一种碲硒纳米材料及其制备方法和应用与流程
本发明涉及纳米材料技术领域,特别是涉及一种碲硒纳米材料及其制备方法和应用。
背景技术:
二维材料,是指电子仅可在两个维度的非纳米尺度(1-100nm)上自由运动(平面运动)的材料。近年来,石墨烯、黑磷(磷烯)、硅烯、锗烯、锑烯,以及金属二硫化物(如二硫化钛、二硫化钼)等一系列只有单原子层厚度的准二维材料相继被发现,它们在生物医学领域(如光热治疗、光动力治疗、载药治疗和光声成像)具有潜在的应用前景。但目前研究的这些材料很少能够兼具高稳定、低毒、强红外吸收和高光热转换效率等特点。因此,对于能进行生物应用的新型光热材料的研发势在必行。
技术实现要素:
鉴于此,本发明提供了一种碲硒纳米材料及其制备方法和应用,所述碲硒纳米材料的光热效果明显、毒性低,制备方法简单。
本发明第一方面提供了一种碲硒纳米材料,包括二维碲硒纳米片,以及包覆在所述二维碲硒纳米片表面的形貌调控材料,所述二维碲硒纳米片的化学通式为tesex,其中,x为se与te的摩尔比,x的取值范围为0<x≤10。
可选地,所述x的取值范围为0.25≤x≤1.5。例如为0.25、0.5、0.75、1或1.2。此取值范围下的碲硒纳米材料,可以兼具良好形貌,以及低毒性、高吸收、强光热转换效应。
本发明中,所述碲硒纳米材料的长宽尺寸为10nm-500nm,厚度为1nm-50nm。可选地,所述碲硒纳米材料的长宽尺寸为50nm-110nm,70nm-150nm。可选地,所述碲硒纳米材料的厚度为10nm-35nm。选择适合的长宽尺寸能够保证其在生物应用时在肿瘤部位具有较好的被动富集效果,避免尺寸过大导致无法进入肿瘤部位,及尺寸过小导致易从肿瘤部位泄露的问题。选择较薄的厚度可以增大所述的比表面积,从而增强其光热效果和负载率。
本发明中,所述形貌调控材料在碲硒纳米材料的制备过程中可以起到二维结构引导剂、晶面阻断剂的作用,可以控制形成晶型稳定剂的特定形貌的二维碲硒纳米片,其包覆还可以提高二维碲硒纳米片的生物相容性和稳定性,提高其在水系体系(如去离子水、生理盐水、磷酸盐等缓冲液、血清、二甲基亚砜水溶液)中的分散性、稳定性。
进一步可选地,所述二维碲硒纳米片和所述形貌调控材料的质量比为1:(0.2-20)。优选为1:(2-10)。
其中,所述形貌调控材料包括聚乙烯吡咯烷酮(pvp)、十六烷基三甲基溴化铵、十二烷基苯磺酸、聚苯乙烯磺酸钠、聚乙二醇胺、聚乳酸-羟基乙酸共聚物、聚乙烯亚胺、聚丙烯酸和聚乙二醇及其衍生物中的一种或多种。
对于所述聚乙二醇胺,可列举甲基聚乙二醇胺(ch3-peg-nh2)、甲氧基聚乙二醇胺(ch3o-peg-nh2,简称为mpeg-nh2)和聚乙二醇二胺(nh2-peg-nh2)中的至少一种。对于所述聚乙二醇衍生物,可列举氨基化聚乙二醇、酯化聚乙二醇、羧基化聚乙二醇、醛基化聚乙二醇和聚乙二醇-聚氨基酸共聚物中的至少一种。
在本发明一实施方式中,所述形貌调控材料为聚乙烯吡咯烷酮(pvp)。其中,所述pvp的重均分子量为10000-50000。例如为20000或40000。
可选地,所述形貌调控材料通过静电作用吸附在所述二维碲硒纳米片的表面。
可选地,所述形貌调控材料上还通过化学键连接有靶向材料。这样可增加所述碲硒纳米材料在进行生物应用时的靶向性。具体地,当所述靶向材料为叶酸,其可通过酰胺键与聚乙二醇胺、氨基化的聚乙二醇等实现连接。
