe-与al/ti之比为4、3、2和1。在fe-与al的孔体积比率保持恒定的情况下,fe-al催化剂样品的表面积显著增加。在较低温度(低于550℃)下,fe-ti催化剂表现出比fe-al催化剂更高的活性。在高于800℃时,fe-al由于硫酸盐的不稳定性而表现出更高的活性。banerjee等人[7]研究了铁铬钙钛矿[fe2(1-x)cr2xo3](x的范围:{0至1})的催化活性。所述催化剂以固态路线制备并且其表面积被发现在14m2/g至15m2/g的范围内。对所有催化剂测试10小时,并且他们发现fe1.8cr0.2o3是活性最大的,并且形成较少的硫酸盐。他们提出低水平的cr-存在增加了催化剂的稳定性并减少了稳定的金属硫酸盐的形成。在2007年ginosar等人[8]研究了载体和催化剂的长期稳定性。在该研究中所使用的催化剂是铂,并且载体是al2o3、tio2和zro2。发现经二氧化钛负载的催化剂在长持续时间(240小时)内比其余的载体稳定。尽管二氧化钛表现出良好的负载,但其在一段时间(240小时)内仍然损失8%的活性。这是由于作为挥发性氧化物从表面损失pt和烧结。在2008年abimanyu等人[9]研究了通过油滴法和凝胶法制备的cu/al2o3、fe/al2o3和cu/fe/al2o3复合颗粒催化剂的活性。cu/fe/al2o3复合材料的催化活性高于cu/al2o3、fe/al2o3。催化活性随着氧化铝颗粒中cu和fe浓度的增加而增加,并且最佳的[cu]与[fe]之比为1:2[10]。karagiannakis等人[11]合成了用于硫酸分解的各种单一的和混合的氧化物材料。这些材料包括通过溶液燃烧合成制备的fe-cr混合氧化物材料以及cu-fe-al体系的二元组合物和三元组合物。在850℃和环境压力下在固定床反应器中以粉末形式测试所述催化剂。对于cu-fe-al体系,发现向fe-氧化物结构中添加cu增强了分解,而向fe-氧化物中添加al和cu两者还改善了稳定性。banerjee等人[12]研究了钴、镍和铜的铁尖晶石用于硫酸分解的活性。这些铁尖晶石是通过甘氨酸-硝酸盐凝胶燃烧法合成的。将化学计算量的起始材料溶解在50ml蒸馏水中,保持燃料-氧化剂摩尔比(1:4),使得氧化价与还原价之比略小于1。在持续搅拌下将混合的硝酸盐甘氨酸溶液在150℃下缓慢加热以除去过量的水。这导致形成高粘性凝胶。随后,将凝胶在300℃下加热,这导致自燃并形成不期望的气态产物,并且形成泡沫状粉末形式的期望产物。将粉末在两种不同的温度(500℃和900℃)下煅烧12小时,以获得cufe2o4、cofe2o4和nife2o4的结晶粉末。发现铜铁氧体是对于反应的活性最大的催化剂,在800℃下的转化率为78%。张等人[13]通过溶胶-凝胶、真空冷冻干燥(vfd)法制备氧化物的复合材料,即cucr2o4和cufe2o4,并通过浸渍法制备负载在sic上的pt。在前一种情况下,他们直接使用复合氧化物作为催化剂,在后一种情况下,载体是非多孔sic。观察到在低于790℃的温度下,pt/sic催化剂表现出更高的活性,在50小时-1的空速下的产率小于50%。在高于850℃的温度下,复合金属氧化物表现出约70%的产率。对于所有的三种催化剂,在850℃的温度下以50小时-1的空速进行催化剂稳定性测试。在三种催化剂中,cufe2o4在操作45小时之后丧失其活性,pt/sic和cucr2o4两者在操作90小时之后表现出为初始活性的几乎20%的活性降低。来自稳定性测试的废催化剂分析表明,三种催化剂通过团聚而损失了其比表面积并且由于形成相应的硫酸盐而损失了活性。尽管这些催化剂在高温下表现出良好的活性,但在酸介质中缺乏良好的稳定性是主要顾虑。karagiannakis等人[14]、giaconia等人[15]使用经fe2o3涂覆的sisic蜂窝体,其中载体具有零孔隙率和非常低的表面积(5.32m2/g)。该催化剂通过重复的浆料浸渍法以将铁(iii)氧化物负载在蜂窝体上制备。活性金属的负载重量百分比在14.9w/w%至18.5w/w%的范围内。在900℃下煅烧之后,将催化剂粉碎并负载到反应器中。在经fe2o3涂覆的sisic蜂窝片段上,在775℃至900℃的温度范围、1至4巴的压力范围和3.2小时-1至49小时-1的whsv下,用96%硫酸作为进料进行催化剂的活性测试。该载体具有低的表面积(5.32m2/g),并且没有孔隙。观察到在最佳操作条件下(whsv6.0小时-1和17.6重量%催化剂负载,在850℃下,在约30%的so3分压下),催化剂表现出约80%的so2转化率和可忽略不计的失活。lee等人[16]研究了在650℃至850℃的温度范围内,在大气压下,在72,000ml/gcat的ghsv下,经1重量%pt/sic涂覆的氧化铝和1重量%pt/al2o3进行的硫酸分解。所述催化剂通过干浸渍法制备。pt/al2o3催化剂在650℃和700℃下由于形成硫酸铝而失活,但是在750℃和850℃下是稳定的,最高产率为60%。通过cvd法用甲基三氯硅烷(mts)使氧化铝载体涂覆有sic,以得到具有高表面积的非腐蚀性载体(sic-al)。从废催化剂的热分析观察到,在氧化铝上涂覆sic抑制了硫酸盐的形成。在650℃、750℃和850℃下,硫酸转化为so2的转化率分别为约28%、48%和71%。废催化剂表面积的减小表明虽然所述催化剂在6小时内是稳定的,但sic涂覆并不能完全阻止硫酸铝的形成,作者认为对催化剂的进一步改进是必要的。
在以上方法中尝试了许多催化剂,但金属氧化物催化剂是有前途的。然而,金属氧化物催化剂倾向于在高温下烧结,导致催化剂不稳定,这也降低了催化剂的活性。此外,使用高活性的铂催化剂是昂贵的,并且过程温度的小波动导致催化剂活性损失,并且从基体表面浸出可能是缺点。
常规使用的碳化硅是极硬的暗的虹彩色晶体,其没有孔隙并且具有通常小于2m2/g的非常小的表面积,其主要用作磨料和耐火材料。它不溶于水,并且在高至800℃时对于酸或碱是惰性的。当在高于1200℃时暴露于空气时,在碳化硅的表面上形成硅氧化物保护层。最近,us4,914,070报道了呈多孔团聚物形式的碳化硅,其比表面积为至少约100m2/g。us5,217,930[17]、us5,460,759[18]和us5,427,761[19]中还描述了这样的高表面积的硅和其他金属或非金属耐火碳化物组合物及其制造,据说其可以用作用于化学、石油和废气消声器反应的催化剂的载体。us6,184,178[20]报道了呈颗粒形式的催化剂载体,其基本上由比表面积为至少5m2/g并且通常为10m2/g至50m2/g,并且根据astmd4179-88a的抗压性为1mpa至20mpa的碳化硅β微晶构成。据说这些载体可以用于化学和石油化学催化反应,例如碳氢化物的氢化、脱氢、异构化、脱环,尽管没有描述具体的方法和催化剂金属。
现有技术中没有报道高表面积的多孔β-碳化硅作为用于催化剂的载体的用途,所述催化剂用于硫酸分解,更准确地说用于三氧化硫分解,或者用于在升高的温度和压力下和在这种分解过程的极端酸性环境中的类似反应。
技术实现要素:
