用于检测罕见序列变体的组合物和方法与流程
本申请要求在2013年12月11日提交的美国临时申请61/914,907、在2014年5月1日提交的美国临时申请61/987,414和在2014年6月11日提交的美国临时申请62/010,975的权益;上述所有美国临时申请均通过引用并入本文。
背景技术:
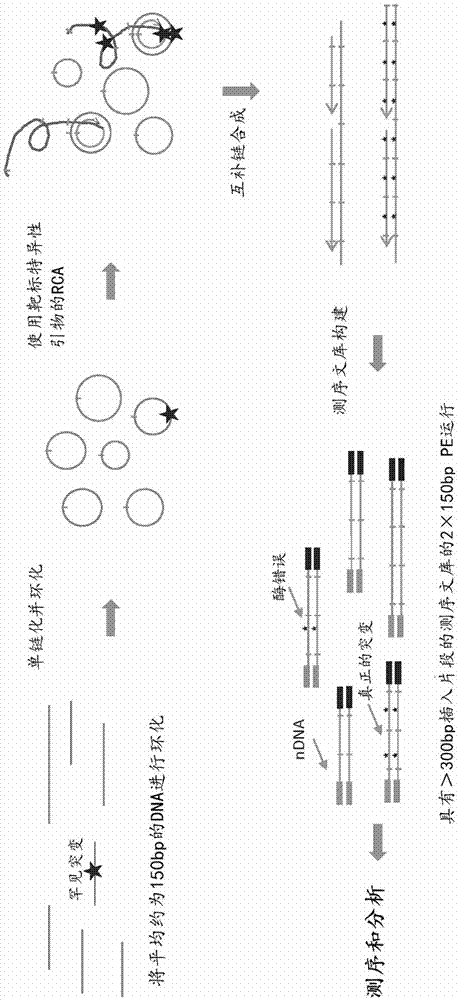
:鉴定复杂群体内的序列变异是一个活跃发展的领域,特别是随着大规模平行核酸测序的出现。然而,由于常用技术的固有误差频率比群体内许多实际序列变异的频率更大,大规模平行测序具有显著的局限性。例如,0.1-1%的误差率已在标准的高通量测序中被报道。当变体频率低,如等于或低于误差率时,对罕见序列变体的检测具有高的假阳性率。检测罕见序列变体的原因有很多。例如,检测罕见特征性序列可用于鉴定和区分有害环境污染物如细菌分类群的存在。表征细菌分类群的常见方式是鉴定高度保守序列如rRNA序列的差异。然而,针对此的典型的基于测序的方法面临与给定样品中如此多数量的不同基因组和成员之间的同源性程度相关的挑战,从而为本已繁琐的程序呈现出复杂的问题。改进的程序将具有加强在多种设置下的污染检测的潜力。例如,用于组装卫星和其他空间飞行器的部件的洁净室可使用本系统和方法进行勘测,以了解存在何种微生物群落,并且开发更好的去污染和清洁技术,从而防止将地球微生物引入其他星球或其样品,或开发用来将由推定的地球外微生物产生的数据与由污染性地球微生物产生的数据进行区分的方法。食品监测应用包括对食品加工厂生产线的定期检测,调查屠宰场,检查餐厅、医院、学校、监狱和其他机构的厨房和食品储存区的食源性病原体。也可以类似地监测水源储备和加工厂。技术实现要素:鉴于以上所述,对改进的检测罕见序列变体的方法存在需求。本发明的组合物和方法满足了该需求,并且还提供了另外的益处。特别是,本发明的各个方面提供了对罕见或低频核酸序列变体(有时称为突变)的高度灵敏的检测。这包括对在正常序列背景中可能含有少量变异序列的样品中的低频核酸变异(包括取代、插入和缺失)的鉴定和阐明,以及对在测序错误背景下的低频变异的鉴定。在一个方面,本发明提供了一种鉴定序列变体,例如核酸样品中的序列变体的方法。在一些实施方案中,多个多核苷酸中的每个多核苷酸具有5’末端和3’末端,并且该方法包括:(a)将所述多个多核苷酸中的单独多核苷酸进行环化以形成多个环状多核苷酸,其中每个环状多核苷酸在5’末端与3’末端之间具有接点(junction);(b)扩增(a)的环状多核苷酸;(c)对扩增的多核苷酸进行测序以生成多个测序读取;(d)鉴定测序读取与参考序列之间的序列差异;和(e)将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定(calling)为序列变体。在一些实施方案中,该方法包括鉴定测序读取与参考序列之间的序列差异,以及将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体,其中:(a)该测序读取对应于该至少两个环状多核苷酸的扩增产物;且(b)该至少两个环状多核苷酸中的每一个包含通过连接相应多核苷酸的5’末端和3’末端而形成的不同的接点。所述多个多核苷酸可以是单链的或双链的。在一些实施方案中,该多核苷酸是单链的。在一些实施方案中,环化是通过对多个多核苷酸进行连接反应而实现的。在一些实施方案中,单独的环状多核苷酸具有在环化的多核苷酸中独特的接点。在一些实施方案中,该序列变体是单核苷酸多态性(SNP)。在一些实施方案中,该参考序列是通过将序列读取彼此进行比对而形成的共有序列。在一些实施方案中,该参考序列是已知的参考序列,例如参考基因组或其部分。在一些实施方案中,环化包括将衔接子多核苷酸连接到多个多核苷酸中的多核苷酸的5’末端、3’末端或5’末端和3’末端两者的步骤。在一些实施方案中,扩增通过使用具有链置换活性的聚合酶而实现,例如在滚环扩增(RCA)中。在一些实施方案中,扩增包括将环状多核苷酸置于含有随机引物的扩增反应混合物中。在一些实施方案中,扩增包括将环状多核苷酸置于含有一种或多种引物的扩增反应混合物中,其中每一种引物通过序列互补性与不同靶序列特异性地杂交。在一些实施方案中,基于判定步骤鉴定微生物污染物。可以在进行或不进行富集的情况下对扩增的多核苷酸进行测序,例如通过在测序之前进行富集步骤而在扩增的多核苷酸中富集一种或多种靶多核苷酸。在一些实施方案中,该富集步骤包括使扩增的多核苷酸与多个与基底附接的探针进行杂交。在一些实施方案中,该富集步骤包括在扩增反应混合物中扩增包含以5’到3’方向取向的序列A和序列B的靶序列,该扩增反应混合物包含:(a)扩增的多核苷酸;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以产生扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。在一个方面,本发明提供了一种鉴定核酸样品中的序列变体的方法,该核酸样品包含少于50ng的多核苷酸,每一个多核苷酸具有5’末端和3’末端。在一些实施方案中,该方法包括:(a)用连接酶环化所述样品中的单独的多核苷酸以形成多个环状多核苷酸;(b)一旦将所述环状多核苷酸与所述连接酶分离,即扩增该环状多核苷酸以形成多联体(concatemer);(c)对该多联体进行测序以生成多个测序读取;(d)鉴定该多个测序读取与参考序列之间的序列差异;和(e)将从所述少于50ng多核酸的核酸样品获得的所述多个读取中以0.05%或更高的频率发生的序列差异判定为序列变体。该多核苷酸可以是单链的或双链的。在一些实施方案中,该多核苷酸是单链的。在一些实施方案中,单独的环状多核苷酸具有在环化的多核苷酸中独特的接点。在一些实施方案中,该序列变体是单核苷酸多态性。在一些实施方案中,该参考序列是通过将测序读取彼此进行比对而形成的共有序列。在一些实施方案中,该参考序列是已知的参考序列,例如参考基因组。在一些实施方案中,扩增通过使用具有链置换活性的聚合酶而实现。在一些实施方案中,扩增包括将环状多核苷酸置于含有随机引物的扩增反应混合物中。在一些实施方案中,扩增包括将环状多核苷酸置于含有一种或多种引物的扩增反应混合物中,其中每一种引物通过序列互补性与不同靶序列特异性地杂交。在一个方面,本发明提供了一种在反应混合物中扩增多个不同多联体的方法,该多联体包含靶序列的两个或更多个拷贝,其中该靶序列包含以5’到3’方向取向的序列A和序列B。在一些实施方案中,该方法包括对反应混合物进行核酸扩增反应,其中该反应混合物包含:(a)多个多联体,其中该多个多联体中单独的多联体包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同的接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过序列B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以产生扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。