本发明第一方面提供的所述碲硒纳米材料,其光热效果明显,光热转换效率高;化学稳定性高,毒性低、生物相容性良好,在光热治疗、光动力治疗、光声成像、载药治疗领域具有广阔的应用前景。
第二方面,本发明提供了一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将碲源、硒源和形貌调控材料加入到溶剂中,得到第一混合液,并调节所述第一混合液的ph至8~10;其中,所述碲源中的碲元素与所述硒源中的硒元素的摩尔比为1:x,x的取值范围为0<x≤10;
(2)将调节ph后的第一混合液置于反应釜中,加入还原剂,得到第二混合液,密封,在160-200℃下反应8-30小时,冷却,得到反应液;
(3)对所述反应液进行固液分离,收集沉淀,所得沉淀即碲硒纳米材料。
其中,步骤(1)中,所述硒源选自亚硒酸钠(na2seo3)、亚硒酸钾(k2seo3)、亚硒酸铵((nh4)2seo3)、亚硒酸氢钠(nahseo3)、亚硒酸氢钾(khseo3)和亚硒酸(h2seo3)中的一种或多种。
所述碲源选自亚碲酸钠(na2teo3)、亚碲酸钾(k2teo3)、亚碲酸(h2teo3)、原碲酸(h6te6o6)、碲酸钾(k2teo4)和二氧化碲(teo2)中的一种或多种。
本发明中,所述形貌调控材料主要用于调控形成型稳定剂的特定形貌的二维碲硒纳米片,并提高其生物相容性和稳定性。可选地,所述形貌调控材料的质量与所述碲源中的碲元素和所述硒源中的硒元素的摩尔之和的比值为(50-1500)g:1mol。
其中,步骤(1)中,调节所述第一混合液的ph时,采用氨水、naoh、koh中的一种或多种进行。
步骤(1)中,所述溶剂为水、乙醇、乙二醇中的一种或多种,但不限于此。
其中,步骤(2)中,所述还原剂为水合肼(n2h4·h2o)、肼(n2h4)、维生素c和亚硫酸钠中的一种或多种。所述还原剂可以以固体形式或其溶液形式加入。
可选地,所述碲源中的碲元素与所述硒源中的硒元素的摩尔之和与所述还原剂的摩尔之比为1:(20-200)。优选为1:(20-50)。
其中,步骤(3)中,所述固液分离具体包括以下操作:向所述反应液中加入水与极性有机溶剂的混合溶剂,进行离心分离。具体地,所述极性有机溶剂为丙酮、正丁醇、异丙醇、四甲基乙二胺中的一种或多种,但不限于此。
本申请步骤(3)中,所述沉淀为初产物,可进一步包括如下的纯化操作:将所述沉淀采用水洗涤多次,并置于去离子水中透析1-7天,冷冻干燥后,得到纯化的碲硒纳米材料。
本发明第二方面提供的碲硒纳米材料的制备方法,原料易得,制备过程简单,易于实现规模化生产。
第三方面,本发明提供了如第一方面所述的碲硒纳米材料或者如第二方面所述的制备方法制得的碲硒纳米材料在制备光声成像药物、光热治疗药物、光动力治疗药物或载药靶向治疗药物中的应用。
本发明提供的碲硒纳米材料的光热效果明显,具有很好的生物相容性,无急性和长期的生物毒性,可以应用于生物医学领域。
例如,具体地,本发明提供了一种纳米光热制剂,所述纳米光热制剂包括二维碲硒纳米片,以及包覆在所述二维碲硒纳米片表面的形貌调控材料,所述二维碲硒纳米片的化学通式为tesex,其中,x为se与te的摩尔比,x的取值范围为0<x≤10。