在本公开内容的一个方面中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个方面中,提供了用于生产催化剂组合物的方法,其包括以下步骤:(a)使至少一种过渡金属盐与选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料接触以获得负载有过渡金属的多孔材料;(b)将负载有过渡金属的多孔材料在250℃至600℃的温度范围内煅烧1小时至6小时的时段并且任选地在900℃至1100℃下加热2小时至5小时以获得催化剂组合物,所述催化剂组合物包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个方面中,提供了用于生产催化剂组合物的方法,其包括以下步骤:(a)使至少一种过渡金属盐与选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料接触并在50℃至150℃下干燥10分钟至5小时;(b)将负载有过渡金属的多孔材料在250℃至600℃的温度范围内煅烧1小时至6小时的时段以获得部分负载有过渡金属的多孔材料;(c)使至少一种过渡金属盐与部分负载有过渡金属的多孔材料接触并在50℃至150℃下干燥10分钟至5小时以获得负载有过渡金属的多孔材料;(d)将负载有过渡金属的多孔材料在250℃至600℃的温度范围内煅烧1小时至6小时的时段并且任选地在900℃至1100℃下加热2小时至5小时以获得催化剂组合物,所述催化剂组合物包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
参照以下描述和所附权利要求将更好地理解本主题的这些和其他特征、方面和优点。提供本发明内容是为了以简化的形式介绍一些概念。本发明内容既不旨在确定所要求保护的主题的关键特征或必要特征,也不旨在用于限制所要求保护的主题的范围。
附图说明
参照附图描述详细的描述。在附图中,附图标记的最左边数字指示该附图标记首次出现的图。贯穿附图,使用相同的标记指代相同的特征和组件。
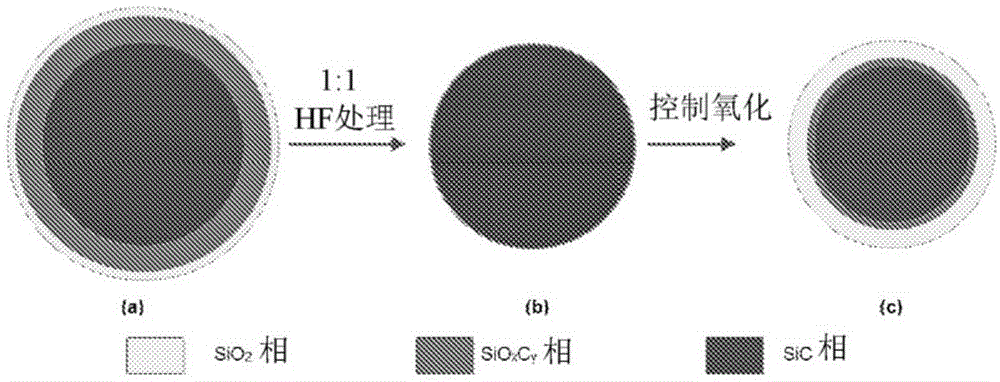
图1a至1c是原样的β-sic的hf处理和氧化的图示。
图2是(a)原样的β-sic(β-sic(r)、(b)经hf处理的β-sic(β-sic(p))、以及(c)在hf处理之后氧化的β-sic(β-sic(pt))的ft-ir光谱的图示。
具体实施方式
本领域技术人员将意识到,除了具体描述的那些之外,本公开内容可以进行变化和修改。应理解,本公开内容包括所有这样的变化和修改。本公开内容还包括本说明书中单独或集合性地提及或指出的所有这样的步骤、特征、组合物和化合物,以及任何或更多个这样的步骤或特征的任何和所有组合。
定义:
为方便起见,在进一步描述本公开内容之前,在此集中了说明书和实施例中采用的某些术语。这些定义应根据本公开内容的其余部分来阅读,并且如本领域技术人员所理解的来理解。本文所使用的术语具有本领域技术人员公认和已知的含义,然而,为了方便和完整起见,下面阐述了特定术语及其含义。
冠词“一个”、“一种”和“一者”用于指代一个或多于一个(即,至少一个)的冠词语法对象。
术语“包括”和“包含”以包含性的开放意义使用,意指可以包括另外的要素。在整个说明书中,除非上下文另有要求,否则词语“包括”以及变体,例如“含有”和“包含”,将被理解为暗示包括所述要素或步骤或者要素或步骤的组,但是不排除任何其他要素或步骤或者要素或步骤的组。
术语“催化剂复合材料”和“催化剂组合物”在本公开内容中可互换使用。
比率、浓度、量和其他数值数据在本文中可以以范围格式呈现。应理解,这样的范围格式仅仅是为了方便和简洁而使用,并且应该被灵活地解释为不仅包括明确叙述为范围限制的数值,而且还包括包含在该范围内的所有单独的数值或子范围,如同每个数值和子范围被明确地叙述一样。
本公开内容一般涉及催化剂组合物,其可用于硫酸分解,更确切地说,可用于在用于氢气生产的硫-碘循环中将三氧化硫分解为二氧化硫和氧气。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中过渡金属选自cu、cr和fe。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含选自cu、cr和fe的氧化物中的过渡金属氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含选自二元氧化物、三元氧化物和尖晶石中的混合过渡金属氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含cu的氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含cr的氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含fe的氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含摩尔比为1:2的cu和fe的二元氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含具有尖晶石结构的fe和cu的氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:包含具有尖晶石结构的cr和cu的氧化物的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的孔体积在0.05cc/g至0.9cc/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的孔体积在0.1cc/g至0.7cc/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的活性表面积在5m2/g至35m2/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的如通过bet多点氮吸附法确定的比表面积在2m2/g至200m2/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的如通过bet多点氮吸附法确定的比表面积在5m2/g至100m2/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中载体材料的如通过bet多点氮吸附法确定的比表面积在10m2/g至60m2/g的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中催化剂组合物的过渡金属含量在0.1重量%至20重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中催化剂组合物的过渡金属含量在0.1重量%至20重量%的范围内,其中催化剂组合物的过渡金属含量在2重量%至10重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