在一些实施方案中,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在第一杂交温度下的第一扩增阶段期间均不与所述多个多联体杂交。在一些实施方案中,扩增包括第一阶段和第二阶段;第一阶段包括在第一温度下的杂交步骤,其间第一和第二引物在引物延伸之前与该多联体杂交;而第二阶段包括在高于第一温度的第二温度下的杂交步骤,其间第一和第二引物与包含延伸的第一或第二引物或其互补体的扩增产物杂交。在一些实施方案中,在第二温度下的5个杂交循环和引物延伸之后,反应混合物中至少5%的扩增多核苷酸包含靶序列的两个或更多个拷贝。在一个相关方面中,本发明提供了一种在反应混合物中扩增多个包含靶序列的不同环状多核苷酸的方法,其中该靶序列包含以5’到3’方向取向的序列A和序列B。在一些实施方案中,该方法包括对反应混合物进行核酸扩增反应,其中该反应混合物包含:(a)多个环状多核苷酸,其中该多个环状多核苷酸中单独的环状多核苷酸包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同的接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过序列B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以产生扩增的多核苷酸;其中序列A和序列B为内源性序列,并且靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。在一些实施方案中,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在第一杂交温度下的第一扩增阶段期间均不与所述多个环状多核苷酸杂交。在一些实施方案中,扩增包括第一阶段和第二阶段;第一阶段包括在第一温度下的杂交步骤,其间第一和第二引物在引物延伸之前与该环状多核苷酸或其扩增产物杂交;第二阶段包括在高于第一温度的第二温度下的杂交步骤,其间第一和第二引物与包含延伸的第一或第二引物或其互补体的扩增产物杂交。在一个方面,本发明提供了一种用于进行本发明的方法的反应混合物。该反应混合物可以包含如本文中关于多种方法中的任一种所描述的各种组分中的一种或多种。在一些实施方案中,该反应混合物是用于扩增多个包含靶序列的两个或更多个拷贝的不同多联体的混合物,其中该靶序列包含以5’到3’方向取向的序列A和序列B,该反应混合物包含:(a)多个多联体,其中该多个多联体中单独的多联体包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同的接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过B与B’之间的序列互补性特异地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以产生扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。在一些实施方案中,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在扩增反应的第一扩增步骤期间均不与所述两个或更多个多联体杂交。在一个方面,本发明提供了可用于本文所述的方法(例如在本发明的各个其他方面中的任意方面中所述的方法)中或由该方法产生的组合物。在一些实施方案中,该组合物包含多个为单链的环化多核苷酸,且基本上不含连接酶。在一些实施方案中,该组合物包含多个多联体,其中该多个多联体对应于一组10000个或更少的靶多核苷酸,并且进一步地,其中该多个多联体中的单独多联体的特征在于:(a)它们包含序列重复(sequencerepeat)的两个或更多个拷贝,其中所有的所述拷贝均对应于相同的靶多核苷酸;且(b)一个单独的多联体中序列重复的两个或更多个拷贝之间的接点与所述组合物中的另一个单独多联体中的不同。在一个方面,本发明提供了一种用于检测序列变体的系统。在一些实施方案中,该系统包含(a)计算机,其被配置为接收对样品进行检测反应的用户请求;(b)扩增系统,其响应于用户请求对样品或其一部分进行核酸扩增反应,其中该扩增反应包括以下步骤:(i)将单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸都在5’末端与3’末端之间具有接点;和(ii)扩增该环状多核苷酸;(c)测序系统,其针对由该扩增系统扩增的多核苷酸生成测序读取,鉴定测序读取与参考序列之间的序列差异,并且将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体;和(d)向接收方发送报告的报告生成器,其中该报告包含关于序列变体检测的结果。在一些实施方案中,该接收方为用户。在一个方面,本发明提供了一种包含代码的计算机可读介质,该代码一旦由一个或多个处理器执行,即实施检测序列变体的方法。在一些实施方案中,该实施的方法包括:(a)接收对样品进行检测反应的客户请求;(b)响应于客户请求对样品或其一部分进行核酸扩增反应,其中该扩增反应包括以下步骤:(i)将单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸都在5’末端与3’末端之间具有接点;和(ii)扩增该环状多核苷酸;(c)进行测序分析,其包括以下步骤:(i)针对扩增反应中扩增的多核苷酸生成测序读取;(ii)鉴定测序读取与参考序列之间的序列差异;以及(iii)将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体;和(d)生成包含关于序列变体检测结果的报告。援引并入本说明书中提及的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别地和单独地表明每个单独的出版物、专利或专利申请通过引用而被并入。附图说明本发明的新颖特征具体示于所附的权利要求书中。通过参考以下对利用了本发明原理的说明性实施方案加以阐述的详细描述及其附图,将会获得对本发明的特征和优点的更好的了解,在附图中:图1描绘了根据本发明的方法的一个实施方案的示意图。将DNA链进行环化,并添加与所研究的基因相对应的靶标特异性引物,以及聚合酶、dNTP、缓冲液等,以使得发生滚环扩增(RCA)从而形成模板DNA(例如,“单体”)的多联体(例如,“多聚体”)。处理该多联体以合成相应的互补链,然后添加衔接子(adapter)以制备测序文库。该生成的文库(随后使用标准技术对其进行测序)通常包含三个种类:不包含罕见序列变体(例如,突变)的nDNA(“正常”DNA);包含酶促测序错误的nDNA;和包含在扩增之前已经存在于样品多核苷酸中的“真正”或实际序列变体的多聚体的DNA。有效罕见突变的多个拷贝的存在使得能够检测和鉴定序列变体。图2描绘了与图1相似的策略,但是添加衔接子以促进多核苷酸的环化。图2还显示了靶标特异性引物的使用。图3与图2相似,除了在扩增中使用了衔接子引物之外。图4描绘了与环化ccDNA的形成相关的三个实施方案。在顶端,在不存在衔接子的情况下对单链DNA(ssDNA)进行环化,中间的方案描绘了使用衔接子,底部的方案使用了两个衔接子寡聚物(在每一端产生不同的序列),并且可进一步包括与两个衔接子都杂交以使两个末端邻近的夹板寡聚体(splintoligo)。