本发明的优点将会在下面的说明书中部分阐明,一部分根据说明书是显而易见的,或者可以通过本发明实施例的实施而获知。
附图说明
图1为本发明实施例1-4制得的碲硒纳米材料的透射电镜图;
图2为本发明实施例4制得的碲硒纳米材料的高分辨电子显微镜图,其中图2中(2)、(3)分别为(1)中不同灰度区域的放大图;
图3为本发明实施例1-4制得的碲硒纳米材料的原子力显微镜图;
图4为本发明实施例1-4制得的碲硒纳米材料的拉曼曲线图;
图5为本发明实施例1-4制得的碲硒纳米材料的x射线衍射图;
图6为本发明实施例1-4制得的碲硒纳米材料与对比例1的单纯碲纳米材料的稳定性比较结果,其中(a)为单纯碲纳米材料在放置不同天数时的状态,(b)为本发明实施例1-4的碲硒纳米材料在放置6天时的状态,(c)为实施例4的碲硒纳米材料在放置不同天数时的状态;
图7为本发明的碲硒纳米材料的光热效应随se元素摩尔比变化的曲线图;
图8为本发明实施例的碲硒纳米材料的生物毒性测试结果图;
图9为本发明实施例4的碲硒纳米材料对肿瘤细胞的体外光热杀伤能力测试结果图;
图10为本发明实施例4的碲硒纳米材料对肿瘤组织的体内光热治疗结果图。
具体实施方式
以下所述是本发明实施例的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明实施例原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明实施例的保护范围。
实施例1
一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将0.45mmol的亚碲酸钠(na2teo3)和0.1125mmol的亚硒酸钠(na2seo3)混合,并溶解于双蒸水中,其中te:se元素的摩尔比为1:0.25;
将400mg的聚乙烯吡咯烷酮(pvp,重均分子量为40000)作为形貌调控剂,溶解于双蒸水,并将其加入到上述亚碲酸钠和亚硒酸钠的混合水溶液中,通过磁力搅拌器搅拌均匀,得到总体积为33ml的均一的第一混合液;然后加入浓氨水调节所述使第一混合液的ph至ph为9.4;
(2)将调节ph后的第一混合溶液置于聚四氟乙烯(pfte)基内胆反应釜内(总容量为60ml),加入水合肼(25wt/%)的水溶液至反应釜内的液体总体积为50ml(其中,水合肼的摩尔数为27mmol),密封,在180℃下反应24小时,之后使其自然冷却,得到反应液;
(3)向所得反应液中加入丙酮与水的混合溶剂,以6000rpm转速离心10分钟,分离出沉淀,并通过去离子水反复洗涤所得沉淀,以除去多余的pvp高分子和绝大多数的无机盐,所得黑褐色沉淀即为碲硒纳米材料;
(4)然后将上述沉淀置于去离子水中透析7天,得到纯化的碲硒纳米材料的水溶液,并通过冷冻干燥的方式得到其固体形式。本发明实施例1制备得到的碲硒纳米材料为表面包覆有pvp的二维碲硒纳米片。
实施例2
一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将0.45mmol的亚碲酸钠(na2teo3)和0.225mmol的亚硒酸钠(na2seo3)混合,并溶解于双蒸水中,其中te:se元素的摩尔比为1:0.5;