中活性材料尺寸在0.1mm至15mm的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中活性材料尺寸在0.1mm至25mm的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及包含多孔β-碳化硅(β-sic)或硅酸盐化的多孔碳化硅(β-sic(pt))的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及包含结晶的多孔β-sic或硅酸盐化的多孔碳化硅(β-sic(pt))的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及呈球形粒料、挤出物或泡沫形式的包含结晶的多孔β-sic或硅酸盐化的多孔碳化硅(β-sic(pt))的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及呈球形粒料、挤出物或泡沫形式的包含结晶的多孔β-sic或硅酸盐化的多孔碳化硅(β-sic(pt))的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内,其中过渡金属选自cu、cr和fe,其中载体材料的孔体积在0.05cc/g至0.9cc/g的范围内,其中载体材料的活性表面积在5m2/g至35m2/g的范围内,其中载体材料的如通过bet多点氮吸附法确定的比表面积在2m2/g至200m2/g的范围内,其中催化剂组合物的过渡金属含量在0.1重量%至20重量%的范围内。
在本公开内容的一个实施方案中,提供了催化剂组合物,其包含过渡金属氧化物,即呈双金属形式或呈尖晶石形式或单独的摩尔比为1:2的铜和铁的氧化物,其用作负载型催化剂以在宽范围的压力(0.1巴至30巴)和温度(450℃至900℃)下将h2so4有效地分解至接近平衡的转化。负载在硅酸盐结晶多孔β-sic(β-sic(pt))上的上述活性材料出乎意料地保持其惰性和结构完整性而没有任何热梯度,并且可以是有效的基体。基体或载体结构选自粉末、微粒、粒料、颗粒、球体、珠、丸、球、条、圆柱体、挤出物和三叶形。
当上述活性材料优选作为负载型催化剂使用时,特定载体必须能够在经受硫酸蒸气气氛时继续起作用,并且具有足够的机械强度,以承受高压和高温并且允许高流量的反应物和产物气体。载体的最重要功能是使分散在表面上的活性组分的微晶迁移的生长速率最小化。如果催化剂在高温下操作,则这些是不可避免的,因为载体的结块使其作为分散剂的作用逐渐减小,这不利地影响催化剂的活性。另外,还重要的是,催化剂载体必须是惰性的,并且能够在腐蚀性硫酸蒸气环境中保持其机械强度、结构完整性,以及在反应的温度和压力范围内具有良好的热稳定性。
已经发现,催化剂体系中采用的许多常用的氧化物载体材料(例如氧化铝、二氧化钛)在450℃至950℃之间和环境中没有表现出商业实用寿命,并因此不被认为是合适的。此外,在该温度范围的较低端操作通常对基体是特别不利的,并且在较高端操作由于烧结而对于活性金属氧化物是危险的。然而,已经发现,在经预处理的多孔β-sic或硅酸盐化的多孔β-sic(β-sic(pt))上负载活性材料表现出良好的稳定性、惰性和有效性。而且,催化剂更经济,并且在经济操作范围内几乎没有热梯度。
在诸如此类的催化反应中,使表面积最大化是非常重要的。在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含以小于约25w/w(重量百分比)的量分散在载体上的呈双金属氧化物混合物形式的铁和铜氧化物混合物。
在本公开内容的一个实施方案中,提供了用于将三氧化硫转化为二氧化硫和氧气的催化剂组合物,其包含以基于载体重量的3%至10%(重量百分比)的量分散在载体上的呈尖晶石形式的铁和铜氧化物混合物。在8%的活性铜-铁尖晶石的水平(基于载体重量的重量百分比)下,催化剂的表面积将为至少10m2/g催化剂。
催化剂组合物可以用于固定床、或者单级或多级操作的单一床的一部分、或者动态床(例如,使用任何形式的催化剂的移动床/流化床)。通过床的硫酸蒸气可以保持在期望的范围(600℃至1000℃),更优选在850℃。
这些催化剂的载体结构呈分开的或离散的结构或颗粒物的形式。如本文所使用的,术语“明晰的(distinct)”或“离散的”结构或颗粒物是指呈分开的材料(例如,颗粒、珠、丸、粒料、圆柱体、三叶形、挤出物、球体或其他圆形形状)形式或者另一种制造构造的载体。或者,分开的材料可以是不规则形状的微粒的形式。优选地,至少大部分(即>50%)的微粒或明晰结构的最大特征长度(即,最长尺寸)小于25毫米,优选小于6毫米。根据一些实施方案,分开的催化剂结构的直径或最长特征尺寸为约0.25mm至约6.4mm(约1/100”至约1/4”),优选地在约0.5mm与约4.0mm之间。在另一些实施方案中,它们在约50微米至6mm的范围内。
本公开内容还涉及用于生产用于在硫-碘循环中分解硫酸的稳定且经济的催化剂的方法。在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其包括以下步骤:(a)使至少一种过渡金属盐与选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料接触以获得负载有过渡金属的多孔材料;(b)将负载有过渡金属的多孔材料在250℃至600℃的温度范围内煅烧1小时至6小时的时段并且任选地在900℃至1100℃下加热2小时至5小时以获得催化剂组合物,所述催化剂组合物包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液接触并均化以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液分部分地接触并通过超声均化,以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液接触,通过超声均化10分钟至1小时,并在50℃至150℃下干燥10分钟至5小时,以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中在煅烧之前将负载有过渡金属的多孔材料在50℃至150℃下空气干燥10分钟至5小时。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,该方法包括:使至少一种过渡金属盐与选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料接触以获得部分负载有过渡金属的多孔材料;将部分负载有过渡金属的多孔材料在50℃至150℃下干燥10分钟至5小时,使至少一种过渡金属盐与部分负载有过渡金属的多孔材料接触以获得负载有过渡金属的多孔材料;将负载有过渡金属的多孔材料在250℃至600℃的温度范围内煅烧1小时至6小时的时段并且任选地在900℃至1100℃下加热2小时至5小时以获得催化剂组合物,所述催化剂组合物包含:选自过渡金属氧化物、混合过渡金属氧化物及其组合的活性材料;以及选自二氧化硅、二氧化钛、氧化锆、碳化物及其组合的载体材料,其中活性材料相对于载体材料的重量比率在0.1重量%至25重量%的范围内。