图5描绘了通过使用“分子钳”对特定靶标进行环化以使得单链DNA的两个末端在空间上邻近以用于连接的实施方案。图6A和6B描绘了利用核酸的封闭末端添加衔接子的两个方案。图7A、7B和7C描绘了用于引发滚环扩增(RC)反应的三种不同方式。图7A显示了使用靶标特异性引物,例如,特定的目标靶基因或靶序列。这通常使得只有靶序列被扩增。图7B描述了使用随机引物,该随机引物通常扩增所有的样品序列,然后在处理过程中通过生物信息学方式分选该样品序列。图7C描述了当使用衔接子时衔接子引物的使用,这通常也产生非靶标特异性扩增。图8描绘了根据一个实施方案,双链DNA环化和扩增以使得两条链都得到扩增的实例。图9A、9B、9C和9D描绘了用以实现互补链合成以供后续测序的多种方案。图9A描述了采用靶链的随机引发,随后进行连接。图9B描述了采用靶链的衔接子引发,类似地继以连接。图9C描述了“环(loop)”衔接子的使用,其中该衔接子具有两个互补的序列部分,以使得它们相互杂交以产生环(例如,茎环结构)。一旦与多联体的末端连接,该环的游离端即作为互补链的引物。图9D显示了使用超分支随机引物以实现第二链合成。图10显示了根据促进对包含至少两个拷贝的靶核酸序列的环状多核苷酸或链的测序的实施方案的PCR方法,其中使用当在靶序列的单体中匹配(aligned)时彼此远离地定向的一对引物(也称为“背靠背(backtoback)”,例如,在两个方向上定向但不位于待扩增区域的末端)。在一些实施方案中,在多联体形成后使用这些引物组,以促进扩增子成为靶序列的更多聚体,例如,二聚体、三聚体等。任选地,该方法可进一步包括大小选择以去除比二聚体更小的扩增子。图11描绘了一个实施方案,其中使用背靠背(B2B)引物和“递升(touchup)”PCR步骤,以使得不太有利于短产物(例如单体)的扩增。在这种情况下,引物具有两个结构域:与靶序列杂交的第一结构域(灰色或黑色箭头),和不与原始靶序列杂交的、为“通用引物”结合域的第二结构域(弯曲矩形;有时也称为衔接子)。在一些实施方案中,采用低温退火步骤进行第一轮PCR,以使得基因特异性序列结合。该低温运行产生各种长度的PCR产物,包括短产物。在少数几轮后,提高退火温度,以使得有利于整个引物、两个结构域的杂交;如图所示,这在模板的末端处被发现,而内部结合较不稳定。因此与在较低的温度下或仅有一个结构域时相比,当在较高的温度下且具有两个结构域时较不利于生成较短的产物。图12A和12B描绘了测序文库构建的两种不同方法。图12A示出了Nextera样品制备系统的实例,通过该系统,DNA可以在一个步骤中同时被片段化以及用测序衔接子被标记。在图12B中,将多联体通过超声处理进行片段化,随后向两个末端都添加衔接子(例如,通过使用KAPABiosystems的试剂盒),并进行PCR扩增。其他方法也是可用的。图13A-C提供了对背靠背(B2B)引物设计相较于传统PCR引物设计的示例性优点的图示。传统PCR引物设计(左)将引物(箭头A和B)置于位于靶序列侧翼的区域中,该区域可以是突变的热点(黑色星号),并且它们一般相距至少60个碱基对(bp),从而产生约100bp的典型足迹。在该图示中,B2B引物设计(右)将引物置于靶序列的一侧。两个B2B引物以相反方向面对,任一个都可以重叠(例如大约或少于约12bp、10bp、5bp或更小)。根据B2B引物的长度,该图示中的总足迹可以是28-50bp。由于足迹较大,片段化事件在传统设计中更可能破坏引物结合,导致序列信息丢失,无论是对于线性片段(13A)、环化DNA(13B),还是对于扩增产物(13C)而言。此外,如图13C所示,B2B引物设计捕获可用于区分不同多核苷酸的接点序列(也被称为“天然条形码”)。图14示出了根据一个实施方案生成用于检测序列变体的模板的方法(例如,使用环化多核苷酸的过程的实施方式示例,在本文中也称为“Nebula”)。将DNA输入变性成为ssDNA,通过连接进行环化,并通过外切核酸酶消化来降解未环化的DNA。通过定量PCR(qPCR)来定量连接效率,比较输入DNA与环化DNA的量,通常产生至少约80%的连接效率。将环化的DNA纯化到交换缓冲液中,接着使用随机引物和Phi29聚合酶进行全基因组扩增(WGA)。将WGA产物纯化,并且将产物片段化(例如通过超声处理)为约400bp或小于约400bp的短片段。通过qPCR定量扩增的DNA的靶标命中率(on-targetrate),其中比较相同量的参考基因组DNA与扩增的DNA,通常显示出约95%或大于约95%的平均靶标命中率。图15示出了使用加尾B2B引物进行扩增以及在较高温度下实施PCR的“递升”第二阶段的其他实施方式。B2B引物包含序列特异性区域(粗黑线)和衔接子序列(空心框)。在较低的第一阶段退火温度下,靶标特异性序列与模板退火,以产生初始单体,并且PCR产物包含串联重复(15A)。在较高温度下的第二扩增阶段,与单独的靶特异性序列杂交相比更有利于靶标特异性序列和衔接子序列的杂交,这降低了优先产生短产物的程度(15B)。当未有利于完整的引物时,与靶标特异性序列的内部退火迅速增加单体的比例(15C,左)。图16示出了通过靶标测序方法检测的背景噪声(变体的频率)之间的比较,该靶标测序方法使用Q30过滤器,要求(底部线)及不要求(顶部线)在待被计为变体的两个不同多核苷酸(例如,通过不同的接点而被鉴定)上存在序列差异。这一验证过滤器的应用在本文中也被称为“Firefly”。人类基因组DNA(12878,CoriellInstitute)被片段化为100-200bp,并包括含有已知SNP(CYP2C19)的基因组DNA(19240,CoriellInstitute)的2%掺入(spike-in)。真正的变体信号(标记的峰)没有显著超出背景(顶部,浅灰色图)。通过应用验证过滤器,背景噪声降低至约0.1(较低的,黑色图)。图17示出了当应用本发明的方法时,在多核苷酸群体中以各种低频率(2%、0.2%和0.02%)被掺入的序列变体的检测,然而其显著高于背景。图18示出了本发明的一个实施方案的连接效率和靶标命中率的分析结果。图19示出了在根据本发明的一个实施方案的方法中,等位基因频率的保持,以及基本上没有偏差。图20示出了根据一个实施方案,对小输入样品中的序列变体的检测结果。图21示出了根据标准测序方法获得的序列变体的检测结果中的高背景的实例,其中未要求在两个不同的多核苷酸上存在序列差异。图22提供的图显示了基因组的GC含量分布与根据本发明实施方案的方法产生的测序结果(“Nebula-Firefly”;左)、使用备选的测序文库构建试剂盒获得的测序结果(Rubicon,RubiconGenomics;中间)以及通常如文献中针对32ng报道的无细胞DNA(cfDNA)(右)的GC含量分布之间的比较。图23提供的图显示了根据一个实施方案的方法从测序读取获得的输入DNA的大小分布。图24提供的图显示了通过根据一个实施方案的随机引物法在多个靶标间的均一扩增。图25示出了在不进行环化的情况下,用于形成具有可鉴别的接点的多核苷酸多聚体的实施方案。将多核苷酸(例如,多核苷酸片段或无细胞DNA)连接以形成具有非天然接点的多聚体,所述非天然接点可用于根据本发明的实施方案区分独立的多核苷酸(在本文中也被称作“自动-标签”)。