将400mg的聚乙烯吡咯烷酮(pvp,重均分子量为40000)作为形貌调控剂,溶解于双蒸水,并将其加入到上述亚碲酸钠和亚硒酸钠的混合水溶液中,通过磁力搅拌器搅拌均匀,得到总体积为33ml的均一的第一混合液;然后加入浓氨水调节所述使第一混合液的ph至ph为9.4;
(2)将调节ph后的第一混合溶液置于聚四氟乙烯(pfte)基内胆反应釜内(总容量为60ml),加入水合肼(25wt/%)的水溶液至反应釜内的液体总体积为50ml(其中,水合肼的摩尔数为27mmol),密封,在180℃下反应24小时,之后使其自然冷却,得到反应液;
(3)向所得反应液中加入丙酮与水的混合溶剂,离心分离出沉淀,并通过去离子水反复洗涤所得沉淀,以除去多余的pvp高分子和绝大多数的无机盐,所得黑褐色沉淀即为碲硒纳米材料;
(4)然后将上述沉淀置于去离子水中透析7天,得到纯化的碲硒纳米材料的水溶液,并通过冷冻干燥的方式得到其固体形式。本发明实施例2制备得到的碲硒纳米材料为表面包覆有pvp的二维碲硒纳米片。
实施例3
一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将0.45mmol的亚碲酸钠(na2teo3)和0.3375mmol的亚硒酸钠(na2seo3)混合,并溶解于双蒸水中,其中te:se元素的摩尔比为1:0.75;
将400mg的聚乙烯吡咯烷酮(pvp,重均分子量为40000)作为形貌调控剂,溶解于双蒸水,并将其加入到上述亚碲酸钠和亚硒酸钠的混合水溶液中,通过磁力搅拌器搅拌均匀,得到总体积为33ml的均一的第一混合液;然后加入浓氨水调节所述使第一混合液的ph至ph为9.4;
(2)将调节ph后的第一混合溶液置于聚四氟乙烯(pfte)基内胆反应釜内(总容量为60ml),加入水合肼(25wt/%)的水溶液至反应釜内的液体总体积为50ml(其中,水合肼的摩尔数为27mmol),密封,在180℃下反应24小时,之后使其自然冷却,得到反应液;
(3)向所得反应液中加入丙酮与水的混合溶剂,离心分离出沉淀,并通过去离子水反复洗涤所得沉淀,以除去多余的pvp高分子和绝大多数的无机盐,所得黑褐色沉淀即为碲硒纳米材料;
(4)然后将上述沉淀置于去离子水中透析7天,得到纯化的碲硒纳米材料的水溶液,并通过冷冻干燥的方式得到其固体形式。本发明实施例3制备得到的碲硒纳米材料为表面包覆有pvp的二维碲硒纳米片。
实施例4
一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将0.45mmol的亚碲酸钠(na2teo3)和0.45mmol的亚硒酸钠(na2seo3)混合,并溶解于双蒸水中,其中te:se元素的摩尔比为1:1;
将400mg的聚乙烯吡咯烷酮(pvp,重均分子量为40000)作为形貌调控剂,溶解于双蒸水,并将其加入到上述亚碲酸钠和亚硒酸钠的混合水溶液中,通过磁力搅拌器搅拌均匀,得到总体积为33ml的均一的第一混合液;然后加入浓氨水调节所述使第一混合液的ph至ph为9.4;
(2)将调节ph后的第一混合溶液置于聚四氟乙烯(pfte)基内胆反应釜内(总容量为60ml),加入水合肼(25wt/%)的水溶液至反应釜内的液体总体积为50ml(其中,水合肼的摩尔数为27mmol),密封,在180℃下反应24小时,之后使其自然冷却,得到反应液;