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液接触并均化以获得部分负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使部分负载有过渡金属的多孔材料与至少一种过渡金属盐的水溶液接触并均化以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液分部分地接触并通过超声均化,以获得部分负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使部分负载有过渡金属的多孔材料与至少一种过渡金属盐的水溶液分部分地接触并通过超声均化,以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使载体材料与至少一种过渡金属盐的水溶液接触,通过超声均化10分钟至1小时,并在50℃至150℃下干燥10分钟至5小时,以获得部分负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中使部分负载有过渡金属的多孔材料与至少一种过渡金属盐的水溶液接触,通过超声均化10分钟至1小时,并在50℃至150℃下干燥10分钟至5小时,以获得负载有过渡金属的多孔材料。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中至少一种过渡金属盐是选自cu、cr和fe的过渡金属的盐。ni盐选自硝酸镍、氯化镍、甲酸镍、乙酸镍和碳酸镍。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中cu、cr和fe的至少一种过渡金属盐选自柠檬酸盐、硝酸盐、氯化物、甲酸盐、乙酸盐和碳酸盐。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中载体材料的孔体积在0.1cc/g至0.7cc/g的范围内。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中载体材料的活性表面积在5m2/g至35m2/g的范围内。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中载体材料是多孔β-碳化硅(sic)或硅酸盐化的多孔β-碳化硅(β-sic)(即β-sic(pt))。
在本公开内容的一个实施方案中,提供了用于生产催化剂组合物的方法,其中载体材料是结晶的多孔β-sic或硅酸盐化的多孔β-碳化硅(β-sic)(即β-sic(pt))。
催化剂组合物可以以各种方式来制造或合成,即,通过沉积、沉淀、浸渍、喷雾干燥、或通过固态途径、或者其中的组合。例如,浸渍可以以以下方式进行。可以将测量体积的包含计算量的各元素化合物的前体的溶液添加至大约相同体积的颗粒尺寸为0.5mm至10mm的催化剂载体中或者过量地添加至所述催化剂载体中。在一个实施方案中,催化剂载体的颗粒尺寸可以为1mm至5mm。在进行中间搅拌的情况下放置2小时之后,溶剂可以蒸发,在343k至393k下干燥并在空气中在550℃下煅烧2小时至5小时。通过以上方法获得的催化剂是负载在β-sic上的金属氧化物,表面积不小于10m2/g。为了制备铜铁氧体,可以按照上述过程单独地或顺序地以所需的摩尔比(fe:cu=1:2)浸渍各金属前体。在煅烧之后,将温度调节在1223k至1273k之间保持2小时至5小时的时段,以完成铁氧化物与铜氧化物之间的反应,以形成铜铁氧体(cufe2o4)。包含在这些催化剂中的元素的量在使样品矿化之后通过原子吸收光谱法(aas)确定。所有都以相对于基体的重量%表示。
大多数已知的金属氧化物催化剂在高温下是有活性的,并且在持续很长时间段的活性之后导致烧结。当关于硫酸分解,并且更准确地关于在硫-碘循环中将so3转化为so2和o2,在873k至1473k,更优选973k至1173k的温度范围内以及在0.1巴至30巴,更优选1巴至20巴的压力范围内长时间地测试时,根据本发明制备的催化剂的活性和稳定性是优异的。根据本发明,在反应器中在大气条件下保持硫酸的空速,在任何地方100ml/g催化剂-小时至500,000ml/g催化剂-小时,优选500ml/g催化剂-小时至72,000ml/g催化剂-小时是合适的。所有实验均在惰性气体氮气的存在下进行。
尽管已经参照本主题的一些实施方案相当详细地描述了本主题,但是其他实施方案也是可能的。
实施例
以下实施例是以举例说明本发明的方式给出的,并且不应被解释为限制本公开内容的范围。应理解,前面的一般描述和以下的详细描述都仅是示例性和说明性的,并且旨在提供对要求保护的主题的进一步说明。由sicat获得的sic(原样的β-sic(r))由光学上不同的相组成。sic粉末的晶粒在外层包含少量的无定形二氧化硅,各向异性sioxcy层夹在本体sic浅表面层与外部sio2层之间,如图1(a)所示。图2(a)示出的原样的sic(β-sic(r))的ft-ir光谱揭示出,在820cm-1至830cm-1处的振动带对应于本体sic层,在900cm-1和1164cm-1处的振动带归因于结晶sioxcy相,以及1200cm-1附近的带对应于无定形二氧化硅。在原样的sic(β-sic(r))中在1080cm-1至1110cm-1范围内没有振动带表明,表面主要是sioxcy层而不是sio2层。当在超声下用hf(用水以1:1稀释)处理原样的sic3分钟至5分钟并随后用大量水洗涤时,导致sioxcy/sio2相的溶解并且留下纯sic相(此后为β-sic(p))(如图1(b)所示),这根据图2(b)中1066cm-1至1164cm-1、1228cm-1处峰的消失也可以证明。当使经hf蚀刻的样品在大气空气中在500℃至750℃的温度范围内进一步氧化2小时至6小时的时段时,sic的浅层被氧化以形成主要具有无定形sio2层的sioxcy/sio2层,如图1(c)所示(此后为β-sic(pt))。图2(c)中的经氧化样品的ft-ir光谱表明,具有在1216cm-1处的肩部的在1098cm-1处的非常强且宽的ir带通常被归因于to和lo模式的si-o-si不对称伸缩振动。在900cm-1至950cm-1处的ir带可以归因于硅烷醇基/si-o-伸缩振动。800cm-1附近的ir带可以归因于si-o-si对称伸缩振动,而460cm-1至480cm-1附近的ir带是由于o-si-o弯曲振动。820cm-1至830cm-1附近的较强吸收带被分配给本体sic。氧化形式的sic形成大量的无定形sio2层,其具有比原样的sic更好的载体与催化剂的相互作用。
实施例1(a)
催化剂载体的预处理