在图25A中,多核苷酸通过平端连接直接彼此连接。在图25B中,多核苷酸通过一个或多个间插衔接子寡核苷酸被连接,该寡核苷酸可以进一步包含条形码序列。然后将多聚体通过多种方法中的任一方法进行扩增,例如通过使用随机引物(全基因组扩增)、衔接子引物或一种或多种靶标特异性引物或引物对。从多个单独的多核苷酸形成具有可鉴别接点的多聚体的过程在本文中也被称为“Eclipse”。图26示出了图25的过程的变化示例。对多核苷酸(例如cfDNA或其他多核苷酸片段)进行末端修复、A加尾以及与衔接子连接(例如,使用标准试剂盒,如KAPABiosystems的试剂盒)。可以补充经内部尿嘧啶(U)标记的载体DNA,以将总DNA输入升高至所需的水平(例如至约20ng或大于约20ng)。待检测的序列变体用“星号”表示。当连接完成时,可通过加入尿嘧啶特异性切割试剂(USER)酶(其为尿嘧啶DNA糖基化酶(UDG)和DNA糖基化酶-裂合酶内切核酸酶VIII的混合物)将载体DNA降解。将产物纯化,以去除载体DNA的片段。扩增(例如,通过PCR,使用针对衔接子序列的引物)经纯化的产物。由于降解以及与至少一个末端的衔接子分离,任何残留的载体DNA不大可能被扩增。可对扩增的产物进行纯化以去除短DNA片段。图27示出了图25的过程的变化示例。靶标特异性扩增引物包含起衔接子作用的共同的5'“尾”(灰色箭头)。进行数个循环(例如,至少约5个、10个或更多个循环)的初始扩增(例如通过PCR)。PCR产物也可以作为引物,与其他PCR产物退火(例如,当退火温度在第二阶段降低时),以产生具有可鉴别接点的多联体。第二阶段可以包括多个循环(例如,5、10、15、20个或更多个循环),并且可包括对有利于多联体形成和扩增的条件的选择或变化。根据该示意图的方法也被称作“RelayAmpSeq”,其尤其可用于区室化的情形(例如,在微滴中)中。图28A-E示出了用于对多核苷酸进行环化的方法的非限制性实例。在图28A中,将双链多核苷酸(例如,dsDNA)变性成为单链,随后直接环化(例如通过CircLigase进行的自连接性连接)。在图28B中,对多核苷酸(例如,DNA片段)进行末端修复和A加尾(向3’端添加腺苷的单碱基延伸),以提高连接效率,接着变性为单链,并环化。在图28C中,对多核苷酸进行末端修复以及A加尾(如果为双链),连接至具有胸苷(T)延伸的衔接子,变性成为单链,并环化。在图28D中,对多核苷酸进行末端修复以及A加尾(如果为双链),两端均连接至具有三个元件(用于连接的T延伸、衔接子之间的互补区和3’尾)的衔接子,将链变性,并且将单链多核苷酸进行环化(由衔接子序列之间的互补区促进)。在图28E中,将双链多核苷酸变性为单链形式,并在使多核苷酸的末端更靠近在一起以促进连接的分子钳的存在下进行环化。图29示出了用于根据本发明的方法鉴定序列变体的扩增系统的工作流程设计示例,特别是针对环化的多核苷酸。图30示出了用于根据本发明的方法鉴定序列变体的扩增系统的工作流程设计示例,特别是针对无环化步骤情况下的线性多核苷酸输入。图31提供了用于按照本发明的方法鉴定序列变体的工作流程示例的概要性图示。根据“Eclipse”(线性多核苷酸分析)分支,分析可包括数字PCR(例如数字微滴PCR,ddPCR)、实时PCR、通过探针捕获的富集(捕获序列)以及对接点序列(自动标签)的分析、基于插入的衔接子序列(条形码插入)的测序或RelayAmp测序。根据“Nebula”(环化多核苷酸分析),分析可包括数字PCR(例如数字微滴PCR,ddPCR)、实时PCR、通过探针捕获的富集(捕获序列)以及对接点序列(自然条形码)的分析、通过探针捕获或靶向扩增(例如B2B扩增)的富集,以及具有验证步骤的序列分析,该验证步骤将序列变体鉴定为在两个不同多核苷酸(例如具有不同接点的多核苷酸)中存在的差异。图32是根据一个实施方案的系统的图示。图33示出根据一个实例的捕获效率和沿着靶区域的覆盖。>90%的靶向碱基被覆盖了超过20x,并且>50%的被靶向碱基具有>50x的覆盖。发明详述除非另有说明,本文公开的一些实施方案的实施采用了本领域技术范围内的免疫学、生物化学、化学、分子生物学、微生物学、细胞生物学、基因组学和重组DNA的常规技术。参见,例如,Sambrook和Green,MolecularCloning:ALaboratoryManual,第四版(2012);CurrentProtocolsinMolecularBiology系列(F.M.Ausubel等编著);MethodsInEnzymology系列(AcademicPress,Inc.),PCR2:APracticalApproach(M.J.MacPherson,B.D.Hames和G.R.Taylor编著(1995));Harlow和Lane编著(1988)Antibodies,ALaboratoryManual,andCultureofAnimalCells:AManualofBasicTechniqueandSpecializedApplications,第6版(R.I.Freshney编著(2010))。术语“约”或“大约”意指如本领域普通技术人员所确定的在特定值的可接受误差范围内,其部分地取决于该值是如何测量或确定的,例如,测量系统的限制。例如,“约”可以是指根据本领域的实践,在1个或大于1个标准差内。或者,“约”可以是指给定值的直至20%、直至10%、直至5%或直至1%的范围。或者,尤其是对于生物系统或过程,该术语可以是指在数值的一个数量级内,优选地在5倍以内,更优选地在2倍以内。在本申请和权利要求书中描述特定值时,除另有说明外,术语“约”应该被认为是指在特定值的可接受误差范围内。术语“多核苷酸”、“核苷酸”、“核苷酸序列”、“核酸”和“寡核苷酸”是可以互换使用的。它们是指任意长度的核苷酸(脱氧核糖核苷酸或核糖核苷酸)或其类似物的聚合形式。多核苷酸可以具有任意的三维结构,并且可以行使任何已知的或未知的功能。以下是多核苷酸的非限制性实例:基因或基因片段的编码或非编码区,通过连锁分析确定的基因座(座位)、外显子、内含子、信使RNA(mRNA)、转运RNA(tRNA)、核糖体RNA(rRNA)、短干扰RNA(siRNA)、短发夹RNA(shRNA)、微小RNA(miRNA)、核糖酶、cDNA、重组多核苷酸、分支多核苷酸、质粒、载体、任意序列的分离的DNA、任意序列的分离的RNA、核酸探针和引物。多核苷酸可以包含一个或多个修饰的核苷酸,例如甲基化核苷酸和核苷酸类似物。如果存在,对核苷酸结构的修饰可以在聚合物组装之前或之后赋予。核苷酸序列可以被非核苷酸组分打断。多核苷酸可以在聚合后被进一步修饰,例如,通过与标记组分缀合。通常,术语“靶多核苷酸”是指具有靶序列的核酸分子起始群体中的核酸分子或多核苷酸,希望确定该靶序列的存在、量和/或核苷酸序列或其中一个或多个的改变。通常,术语“靶序列”是指在核酸单链上的核酸序列。靶序列可以是基因的一部分,调控序列,基因组DNA,cDNA,包括mRNA、miRNA、rRNA在内的RNA,等等。靶序列可以是来自样品的靶序列或次级靶标如扩增反应的产物。通常,“核苷酸探针”、“探针”或“标签寡核苷酸”是指用于通过与相应的靶序列杂交而在杂交反应中检测或鉴定其相应的靶多核苷酸的多核苷酸。