(3)向所得反应液中加入丙酮与水的混合溶剂,离心分离出沉淀,并通过去离子水反复洗涤所得沉淀,以除去多余的pvp高分子和绝大多数的无机盐,所得黑褐色沉淀即为碲硒纳米材料;
(4)然后将上述沉淀置于去离子水中透析7天,得到纯化的碲硒纳米材料的水溶液,并通过冷冻干燥的方式得到其固体形式。本发明实施例4制备得到的碲硒纳米材料为表面包覆有pvp的二维碲硒纳米片。
实施例5
一种碲硒纳米材料的制备方法,包括以下步骤:
(1)将0.4mmol的亚碲酸(h2teo3)和0.6mmol的亚硒酸氢钠(nahseo3)混合,并溶解于双蒸水中,其中te:se元素的摩尔比为1:1.5;
将500mg的十六烷基三甲基溴化铵(ctab)作为形貌调控剂,溶解于双蒸水,并将其加入到上述亚碲酸和亚硒酸氢钠的混合水溶液中,通过磁力搅拌器搅拌均匀,得到总体积为45ml的均一的第一混合液;然后加入浓氨水调节所述使第一混合液的ph至ph为10;
(2)将调节ph后的第一混合溶液置于聚四氟乙烯基内胆反应釜内(总容量为60ml),加入50mmol的亚硫酸钠至反应釜内的液体总体积为50ml,密封,在170℃下反应12小时,之后使其自然冷却,得到反应液;
(3)向所得反应液中加入丙酮与水的混合溶剂,以5000rpm转速离心15分钟,分离出沉淀,并通过去离子水反复洗涤所得沉淀,以除去多余的pvp高分子和绝大多数的无机盐,所得黑褐色沉淀即为碲硒纳米材料;
(4)然后将上述沉淀置于去离子水中透析5天,得到纯化的碲硒纳米材料的水溶液,并通过冷冻干燥的方式得到其固体形式。本发明实施例5制备得到的碲硒纳米材料为表面包覆有ctab的二维碲硒纳米片。
对比例1
制备单纯的碲纳米材料,其与实施例4的区别在于:步骤(1)中,未加入亚硒酸钠。
对以上实施例1-4的各样品进行形貌分析,具体如下:
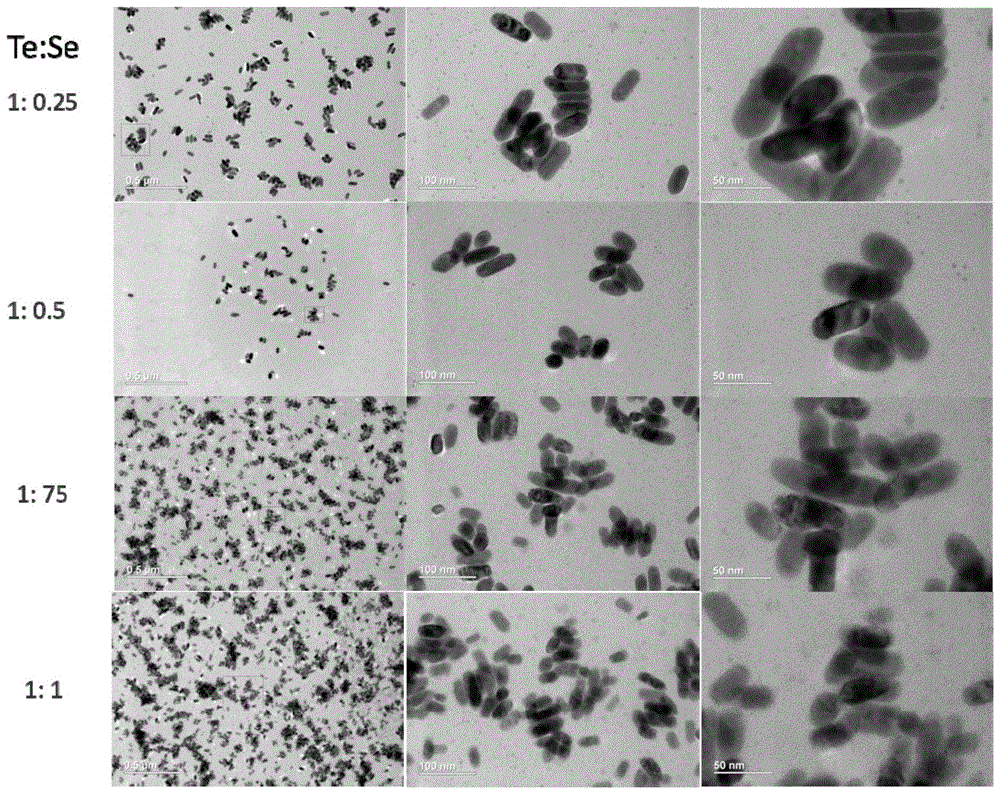
图1为本发明实施例1-4制备得到的碲硒纳米材料的透射电镜图,其中各行从左到右的刻度尺分别是500nm、100nm、50纳米。从图1中可以获知,实施例1所得碲硒纳米材料的长度约为100nm,宽度约为60nm,厚度约为32nm。实施例2所得碲硒纳米材料的长度约为85nm,宽度约为40nm,厚度约为25nm。实施例3所得碲硒纳米材料的长度约为75nm,宽度为40nm,厚度约为25nm。实施例4所得碲硒纳米材料的长度约为72nm,宽度约为38nm,厚度约为20nm。可见,对于某一特定te:se比例的实施例,所制得的样品的尺寸均一,形貌相似;当te:se比例从1:0.25变到1:1,碲硒纳米材料的尺寸越来越小。
以本发明实施例4制得的碲硒纳米材料为例,对其进行高分辨电子显微镜表征,结果如图2所示。图2中(2)对应(1)中灰度较深的pvp的放大;(3)对应(1)中灰度较浅的二维碲硒纳米片的放大。由图2中(3)可以获知,二维碲硒纳米片沿着<0001>和<1210>侧向生长,且沿着<1010>方向垂直堆积,这进一步验证了其为二维纳米片状,而非纳米棒。