通过使用称为预处理方法(ptm)的合成方法获得催化剂载体。碳化硅(β-sic)挤出物(直径2mm)由sicatsarl(法国)提供,并且此后称为原样的β-sic(r)或β-sic。在室温下在超声下用在水中的1:1hf溶液蚀刻β-sic(r)样品3分钟至5分钟以从β-sic的表面除去sioxcy/sioz。将样品过滤并用大量去离子水洗涤直至滤液ph值达到6.5至7,然后将样品在120℃下在真空下干燥3小时至5小时,此后称为β-sic(p)或简言之不含二氧化硅的β-sic。随后将经干燥的样品(β-sic(p))在大气空气中在700℃至1000℃之间氧化2小时至6小时的时段,以获得经预处理的β-sic或简言之β-sic(pt)。
实施例1(b)
催化剂fe2o3/β-sic(r)的制备(用于比较)
将1.713g铁前体(柠檬酸铁铵)溶解在10ml蒸馏水中,然后添加10g经预干燥并脱气的2mm尺寸的β-sic(r)挤出物。然后,将所得混合物超声约30分钟,使得全部β-sic(r)完全浸入溶液中。半小时之后,从溶液中分离出β-sic(r)并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部铁溶液被β-sic(r)吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。最终的催化剂是负载在β-sic(r)上的5%fe2o3。还通过类似方法制备了2%至15%(w/w)的负载型铁氧化物催化剂。
实施例1(c)
催化剂fe2o3/β-sic(p)的制备
使用与实施例1(b)中使用的相同的方案制备负载有fe2o3的β-sic(p),其中在该实施例中使用β-sic(p)载体代替β-sic(r)载体。
实施例1(d)
催化剂fe2o3/β-sic(pt)的制备(用于比较)
使用与实施例1(b)中使用的相同的方案制备负载有fe2o3的β-sic(pt),其中使用β-sic(pt)载体代替β-sic(r)载体。
实施例2(a):
催化剂cu2o/β-sic(r)的制备(用于比较)
将1.8741g铜前体(cu(no3)2.3h2o)溶解在10ml蒸馏水中,然后添加10g经预干燥并脱气的2mm尺寸的β-sic(r)挤出物。然后,将所得混合物超声约30分钟,使得全部β-sic(r)完全浸入溶液中。半小时之后,从溶液中分离出β-sic(r)并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部铜溶液被β-sic(r)吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。最终的催化剂是负载在β-sic(r)上的5%cu2o。还通过类似方法制备了2%至15%(w/w)的负载型铜(i)氧化物催化剂。
实施例2(b)
催化剂cu2o/β-sic(pt)的制备(用于比较)
使用与实施例1(b)中使用的相同的方案制备5%cu2o/β-sic(pt)催化剂,其中在该实施例中使用β-sic(pt)载体代替β-sic(r)载体。使用类似方法,还制备了在β-sic(pt)载体上的2%至15%(w/w)的负载型铜(i)氧化物催化剂。
实施例3(a):
催化剂cr2o3/β-sic(r)的制备(用于比较)
将1.101g铬酸铵(cu(no3)2.3h2o)溶解在10ml蒸馏水中,然后添加10g经预干燥并脱气的2mm尺寸的β-sic(r)挤出物。然后,将所得混合物超声约30分钟,使得全部β-sic(r)完全浸入溶液中。半小时之后,从溶液中分离出β-sic(r)并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部铬酸铵溶液被β-sic(r)吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。最终的催化剂是负载在β-sic(r)上的5%cr2o3。还通过类似方法制备了在β-sic(r)载体上的2%至15%(w/w)的负载型铬(iii)氧化物催化剂。
实施例3(b)
催化剂cr2o3/β-sic(pt)的制备(用于比较)
使用与实施例3(a)中使用的相同的方案制备5%cr2o3/β-sic(pt)催化剂,其中使用β-sic(pt)载体代替β-sic(r)载体。使用类似方法,还制备了负载在β-sic(pt)上的2%至15%(w/w)的负载型cr2o3催化剂。
实施例4(a):
催化剂cufe2o4/β-sic(r)的制备
将1.176g硝酸铵(fe(no3).9h2o)和0.5049g硝酸铜(cu(no3)2.3h2o)溶解在15ml蒸馏水中,然后添加10g经预干燥并脱气的2mm尺寸的β-sic(r)挤出物。然后将所得混合物超声约30分钟,使得全部β-sic(r)完全浸入溶液中。半小时之后,从溶液中分离出β-sic(r)并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部溶液被β-sic(r)吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。然后,在中间混合固体的情况下将炉温逐渐升至1000℃并在1000℃下保持3小时。所获得的催化剂为负载在β-sic(r)上的5%cufe2o4的催化剂。
实施例4(b):
催化剂cufe2o4/β-sic(p)的制备
使用与实施例4(a)中使用的相同的方案制备5%cufe2o4/β-sic(p)催化剂,其中在该实施例中使用β-sic(p)作为载体代替β-sic(r)。还通过类似方法制备了2%至15%(w/w)的cufe2o4/β-sic(p)催化剂。
实施例4(c):
催化剂cufe2o4/β-sic(pt)的制备
使用与实施例4(a)中使用的相同的方案制备5%cufe2o4/β-sic(pt)催化剂,其中使用β-sic(pt)作为载体代替β-sic(r)。通过类似方法制备了2%至15%(w/w)的cufe2o4/β-sic(pt)催化剂。
实施例5(a)
催化剂cucr2o4/β-sic(r)的制备
使用孔体积法或干浸渍法将铬酐和硝酸铜的水溶液浸渍至β-sic(r)中。在该方法中,将铬酐和硝酸铜(化学计量比例)的6ml水溶液添加至10gβ-sic(r)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时,以获得cucr2o4/β-sic(r)。
实施例5(b)
催化剂cucr2o4/β-sic(pt)的制备
使用与实施例5(a)中使用的相同的方案制备cucr2o4/β-sic(pt)催化剂,其中使用β-sic(pt)作为载体代替β-sic(r)。通过类似方法制备了2%至15%(w/w)的cucr2o4/β-sic(pt)催化剂。
实施例6(a)
催化剂fecr2o4/β-sic(r)的制备
使用孔体积法或干浸渍法将铬酐和硝酸铁的水溶液浸渍至β-sic(r)中。在该方法中,将铬酐和硝酸铁(化学计量比例)的6ml水溶液添加至10gβ-sic(r)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时,以获得fecr2o4/β-sic(r)。
实施例6(b)
催化剂fecr2o4/β-sic(pt)的制备