因此,核苷酸探针可与一个或多个靶多核苷酸杂交。标签寡聚核苷酸可以与样品中的一个或多个靶多核苷酸完美互补,或者包含不与样品中的一个或多个靶多核苷酸中相应的核苷酸互补的一个或多个核苷酸。“杂交”是指这样的反应,在该反应中,一个或多个多核苷酸发生反应以形成复合体,该复合体通过核苷酸残基的碱基之间的氢键键合而被稳定化。该氢键键合可以通过WatsonCrick碱基配对、Hoogstein结合或根据碱基互补性以任意其他序列特异性方式而发生。该复合体可以包含形成双链体结构的两条链、形成多链复合体的三条或更多条链、单一的自杂交链、或它们的任意组合。杂交反应可以构成更广泛的过程中的步骤,例如PCR的起始,或内切核酸酶对多核苷酸的酶切。与第一序列互补的第二序列被称作第一序列的“互补体(complement)”。如用于多核苷酸的术语“可杂交的”是指多核苷酸形成复合体的能力,该复合体通过杂交反应中核苷酸残基的碱基之间的氢键键合而被稳定化。“互补性”是指核酸与另一核酸序列通过经典Watson-Crick或其他非经典类型的方式形成氢键的能力。互补性百分比表示核酸分子中能够与第二核酸序列形成氢键(例如,Watson-Crick碱基配对)的残基的百分比(例如,10个中有5、6、7、8、9、10个分别为50%、60%、70%、80%、90%和100%互补)。“完美互补”是指核酸序列的所有连续残基将与第二核酸序列中相同数目的连续残基氢键键合。本文使用的“基本上互补”是指互补性程度在8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50个或更多个核苷酸的区域中至少为60%、65%、70%、75%、80%、85%、90%、95%、97%、98%、99%或100%,或者是指在严格条件下杂交的两个核酸。序列同一性,例如为了评估互补性百分比,可以通过任何合适的比对算法进行测量,包括但不限于Needleman-Wunsch算法(参见,例如,EMBOSSNeedle比对器,可从www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html获得,任选地具有默认设置)、BLAST算法(参见,例如,BLAST比对工具,可从blast.ncbi.nlm.nih.gov/Blast.cgi获得,任选地具有默认设置)或者Smith-Waterman算法(参见,例如,EMBOSSWater比对器,可www.ebi.ac.uk/Tools/psa/emboss_water/nucleotide.html可获得,任选地具有默认设置)。最优比对可以使用所选定算法的任意合适的参数(包括默认参数)来评估。通常,杂交的“严格条件”是指在该条件下,具有与靶序列的互补性的核酸主要与靶序列杂交,并且基本上不与非靶序列杂交。严格条件通常是序列依赖性的,并且根据许多因素而不同。通常,序列越长,该序列与其靶序列特异性杂交时的温度越高。严格条件的非限制性实例在下列中有详细描述:Tijssen(1993),LaboratoryTechnniquesInBiochemistryAndMolecularBiology-HybridizationWithNucleicAcidProbes的第一部分,第二章,“Overviewofprinciplesofhybridizationandthestrategyofnucleicacidprobeassay”,Elsevier,N.Y.。在一个方面,本发明提供了一种鉴定序列变体,例如核酸样品中的序列变体的方法。在一些实施方案中,多个多核苷酸中的每个多核苷酸具有5’末端和3’末端,并且该方法包括:(a)将所述多个多核苷酸中的单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸在5’末端与3’末端之间具有接点;(b)扩增(a)的环状多核苷酸;(c)对扩增的多核苷酸进行测序以生成多个测序读取;(d)鉴定测序读取与参考序列之间的序列差异;和(e)将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体。在一些实施方案中,该方法包括鉴定测序读取与参考序列之间的序列差异,以及将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体,其中:(a)该测序读取对应于至少两个环状多核苷酸的扩增产物;且(b)该至少两个环状多核苷酸中的每一个包含通过连接相应多核苷酸的5’末端和3’末端而形成的不同的接点。通常,术语“序列变体”是指序列中相对于一个或多个参考序列的任何变异。一般而言,对于参考序列已知的个体的给定群体,序列变体以比参考序列更低的频率发生。例如,特定的细菌属可能具有针对16SrRNA基因的共有参考序列,但是该属内的单独的种可能在基因(或其一部分)内具有一个或多个序列变体,其对鉴定细菌群体中的该种是有用的。作为另一个实例,当最佳地比对时,同一个种的多个个体的序列(或同一个体的多个测序读取)可以产生共有序列,并且相对于该共有序列的序列变体可被用于鉴定该群体中指示危险的污染的突变体。通常,“共有序列”是指这样的核苷酸序列,其反映当对一系列相关核酸进行大量的数学和/或序列分析(例如按照多种序列比对算法中的任意一种的最佳序列比对)时,序列中每个位置处的最常见碱基选择。多种比对算法是可以使用的,其中的一些在本文中进行了描述。在一些实施方案中,参考序列是单一的已知参考序列,例如单个个体的基因组序列。在一些实施方案中,参考序列是通过比对多个已知序列(例如作为参考群体的多个个体的基因组序列,或来自同一个体的多核苷酸的多个测序读取)而形成的共有序列。在一些实施方案中,参考序列是通过最佳地比对来自所分析的样品的序列而形成的共有序列,从而序列变体代表同一样品中相对于相应序列的变异。在一些实施方案中,序列变体在群体中以低频率发生(也称为“罕见”序列变体)。例如,序列变体可以以约为或低于约5%、4%、3%、2%、1.5%、1%、0.75%、0.5%、0.25%、0.1%、0.075%、0.05%、0.04%、0.03%、0.02%、0.01%、0.005%、0.001%或更低的频率发生。在一些实施方案中,序列变体以约为或低于约0.1%的频率发生。序列变体可以是相对于参考序列的任何变异。序列变异可以由一个核苷酸或多个核苷酸(例如2、3、4、5、6、7、8、9、10个或更多个核苷酸)的改变、插入或缺失组成。当序列变体包含两个或更多个核苷酸差异时,不同的核苷酸可以是彼此连续的,或不连续的。序列变体类型的非限制性实例包括单核苷酸多态性(SNP)、缺失/插入多态性(DIP)、拷贝数变体(CNV)、短串联重复(STR)、简单序列重复(SSR)、可变数目串联重复(VNTR)、扩增片段长度多态性(AFLP)、基于反转录转座子的插入多态性和序列特异性扩增多态性。可以进行本文所述的方法的核酸样品可以源自任何合适的来源。在一些实施方案中,所使用的样品是环境样品。环境样品可以来自任何环境来源,例如,天然存在的或人造的大气、水系、土壤或任意其他目的样品。在一些实施方案中,环境样品可以获自,例如,大气病原体收集系统、地表下沉积物、地下水、地层深处古代水、草原的植物根-土壤界面、沿岸水和污水处理厂。