图3为本发明实施例1-4制备得到的碲硒纳米材料的afm图,其中,从左到右的四列图分别对应te:se比例为1:0.25、1:0.5、1:0.75、1:1的实施例;第二行图为第一行图中划线区域对应的厚度分布。从图3中可看出,当te:se比例为1:0.25时,其长度、厚度分别约为105、30nm;当te:se=1:0.5时,其长度、厚度分别约为90、25nm;当te:se=1:0.75时,其长度、厚度分别约为75、25nm;以及当te:se=1:1时,其长度、厚度分别约为72、22nm,且对于同一te:se比例的碲硒纳米材料,其尺寸分布均匀。
图4为本发明实施例1-4制得的碲硒纳米材料的拉曼曲线图,并与商品化碲块体、对比例1中的单纯碲纳米材料进行对比。由图4可知,本发明提供的碲硒纳米材料在100-150cm-1区间呈现出与单纯碲纳米材料及碲块体截然不同的谱峰,该区域的特征峰表明本发明提供的制备方法成功得到了新型碲硒纳米材料。
图5为本发明实施例1-4制得的碲硒纳米材料的x射线衍射图,并与商用碲块体、商品化硒块体进行对比。由图5可以看出,本发明实施例1-4提供的碲硒纳米材料呈现出te元素和se元素的特征衍射峰,这说明本发明成功得到含te、se的新型材料。
效果实施例
为对本发明技术方案带来的有益效果进行有力支持,特提供以下性能测试:
(1)稳定性的考察:
将本发明实施例1-4制得的碲硒纳米材料,以及对比例1的单纯碲纳米材料分别分散在生理盐水中,刚配置好后的分散液呈黑褐色,材料的浓度为250ppm。将它们分别与生理盐水进行比较,考察放置不同天数后的状态,结果如图10所示。其中,图6中(a)为单纯碲纳米材料在放置不同天数时的状态,(b)为本发明实施例1-4的碲硒纳米材料在放置6天时的状态,(c)为实施例4的碲硒纳米材料在放置不同天数时的状态。
由图6中(a)可知,对比例1的单纯碲纳米材料在放置第3天即发生显著降解(分散液的颜色变淡),放置第6天时已接近生理盐水。而本发明实施例1-4的碲硒纳米材料在放置6天时的稳定性仍较好(参见图6中(b))。以实施例4的碲硒纳米材料为代表,由图6中(c)可知,即使在放置第30天时,仍没有显著降解,说明其稳定性很好。以上结果表明,本发明提供的碲硒纳米材料在溶液中的稳定性极好,为其进行光热治疗、光动力治疗、光声成像等应用奠定了基础。
(2)光热效应的测定
将本发明制得的一系列不同te:se比例的碲硒纳米材料分散于水中,得到浓度为0.1mg/ml的分散液,将分散液装入比色皿中,采用功率密度为1.0w/cm2、波长为808nm的激光垂直照射比色皿5min,采用红外测温仪测量分散液的平衡温度,结果如图7所示。
由图7可以看出,随着se元素的摩尔比(即se与se+te的摩尔数之比)的逐渐升高,本发明实施例提供的碲硒纳米材料的光热效应逐渐降低,但是总体而言,当se与te元素的摩尔比不超过50%时,本发明实施例提供的碲硒纳米材料始终表现出较高的光热效应。
此外,以实施例4的碲硒纳米材料(te:se=1:1)为例,本发明还定量测定其光热转化效率可达50%。
(3)生物毒性测试
将人肝癌smmc-7721细胞接种于96孔板,用rpmi1640培养基贴壁培养约12小时后,将rpmi1640培养基分别置换为分散有实施例1-4制得的碲硒纳米材料,以及对比例1单纯碲纳米材料的rpmi1640培养基(其中各处理组的孔内所加材料的分散浓度如下图7所示),再培养36小时,之后弃去各孔内的培养基,并用适量的磷酸盐缓冲液冲洗细胞,之后再用cck8试剂盒测定细胞的活力,结果如图8所示。