使用与实施例6(a)中使用的相同的方案制备fecr2o4/β-sic(pt)催化剂,其中使用β-sic(pt)作为载体代替β-sic(r)。
实施例7
催化剂cufe2o4/al2o3的制备
将1.176g硝酸铵(fe(no3).9h2o)和0.5049g硝酸铜(cu(no3)2.3h2o)溶解在15ml蒸馏水中,然后添加10g经预干燥并脱气的1mm直径的氧化铝挤出物。然后将所得混合物超声约30分钟,使得全部氧化铝完全浸入溶液中。半小时之后,从溶液中分离出氧化铝并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部溶液被氧化铝吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。然后,在进行中间混合的情况下将所得的经煅烧材料的温度逐渐升至1000℃并加热3小时。所获得的催化剂为负载在氧化铝(al2o3)上的5%cufe2o4的催化剂。
实施例8
催化剂fe2o3/al2o3的制备
将1.713g铁前体(柠檬酸铁铵)溶解在10ml蒸馏水中,然后添加10g经预干燥并脱气的1mm直径的氧化铝挤出物。然后,将所得混合物超声约30分钟,使得全部氧化铝完全浸入溶液中。半小时之后,从溶液中分离出氧化铝挤出物并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部铁溶液被氧化铝挤出物吸收。最后,将经浸渍的基体在100℃下空气干燥1小时,然后在500℃下煅烧2小时。最终的催化剂是负载在al2o3上的5%fe2o3。还通过类似方法制备了负载在氧化铝上的2%至15%(w/w)的负载型铁氧化物和铜氧化物催化剂。
实施例9(a)
cofe2o4催化剂的制备。
在典型的方法中,将0.20mfe(no3)3溶液与0.10mco(no3)2溶液混合在一起。然后,向混合溶液中添加适量的6mnaoh溶液以将ph调节至8至14,并向所得溶液中添加去离子水直至溶液的体积为约160ml。将混合物强烈搅拌30分钟,然后转移至300ml衬有teflon的高压釜中。将高压釜密封并在200℃下保持48小时。反应完成之后,将所得固体产物过滤并用水和无水醇洗涤数次。最后,将经过滤的样品在120℃下干燥4小时,以获得cofe2o4尖晶石催化剂。
实施例9(b)
催化剂cofe2o4/β-sic(pt)的制备
将1.135g柠檬酸铁铵溶解在10ml蒸馏水中,并添加10g经预干燥并脱气的2mm直径的β-sic(pt)挤出物。然后将所得混合物超声约30分钟,使得全部β-sic(pt)完全浸入溶液中。半小时之后,从溶液中分离出β-sic挤出物并在80℃下干燥30分钟,然后再次添加至剩余的溶液中,使得全部溶液被β-sic(pt)吸收。然后将样品在空气中干燥5小时,并在400℃下在炉中煅烧3小时。然后再次从炉中移出样品并冷却至室温,随后用10ml硝酸钴溶液(在10ml水中的0.619gco(no3)2.6h2o)浸渍。再次重复相同的过程并在900℃温度下煅烧3小时,然后将炉温逐渐升至1000℃,保持4小时以完成固态反应。所得催化剂称为cofe2o4/β-sic(pt)。
实施例10(a)
nife2o4催化剂的制备
通过以1:2的摩尔比(即,分别为0.10m、0.2m)混合等体积的ni(no3)2·6h2o和fe(no3)3·9h2o溶液,通过水热法制备nife2o4催化剂。向混合的盐溶液中逐滴添加6mnaoh溶液,直至最终ph值达到指定值以形成掺合物。将掺合物转移至具有不锈钢壳的teflon高压釜(300ml)中,并向teflon高压釜中添加少量去离子水直至总体积的80%。将高压釜加热至200℃,保持48小时,使其自然冷却至室温。将最终产物过滤并用去离子水和纯醇洗涤数次以除去可能的残余物,然后在120℃下干燥4小时以获得nife2o4催化剂。
实施例10(b)
nife2o4/β-sic(pt)催化剂的制备
如实施例9(b)中给出的,在β-sic(pt)挤出物上依次顺序沉积柠檬酸铁铵(在10ml中1.135g)和硝酸镍溶液(在10ml水中0.619gni(no3)2.6h2o)。在空气中煅烧之后,将样品温度保持在900℃以完成镍氧化物与铁(iii)氧化物之间的固态反应以形成载体的镍铁氧体晶体。因此形成的催化剂被称为负载在β-sic(pt)上的nife2o4。
实施例11(a)
znfe2o4催化剂的制备
通过使用其中将化学计算量的硝酸锌和硝酸铁溶解在去离子水中的水热法制备znfe2o4尖晶石。然后向盐溶液中添加适量的6mnaoh溶液以调节ph=10至12。然后将所得混合物转移至teflon不锈钢高压釜中,并将温度保持在200℃下24小时。反应完成之后,将所得固体产物过滤并用大量水和醇洗涤数次。最后将经过滤的样品在120℃下空气干燥4小时,以获得znfe2o4尖晶石催化剂。
实施例11(b)
znfe2o4/β-sic(pt)催化剂的制备
将10ml柠檬酸铁铵(0.1104m)添加至10gβ-sic(pt)挤出物中。然后将所得混合物摇动几分钟,使得全部陶瓷正好浸入溶液中并放置半小时。在这之后,从剩余溶液中分离出碳化硅挤出物并在80℃下在烘箱中干燥2小时,然后再次添加至剩余的溶液中,使得全部铁溶液被β-sic(pt)挤出物吸收。首先将经浸渍的负载型催化剂在100℃下干燥2小时,在马弗炉中在400℃下煅烧3小时并冷却至室温。再用10ml硝酸锌溶液(在10ml水中0.615g)重复相同的过程。最后,将催化剂在900℃下煅烧2小时,然后在炉内逐渐将温度升至1000℃,保持3小时,以完成最终的固态反应,以获得负载在β-sic(pt)上的znfe2o4。
实施例12(a)
催化剂nicr2o4的制备
使用nio和α-cr2o3作为起始材料通过固态路线合成nicr2o4催化剂。使用研钵和研杵将nio和α-cr2o3样品的1:1摩尔混合物充分混合并且加热至650℃保持6小时,然后在进行中间混合的情况下在12小时内逐渐加热至900℃以完成两种氧化物之间的均相反应。最后,将样品在900℃下进一步保持5小时以获得nicr2o4催化剂。
实施例12(b)
催化剂nicr2o4/β-sic(pt)的制备
使用孔体积法或干浸渍法将铬酐和硝酸镍的水溶液浸渍至β-sic(pt)中。在该方法中,将铬酐和硝酸镍(化学计量比例)的6ml水溶液添加至10gβ-sic(pt)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时以获得nicr2o4/β-sic(pt)。
实施例13(a)
催化剂zncr2o4的制备
将0.025摩尔的zn(no3)2.6h2o和0.05摩尔的cr(no3)3.9h2o溶解在90ml蒸馏水中以形成澄清的水溶液。向剧烈搅拌的水溶液中缓慢滴入4mnaoh溶液以将ph调节至7至12以获得悬浮体。将所获得的悬浮体转移至衬有teflon的300ml容量的高压釜中,并加热至200℃保持48小时。然后将产物过滤并用大量去离子水和醇洗涤。然后将经洗涤的产物在120℃下干燥4小时以获得绿色粉末(zncr2o4)。