来自样品的多核苷酸可以是多种多核苷酸中的任意一种,包括但不限于DNA、RNA、核糖体RNA(rRNA)、转运RNA(tRNA)、微小RNA(miRNA)、信使RNA(mRNA)、以上任意一种的片段或以上任意两种或更多种的组合。在一些实施方案中,样品包含DNA。在一些实施方案中,样品包含基因组DNA。在一些实施方案中,样品包含线粒体DNA、叶绿体DNA、质粒DNA、细菌人工染色体、酵母人工染色体、寡核苷酸标签或其组合。在一些实施方案中,样品包含通过扩增生成的DNA,例如通过使用任何合适的引物组合和DNA聚合酶进行的引物延伸反应,包括但不限于聚合酶链反应(PCR)、逆转录及其组合。当引物延伸反应的模板是RNA时,逆转录的产物被称为互补DNA(cDNA)。在引物延伸反应中有用的引物可以包含对一个或多个靶标具有特异性的序列、随机序列、部分随机序列及其组合。通常,样品多核苷酸包含在样品中存在的任意多核苷酸,其可包括或可不包括靶多核苷酸。该多核苷酸可以是单链的、双链的或其组合。在一些实施方案中,经历本发明的方法的多核苷酸是单链多核苷酸,其可存在或可不存在双链多核苷酸。在一些实施方案中,该多核苷酸是单链DNA。单链DNA(ssDNA)可以是以单链形式分离的ssDNA,或者是以双链形式分离并且随后制成单链以用于本发明方法的一个或多个步骤的DNA。在一些实施方案中,多核苷酸不经提取步骤和/或不经纯化步骤而经历后续步骤(例如环化和扩增)。例如,流体样品可以不经提取步骤而被处理以去除细胞,从而产生纯化的液体样品和细胞样品,随后从纯化的流体样品中分离DNA。多种用于分离多核苷酸的程序是可用的,例如通过沉淀或与基底的非特异性结合,随后洗涤基底以释放结合的多核苷酸。当不经细胞提取步骤而从样品中分离多核苷酸时,多核苷酸大部分将是细胞外的或“无细胞的”多核苷酸,其可对应于死亡或受损的细胞。这类细胞的身份可被用于表征它们所源自的细胞或细胞群体,例如在微生物群落中。如果对样品进行处理以提取多核苷酸,例如从样品中的细胞中提取,则多种提取方法是可用的,例如,核酸可通过用苯酚、苯酚/氯仿/异戊醇或类似的制剂(包括TRIzol和TriReagent)进行有机萃取而纯化。纯化技术的其他非限制性实例包括:(1)有机萃取然后乙醇沉淀,例如,使用苯酚/氯仿有机试剂(Ausubel等,1993),使用或不使用自动核酸提取器,例如可从AppliedBiosystems(Fostercity,Calif)获得的341型DNA提取器;(2)固定相吸附法(美国专利号5,234,809;Walsh等,1991);和(3)盐诱导核酸沉淀法(Miller等,1988),该沉淀方法一般被称作“盐析”法。核酸分离和/或纯化的另一个实例包括使用核酸能够特异性或非特异性结合的磁性颗粒,然后使用磁体分离珠子,并洗涤和从珠子中洗脱核酸(参见,例如,美国专利号5,705,628)。在一些实施方案中,在上述分离方法之前可先进行酶消化步骤以帮助从样品中去除不需要的蛋白质,例如用蛋白酶K或其他类似的蛋白酶进行消化。参见,例如,美国专利号7,001,724。如果需要,可向裂解缓冲液中添加RNase抑制剂。对于特定的细胞或样品类型,可能需要在方案中增加蛋白质变性/消化步骤。纯化方法可以针对分离DNA、RNA或此两者。当DNA和RNA在提取程序过程中或之后被一起分离时,可使用进一步的步骤来彼此单独地纯化一者或两者。也可生成提取的核酸的亚级分,例如,根据大小、序列或其他物理或化学特性进行纯化。除了初始核酸分离步骤外,核酸的纯化还可以在所公开的方法的任意步骤之后进行,例如用于去除过量的或不需要的试剂、反应物或产物。多种用来确定样品中的核酸量和/或核酸纯度的方法是可用的,例如通过吸光度(例如,在260nm、280nm处的光吸收,和它们的比值)和标记物的检测(例如,荧光染料和嵌入剂,例如SYBR绿、SYBR蓝、DAPI、碘化丙啶、Hoechst染色剂、SYBR金、溴化乙锭)。根据一些实施方案,将来自样品的多个多核苷酸中的多核苷酸进行环化。环化可包括将多核苷酸的5’末端连接到同一多核苷酸的3’末端,连接到样品中的另一多核苷酸的3’末端,或连接到来自不同来源的多核苷酸(例如,人工多核苷酸,如寡核苷酸衔接子)的3’末端。在一些实施方案中,多核苷酸的5’末端被连接到同一多核苷酸的3’末端(也称为“自连接”)。在一些实施方案中,选择环化反应的条件以利于在特定长度范围内的多核苷酸的自连接,以便生成具有特定平均长度的环化多核苷酸群体。例如,可以选择环化反应条件以利于长度短于约5000、2500、1000、750、500、400、300、200、150、100、50个或更少的核苷酸的多核苷酸的自连接。在一些实施方案中,有利于长度为50-5000个核苷酸、100-2500个核苷酸或150-500个核苷酸的片段,以使得环化多核苷酸的平均长度落入相应的范围内。在一些实施方案中,80%或更多的环化片段的长度为50-500个核苷酸,例如长度为50-200个核苷酸。可以优化的反应条件包括分配给连接反应的时间长度、各种试剂的浓度和待连接的多核苷酸的浓度。在一些实施方案中,环化反应保持环化前存在于样品中的片段长度的分布。例如,环化前样品中的片段长度以及环化多核苷酸的片段长度的平均值、中值、众数(mode)和标准差中的一个或多个在彼此的75%、80%、85%、90%、95%或更高的百分比以内。使用了一个或多个衔接子寡核苷酸,而非优先形成自连接环化产物,从而样品中多核苷酸的5’末端和3’末端通过一个或多个间插衔接子寡核苷酸被连接,以形成环状多核苷酸。例如,多核苷酸的5’末端能够被连接到衔接子的3’末端,并且同一衔接子的5’末端能够被连接到同一多核苷酸的3’末端。衔接子寡核苷酸包括具有序列的任意寡核苷酸,该序列的至少一部分是已知的,它能够与样品多核苷酸连接。衔接子寡核苷酸可以包含DNA、RNA、核苷酸类似物、非典型核苷酸、标记的核苷酸、修饰的核苷酸或它们的组合。衔接子寡核苷酸可以是单链的、双链的或部分双链体。通常,部分双链体衔接子包含一个或多个单链区域和一个或多个双链区域。双链衔接子可包含彼此相互杂交的两个单独的寡核苷酸(也称作“寡核苷酸双链体”),并且杂交可留下一个或多个平端、一个或多个3’突出端、一个或多个5’突出端、一个或多个由错配的和/或不配对的核苷酸所导致的凸起,或它们的任意组合。当衔接子的两个杂交区域被非杂交区域彼此隔开时,会产生“泡”结构。不同类型的衔接子能够组合使用,例如不同序列的衔接子。不同的衔接子可在顺次反应中或同时地与样品多核苷酸连接。在一些实施方案中,将相同的衔接子添加至靶多核苷酸的两个末端。例如,可将第一和第二衔接子添加至同一反应中。衔接子可在与样品多核苷酸组合前被操作。例如,可以添加或去除末端磷酸。当使用衔接子寡核苷酸时,该衔接子寡核苷酸可包含多种序列元件中的一个或多个,包括但不限于,一个或多个扩增引物退火序列或其互补体、一个或多个测序引物退火序列或其互补体、一个或多个条形码序列、一个或多个在多个不同衔接子或不同衔接子的子集之间共有的共同序列、一个或多个限制酶识别位点、一个或多个与一个或多个靶多核苷酸突出端互补的突出端、一个或多个探针结合位点(例如,用于连接到测序平台,例如用于大规模平行测序的流动池,如Illumina,Inc.