从图8可以看出,本发明实施例提供的碲硒纳米材料从低浓度的10ppm至高浓度的400ppm均没有对smmc-7721细胞产生毒性,这说明本发明实施例提供的碲硒纳米材料的生物毒性较低,为其进行光热治疗、光动力治疗、光声成像等应用奠定了基础。
(4)对肿瘤的体外光热杀伤能力测试
将人肝癌smmc-7721细胞接种于96孔板,随后采用本发明提供的碲硒纳米材料(以te:se=1:1的实施例4为代表),对肿瘤细胞进行以下三个维度的光热杀伤能力的测试:
a.浓度梯度:采用含不同分散浓度的碲硒纳米材料(te:se=1:1)的rpmi1640培养基(浓度分别为0、50ppm、100ppm、200ppm)孵育细胞4小时,然后固定激光光照功率(波长为808nm,功率密度为1w/cm2),进行激光照射10分钟。待激光照射结束后,将各孔内的培养基置换为新鲜无碲硒纳米片的rpmi1640培养基,再孵育6小时,用calceinam/pi法测定细胞的存活率。其中,还以一直用单纯的rpmi1640培养基孵育且无激光照射的细胞进行对照。
b.光照时间梯度:固定激光光照功率(波长为808nm,功率密度为1w/cm2)以及固定碲硒纳米材料的浓度为50ppm,按与上述a中类似的方法测定不同激光照射时间(照射时间分别为0、2min、5min、10min)对癌细胞的光热杀伤效率。
c.功率梯度:固定碲硒纳米材料的浓度为50ppm以及固定激光照射的波长为808nm、照射时间为10分钟,按上述方法测定不同激光照射功率(功率分别为0、0.5w/cm2、1.0w/cm2、1.5w/cm2)对癌细胞的光热杀伤效率。
以上测试的结果如图9所示,其中,图中绿色荧光代表活细胞,红色荧光代表死细胞(以图9中第2、3行中第3列为例,灰度较浅(黑白图呈白色)的为活细胞)。从图9中可以获知,未加入含本发明实施例碲硒纳米材料的培养基,其癌细胞无死亡,而加入本发明实施例碲硒纳米材料的培养基,其中癌细胞有不同程度的死亡,且随着培养基中碲硒纳米材料浓度、激光照射时间或激光照射功率的增大,癌细胞的死亡率升高,因此说明本发明实施例提供的碲硒纳米材料对体外癌细胞具有较高的光热杀伤效率高。
(5)体内水平的肿瘤光热治疗
在balb/c小鼠的后肢右侧背部进行皮下植瘤(人肺癌a549细胞的植入量为5×106细胞/只小鼠),大约两周后肿瘤的体积生长至75-200立方毫米,然后开始进行光热杀伤实验:将本发明提供的碲硒纳米材料(以te:se=1:1的实施例4为代表)分散于100μl生理盐水中,通过尾静脉注射到荷瘤小鼠体内(注射剂量为2mg/kg小鼠)。注射4小时后,将小鼠麻醉,并进行光热治疗(激光功率密度为1w/cm2,波长为808nm,照射时间为10分钟),设置治疗的当天为第0天,治疗后持续观察9周,并测量肿瘤的尺寸,计算肿瘤的体积。同时设置阳性对照组(向另外的荷瘤小鼠体内注射等量的pvp溶液,并进行相同的激光照射),以及阴性对照组(向另外的荷瘤小鼠体内仅注射单纯的生理盐水,不进行激光照射),结果如图10所示。
从图10中可以获知,经过本发明的碲硒纳米材料光热治疗后,荷瘤小鼠的肿瘤体积相对于两个对照组显著减小。这说明本发明的碲硒纳米材料可以用于肿瘤的高效光热治疗。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!