实施例13(b)
zncr2o4/β-sic(pt)催化剂的制备
使用孔体积法或干浸渍法将铬酐和硝酸镍锌的水溶液浸渍至β-sic(pt)中。在该方法中,将铬酐和硝酸锌(化学计量比例)的6ml水溶液添加至10gβ-sic(pt)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时以获得zncr2o4/β-sic(pt)。
实施例14
cr2o3催化剂的制备
铬(iii)氧化物催化剂如下制备:将硫酸铬与3%重量%的聚乙烯醇混合并制成球形粒料。将这些粒料在1000℃下在空气中煅烧5小时以分解成铬氧化物。
实施例15
cu2o催化剂的制备
氧化亚铜如下制备:将硫酸铜与3%重量%的聚乙烯醇混合并制成球形粒料。将这些粒料在1000℃下在空气中煅烧5小时以分解成铜(i)氧化物。
实施例16(a)
催化剂pt/al2o3的制备。
使用孔体积法或干浸渍法将氯铂酸的水溶液浸渍至氧化铝(al2o3)中。计算溶液中的铂(pt)浓度以在载体上获得期望的pt含量,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在500℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时,并在350℃下在氮气中的10%氢气流(1升/小时.g催化剂)中还原3小时以获得1%pt/al2o3。
实施例16(b)
催化剂pt/β-sic(pt)的制备
使用孔体积法或干浸渍法将氯铂酸的水溶液浸渍至碳化硅(β-sic(pt))中。计算溶液中的铂(pt)浓度以在载体上获得期望的pt含量,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在500℃下在干燥空气流(1升/小时.g催化剂)中煅烧3小时,并在350℃下在氮气中的10%氢气流(1升/小时.g催化剂)中还原3小时以获得1%pt/β-sic(pt)。
实施例17
cufecrob/β-sic(pt)催化剂的制备
使用孔体积法或干浸渍法将铬酐、柠檬酸铁铵和硝酸铜的水溶液浸渍至β-sic(pt)中。在该方法中,将摩尔比为1:1:1(化学计量比例)的铬酐、柠檬酸铁铵和硝酸铜的6ml水溶液添加至10g的β-sic(pt)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧5小时以获得其中cu:fe:cr的元素比被认为是1:1:1的cufecrob/β-sic(pt)。
实施例18
cufecroc/β-sic(pt)催化剂的制备
使用孔体积法或干浸渍法将硝酸铜、柠檬酸铁铵和铬酐的水溶液浸渍至β-sic(pt)中。在该方法中,将摩尔比为1:1:4(化学计量比例)的硝酸铜、柠檬酸铁铵和铬酐的6ml水溶液添加至10g的β-sic(pt)中,然后使固体成熟12小时。然后将固体在120℃下烘箱干燥12小时,并在900℃下在干燥空气流(1升/小时.g催化剂)中煅烧5小时以获得其中cu:fe:cr的元素比被认为是1:1:4的cufecrob/β-sic(pt)。
实施例19(制备的催化剂的活性测试)
方法1:如下所述在固定床反应器中测试由以上实施例1至6获得的催化剂。将1g催化剂装入玻璃管反应器的中间,并通过注射泵将经预热的n2惰性气体连同液体h2so4(98重量%)连同n2惰性气体泵送至初级分解器中,在其中将温度保持在973k。将硫酸的空速保持在500ml/g.催化剂-小时与50,000ml/g.催化剂-小时之间。将反应器温度保持在1000k与1223k之间并将压力保持在大气压下。对于高压实验(即,压力在1巴至20巴之间),使用hastelloy反应器。使经催化剂分解的产物(痕量的h2so4、so3、h2o、so2和o2)通过一系列吸收器,在吸收器中除了n2和o2之外的所有气体都被吸收以用于定量分析。使用气相色谱仪和氧气分析仪对未吸收的氧气进行定量。
方法2:在双级固定床反应器中测试由以上实施例1至6获得的催化剂。在典型的实验中,借助于注射泵将在室温下的液体硫酸以限定的流量连同惰性载气氮气通过质量流量控制器(mfc)进给至第一级分解器。在整个实验中使第一级保持在973k以确保硫酸的完全分解。热分解的so3、h2o和n2在到达第二级反应器中的催化剂床之前流过充当预热段的热陶瓷珠。将催化分解的产物(so2、o2、h2o、n2和未分解的so3)冷却并捕集在两个串联的瓶中,瓶中填充有i2/i-水溶液以测量so3和so2的浓度。在气相色谱仪(nucon,型号5765,配备有tcd和填充有carbosphere的gc柱)和在线氧分析仪中分析未吸收的气体。
表1:各种负载型催化剂在硫酸分解反应中的活性测试。
表2:大多数活性催化剂的催化剂稳定性测试
如表1的实施例1(b)、1(c)和1(d)所示,将铁(iii)氧化物负载在三种不同的经表面处理的β-sic上。在不同温度下在固定床反应器中测量催化剂活性。显然,与原样的碳化硅或纯碳化硅相比,由经预处理的载体制备的催化剂具有最高的转化率。这种高活性归因于铁(iii)氧化物在富含sio2的载体上的高度分散。类似地,在所有催化剂中,实施例4(c)、实施例5和实施例6在所考虑的温度范围内表现出最高的活性,其也具有经预处理或硅酸盐化的β-sic载体。尽管这些经预处理的载体催化剂与通过原样的催化剂载体制备的催化剂相比表现出略微高的转化率,但是催化剂的稳定性出乎意料地在多孔β-sic的硅酸盐化催化剂载体的情况下增加。在10小时至300小时的时段测试各种催化剂的稳定性并示于表2中。看起来,负载在经预处理的碳化硅上的催化剂比负载在原样的sic或其他载体上的催化剂有活性得多且稳定得多。在测试的前25个小时期间,具有所有类型的β-sic载体的催化剂对于硫酸分解表现出相似的活性,而载体经过预处理的催化剂,实施例4(c)、2(b)和1(d)(即,催化剂cufe2o4/β-sic(pt)、cu2o/β-sic(pt)和fe2o3/β-sic(pt)),在长达300小时的操作中保持其活性。
无需进一步详细说明,相信本领域技术人员可以使用前面的描述来最大限度地利用本发明。因此,前述优选的具体实施方案被解释为仅是说明性的,并且无论如何不以任何方式限制本公开内容的其余部分。通过用本发明的一般或具体描述的反应物和/或操作条件替换前述实施例中使用的反应物和/或操作条件,可以同样成功地重复前述实施例。根据前面的描述,本领域技术人员可以容易地确定本发明的基本特征,并且在不脱离其精神和范围的情况下,可以对本发明作出各种改变和修改以使其适于各种用途和条件。
虽然已经参照本主题的一些实施方案相当详细地描述了本主题,但是其他实施方案也是可能的。
参考文献
[1]dokiyam,kameyamat,fukudak,koteray.thestudyofthermochemicalhydrogenpreparation.iii.anoxygen-evolvingstepthroughthethermalsplittingofsulfuricacid.bullchemsocjpn1977;50:2657-60.