开发的流动池)、一个或多个随机的或接近随机的序列(例如,在一个或多个位置处从一组两个或更多个不同核苷酸中随机选择的一个或多个核苷酸,其中在一个或多个位置处选择的不同核苷酸中的每一个均在包含随机序列的衔接子的集合中被体现),或它们的组合。在一些情况下,衔接子可用于纯化含有衔接子的那些环,例如通过使用以包含衔接子互补序列的寡核苷酸涂覆的珠子(为了易于处理,特别是磁珠),该珠子可以通过与之杂交而“捕获”具有正确衔接子的闭合环,洗掉那些不包含衔接子的环和任何未连接的组分,然后从珠子上释放所捕获的环。此外,在一些情况下,杂交的捕获探针和目标环的复合体可直接用于生成多联体,例如通过直接滚环扩增(RCA)。在一些实施方案中,环中的衔接子也可用作测序引物。两个或更多个序列元件可以是彼此不邻近的(例如被一个或多个核苷酸隔开)、彼此邻近的、部分重叠的或完全重叠的。例如,扩增引物退火序列也可作为测序引物退火序列。序列元件可位于或靠近3’末端、位于或靠近5’末端或在衔接子寡核苷酸内部。序列元件可以是任何合适的长度,例如约为或少于约3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50个或更多个核苷酸的长度。衔接子寡核苷酸可具有任意合适的长度,至少足以容纳其所包含的一个或多个序列元件。在一些实施方案中,衔接子的长度为约或少于约10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、90、100、200个或更多个核苷酸。在一些实施方案中,衔接子寡核苷酸的长度在约12到40个核苷酸的范围内,例如长度为约15到35个核苷酸。在一些实施方案中,与来自一个样品的片段化的多核苷酸连接的衔接子寡核苷酸包含一个或多个所有衔接子寡核苷酸所共有的序列和对于与该特定样品的多核苷酸连接的衔接子而言独特的条形码,以使得该条形码序列可用于将来源于一个样品或衔接子连接反应的多核苷酸与来源于另一个样品或衔接子连接反应的多核苷酸进行区分。在一些实施方案中,衔接子寡核苷酸包含与一个或多个靶多核苷酸突出端互补的5’突出端、3’突出端或此两者。互补突出端在长度上可以是一个或多个核苷酸,包括但不限于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15个或更多个核苷酸的长度。互补突出端可包含固定的序列。衔接子寡核苷酸的互补突出端可以包含一个或多个核苷酸的随机序列,以使得在一个或多个位置处从一组两个或更多个不同核苷酸中随机选择一个或多个核苷酸,其中在一个或多个位置处选择的不同核苷酸中的每一个都在具有包含随机序列的互补突出端的衔接子的集合中被体现。在一些实施方案中,衔接子突出端与通过限制性内切核酸酶消化产生的靶多核苷酸突出端互补。在一些实施方案中,衔接子突出端由腺嘌呤或胸腺嘧啶组成。多种环化多核苷酸的方法是可用的。在一些实施方案中,环化包含酶反应,例如使用连接酶(例如RNA或DNA连接酶)。多种连接酶是可用的,包括但不限于,CircligaseTM(Epicentre;Madison,WI)、RNA连接酶、T4RNA连接酶1(ssRNA连接酶,其作用于DNA和RNA两者)。此外,如果不存在dsDNA模板,T4DNA连接酶也可以连接ssDNA,尽管这通常是缓慢的反应。连接酶的其他非限制性实例包括:NAD-依赖性连接酶,包括TaqDNA连接酶、丝状栖热菌(Thermusfiliformis)DNA连接酶、大肠杆菌DNA连接酶、TthDNA连接酶、水管致黑栖热菌(Thermusscotoductus)DNA连接酶(I和II)、热稳定的连接酶、Ampligase热稳定的DNA连接酶、VanC-型连接酶、9°NDNA连接酶、TspDNA连接酶和通过生物勘探发现的新型连接酶;ATP-依赖性连接酶,包括T4RNA连接酶、T4DNA连接酶、T3DNA连接酶、T7DNA连接酶、PfuDNA连接酶、DNA连接酶1、DNA连接酶III、DNA连接酶IV和通过生物勘探发现的新型连接酶;以及野生型、突变体同种型,及它们的遗传工程变体。当需要自连接时,可调节多核苷酸和酶的浓度以促进分子内环而非分子间结构的形成。反应温度和时间也可调整。在一些实施方案中,使用60℃来促进分子内环的形成。在一些实施方案中,反应时间为12-16小时。反应条件可以是所选择的酶的制造商所规定的条件。在一些实施方案中,可以包括外切核酸酶步骤以在环化反应后消化任何未连接的核酸。也就是说,闭合环不含游离5’或3’末端,因此引入5’或3’外切核酸酶不会消化闭合环但会消化未连接的组分。这尤其可用于多重系统中。一般地,将多核苷酸末端彼此连接以形成环状多核苷酸(直接地,或者是使用一个或多个中间衔接子寡核苷酸)会产生具有接点序列的接点。当多核苷酸的5’末端和3’末端通过衔接子多核苷酸连接时,术语“接点”可以是指多核苷酸与衔接子之间的接点(例如5’末端接点或3’末端接点之一),或指如通过衔接子多核苷酸形成并包含衔接子多核苷酸的、多核苷酸的5’末端与3’末端之间的接点。当多核苷酸的5’末端和3’末端在不使用间插衔接子的情况下连接时(例如,单链DNA的5’末端和3’末端),术语“接点”是指这两个末端被连接的点。接点可以根据包含接点的多核苷酸的序列(也称为“接点序列”)被鉴定。在一些实施方案中,样品包含具有通过以下过程形成的末端混合物的多核苷酸:自然降解过程(例如细胞裂解、细胞死亡和使DNA从细胞释放到其周围环境中的其它过程,DNA在该周围环境中可进一步被降解,例如在无细胞多核苷酸中),作为样品处理(例如,固定、染色和/或存储过程)的副产物的片段化,以及通过不限定特定靶序列的切割DNA的方法进行的片段化(例如,机械片段化,如超声处理;非序列特异性核酸酶处理,如DNaseI、片段化酶(fragmentase))。当样品包含具有末端混合物的多核苷酸时,两个核苷酸具有相同5’末端或3’末端的可能性是低的,并且两个核苷酸独立地具有相同的5’末端和3’末端两者的可能性极低。因此,在一些实施方案中,甚至在两个多核苷酸包含具有相同靶序列的部分时,也可以使用接点来区分不同的多核苷酸。当多核苷酸末端在不使用间插衔接子的情况下连接时,接点序列可通过与参考序列比对而被鉴定。例如,当两个组分序列的顺序相对于参考序列似乎被反转时,则似乎发生反转的点可以指示在该点具有接点。当多核苷酸末端通过一个或多个衔接子序列连接时,接点可通过与已知衔接子序列的邻近而被鉴定,或者在测序读取的长度足以从环化多核苷酸的5’和3’末端均获得序列的情况下通过上述比对进行鉴定。在一些实施方案中,特定接点的形成是十分罕见的事件,以至于其在样品的环化多核苷酸之中是独特的。图4示出了环化多核苷酸的方法的三个非限制性实例。在最上方,在不存在衔接子的情况下对多核苷酸进行环化,中间的方案描述了使用衔接子,而最下方的方案使用了两个衔接子。当使用两个衔接子时,其中一个能够与多核苷酸的5’末端连接,而第二衔接子能够与同一多核苷酸的3’末端连接。在一些实施方案中,衔接子连接可包括使用两个不同的衔接子以及与这两个衔接子互补的“夹板(splint)”核酸以促进连接。也可使用叉状或“Y”形衔接子。当使用两个衔接子时,在两个末端具有相同衔接子的多核苷酸可由于自退火而在后续步骤中被去除。图6示出了环化多核苷酸如单链DNA的方法的其它非限制性示例。