[2]normanj,myselsk,sharpr,williamsond.studiesofthesulfur-iodinethermochemicalwater-splittingcycle.intjhydrogenenergy1982;7:545-56.doi:10.1016/0360-3199(82)90035-0.
[3]ishikawah,ishiie,ueharai,nakanem.catalyzedthermaldecompositonofh2so4andproductionofhbrbythereactionofso2withbr2andh2o.intjhydrogeaenergy1982;7:237-46.doi:10.1016/0360-3199(82)90087-8.
[4]tagawah,endot.catalyticdecompositionofsulfuricacidusingmetaloxidesastheoxygengeneratingreactioninthermochemicalwatersplittingprocess.intjhydrogenenergy1989;14:11-7.doi:10.1016/0360-3199(89)90151-1.
[5]barbarossav,bruttis,diamantim,saus,demariag.catalyticthermaldecompositionofsulphuricacidinsulphur-iodinecycleforhydrogenproduction.intjhydrogenenergy2006;31:883-90.doi:10.1016/j.ijhydene.2005.08.003.
[6]kimt,gongg,gwonb,leek-y,jeonh-y,shinc-h,etal.catalyticdecompositionofsulfurtrioxideonthebinarymetaloxidecatalystsoffe/alandfe/ti.applcatalagen2006;305:39-45.doi:10.1016/j.apcata.2006.02.052.
[7]banerjeea,paim,bhattacharyak,tripathia,kamblev,bharadwajs,etal.catalyticdecompositionofsulfuricacidonmixedcr/feoxidesamplesanditsapplicationinsulfur-iodinecycleforhydrogenproduction.intjhydrogenenergy2008;33:319-26.doi:10.1016/j.ijhydene.2007.07.017.
[8]ginosardm,petkoviclm,glennaw,burchkc.stabilityofsupportedplatinumsulfuricaciddecompositioncatalystsforuseinthermochemicalwatersplittingcycles.intjhydrogenenergy2007;32:482-8.doi:10.1016/j.ijhydene.2006.06.053.
[9]abimanyuh,jungk-d,junk-w,kimj,yooks.preparationandcharacterizationoffe/cu/al2o3-compositegranulesforso3decompositiontoassisthydrogenproduction.applcatalagen2008;343:134-41.doi:10.1016/j.apcata.2008.03.033.
[10]mallannab,kwangn/e,jungd.synthesisofcu/fe/ti/al2o3compositegranulesforso3decompositioninsicycle.ratio2009:248-52.doi:10.1007/s10562-008-9747-3.
[11]karagiannakisg,agrafiotiscc,zygogiannia,pagkourac,konstandopoulosag.hydrogenproductionviasulfur-basedthermochemicalcycles:part1:synthesisandevaluationofmetaloxide-basedcandidatecatalystpowdersfbrthesulfuricaciddecompositionstep.intjhydrogenenergy2010:1-14.doi:10.1016/j.ijhydene.2010.11.083.
[12]banerjeeam,paimr,meenass,tripathiak,bharadwajsr.catalyticactivitiesofcobalt,nickelandcopperferrospinelsforsulfuricaciddecomposition:thehightemperaturestepinthesulfurbasedthermochemicalwatersplittingcycles.intjhydrogenenergy2011;36:4768-80.doi:10.1016/j.ijhydene.2011.01.073.
[13]zhangp,sut,chenqh,wanglj,chensz,xujm.catalyticdecompositionofsulfuricacidoncompositeoxidesandpt/sic.intjhydrogenenergy2012;37:760-4.doi:10.1016/j.ijhydene.2011.04.064.
[14]karagiannakisg,agrafiotiscc,zygogiannia,pagkourac,konstandopoulosag.hydrogenproductionviasulfur-basedthermochemicalcycles:part1:synthesisandevaluationofmetaloxide-basedcandidatecatalystpowdersforthesulfuricaciddecompositionstep.intjhydrogenenergy2011;36:2831-44.doi:10.1016/j.ijhydene.2010.11.083.
[15]giaconiaa,saus,felicic,tarquinip,karagiannakisg,pagkourac,etal.hydrogenproductionviasulfur-basedthermochemicalcycles:part2:performanceevaluationoffe2o3-basedcatalystsforthesulfuricaciddecompositionstep.intjhydrogenenergy2011;36:6496-509.doi:10.1016/j.ijhydene.2011.02.137.
[16]leesy,jungh,kimwj,shulyg,jungk-d.sulfuricaciddecompositiononpt/sic-coated-aluminacatalystsforsicyclehydrogenproduction.intjhydrogenenergy2013;38:6205-9.doi:10.1016/j.ijhydene.2013.01.107.
[17]dominiquedubotslf.binodalmetalliccarbideanditsuseasacatalyst.us5217930,1993.
[18]doniniquedubotslf.unitedstatespatent1191.us5460759,n.d.
[19]grindattob,jourdana,prinm.processfortheproductionofmetalcarbideshavingalargespecificsurfaceunderatmosphericpressureinertgasscavenging.us5427761,n.d.
[20]baluaisg,ollivierb.catalystsupporrwithbaseofsiliconcarbidewithhighspecificsurfaceareaingranulatedformhavingimprovedmechanicalcharacteristics.6184178b1,2001.
- 还没有人留言评论。精彩留言会获得点赞!