衔接子可被不对称地添加至多核苷酸的5’末端或3’末端。如图6A所示,单链DNA(ssDNA)在3’末端具有游离羟基,并且衔接子具有封闭的3’末端,以使得在连接酶的存在下,优选的反应将ssDNA的3’末端连接至衔接子的5’末端。在该实施方案中,在分子内连接形成环之前使用试剂(诸如聚乙二醇(PEG))来驱动单一ssDNA片段和单一衔接子的分子间连接可能是有用的。也可以进行末端的反向顺序(封闭的3’、游离的5’,等等)。一旦线性连接完成,即可用酶处理所连接的片段以去除封闭部分,例如通过使用激酶或其他合适的酶或化学品。一旦去除了封闭部分,环化酶(如CircLigase)的添加就允许进行分子内反应以形成环化的多核苷酸。如图6B所示,通过使用其中一条链具有封闭的5’或3’末端的双链衔接子,能够形成双链结构,其在连接后产生具有切口的双链片段。之后能够分离这两条链,去除封闭部分,并且使单链片段进行环化以形成环化的多核苷酸。在一些实施方案中,使用分子钳使多核苷酸(例如,单链DNA)的两个末端靠在一起以提高分子内环化率。图5显示了一种这样的过程的示例图示。这可通过使用或不使用衔接子而完成。分子钳的使用在平均多核苷酸片段的长度大于约100个核苷酸的情况下可能尤其有用。在一些实施方案中,分子钳探针包含三个结构域:第一结构域、间插结构域和第二结构域。第一和第二结构域将首先通过序列互补性与靶多核苷酸中的相应序列杂交。分子钳探针的间插结构域不明显与靶序列杂交。分子钳与靶多核苷酸的杂交因此使靶序列的两个末端更加靠近,这促进了在环化酶的存在下靶序列的分子内环化。在一些实施方案中,这另外有用,因为分子钳也能作为扩增引物。环化之后,反应产物可在扩增或测序之前被纯化以提高可参与后续步骤的环化多核苷酸的相对浓度或纯度(例如,通过环状多核苷酸的分离或反应中一种或多种其他分子的去除)。例如,可处理环化反应或其组分以去除单链(未环化的)多核苷酸,例如通过用外切核酸酶处理。作为进一步的实例,可对环化反应或其部分进行尺寸排阻色谱法,从而保留及丢弃小试剂(例如未反应的衔接子),或在单独的体积中保留并释放环化产物。多种用于清理连接反应的试剂盒是可用的,例如由ZymoReaserch制造的Zymo寡核苷酸纯化试剂盒所提供的试剂盒。在一些实施方案中,纯化包括用于去除或降解在环化反应中使用的连接酶和/或将环化多核苷酸从该连接酶中纯化的处理。在一些实施方案中,用于降解连接酶的处理包括用蛋白酶(如蛋白酶K)进行的处理。蛋白酶K处理可遵循制造商的方案或标准方案(例如,如Sambrook和Green,MolecularCloning:ALaboratoryManual,第4版(2012)所提供的)。蛋白酶处理之后还可进行提取和沉淀。在一个实例中,环化多核苷酸如下纯化:在0.1%SDS和20mMEDTA的存在下进行蛋白酶K(Qiagen)处理,用1:1苯酚/氯仿和氯仿萃取,并用乙醇或异丙醇沉淀。在一些实施方案中,沉淀在乙醇中进行。环化之后可直接对环化多核苷酸进行测序。或者,测序之前可进行一个或多个扩增反应。通常,“扩增”是指形成靶多核苷酸或其部分的一个或多个拷贝的过程。多种扩增多核苷酸(例如DNA和/或RNA)的方法是可用的。扩增可以是线性的,指数式的,或在多阶段扩增过程中涉及线性和指数阶段两者。扩增方法可包括温度的改变,例如热变性步骤,或者可以是不需要热变性的等温过程。聚合酶链反应(PCR)采用变性、引物对与相反链的退火和引物延伸的多个循环,以指数式增加靶序列的拷贝数。退火的核酸链的变性可以通过如下来实现:施加热、提高局部金属离子浓度(例如,美国专利号6,277,605)、超声辐射(例如,WO/2000/049176)、施加电压(例如,美国专利号5,527,670、美国专利号6,033,850、美国专利号5,939,291和美国专利号6,333,157)和与结合到磁响应性材料上的引物相组合地施加电磁场(例如,美国专利号5,545,540)。在被称作RT-PCR的变化形式中,使用逆转录酶(RT)由RNA制备互补DNA(cDNA),之后通过PCR扩增cDNA以生成DNA的多个拷贝(例如,美国专利号5,322,770和美国专利号5,310,652)。等温扩增方法的一个实例是链置换扩增,通常称为SDA,其使用以下过程的循环:使引物序列对与靶序列的相反链退火,在dNTP存在下进行引物延伸以生成双链体半硫代磷酸化的引物延伸产物,内切核酸酶介导的对半修饰限制内切核酸酶识别位点形成切口,和聚合酶介导的从切口的3’端进行引物延伸以取代已存在的链并产生用于下一轮引物退火、切口形成和链置换的链,从而引起产物的几何扩增(例如,美国专利号5,270,184和美国专利号5,455,166)。嗜热SDA(tSDA)在基本相同的方法中在更高的温度下使用嗜热内切核酸酶和聚合酶(欧洲专利号0684315)。其他扩增方法包括滚环扩增(RCA)(例如Lizardi,“RollingCircleReplicationReporterSystems”美国专利号5,854,033);解旋酶依赖性扩增(HDA)(例如,Kong等,“HelicaseDependentAmplificationNucleicAcids”美国专利申请公开号US2004-0058378A1);和环介导的等温扩增(LAMP)(例如Notomi等,“ProcessforSynthesizingNucleicAcid”美国专利号6,410,278)。在一些情况下,等温扩增采用通过RNA聚合酶从启动子序列起进行转录,例如可以引入寡核苷酸引物中。基于转录的扩增方法包括基于核酸序列的扩增,也称为NASBA(例如美国专利号5,130,238);依赖于使用RNA复制酶(通常称为Qβ复制酶)扩增探针分子本身的方法(例如,Lizardi,P.等(1988)BioTechnol.6,1197-1202);自动维持序列复制(例如,Guatelli,J.等(1990)Proc.Natl.Acad.Sci.USA87,1874-1878;Landgren(1993)TrendsinGenetics9,199-202;和HELENH.LEE等,NUCLEICACIDAMPLIFICATIONTECHNOLOGIES(1997));和生成额外的转录模板的方法(例如,美国专利号5,480,784和美国专利号5,399,491)。另外的等温核酸扩增方法包括使用含有非典型核苷酸(例如,尿嘧啶或RNA核苷酸)的引物并结合使用在非典型核苷酸处切割核酸的酶(例如DNA糖基化酶或RNaseH),以暴露针对额外的引物的结合位点(例如,美国专利号6,251,639、美国专利号6,946,251和美国专利号7,824,890)。等温扩增过程可以是线性的或指数式的。在一些实施方案中,扩增包括滚环扩增(RCA)。典型的RCA反应混合物包含一种或多种引物、聚合酶和dNTPs,并且生成多联体。一般来说,RCA反应中的聚合酶是具有链置换活性的聚合酶。多种这样的聚合酶是可用的,其非限制性实例包括外切核酸酶-DNA聚合酶I大(Klenow)片段、Phi29DNA聚合酶、TaqDNA聚合酶等。通常,多联体是包含来自模板多核苷酸的靶序列的两个或更多个拷贝(例如靶序列的约或多于约2、3、4、5、6、7、8、9、10个或更多个拷贝;在一些实施方案中,约为或多于约2个拷贝)的多核苷酸扩增产物。扩增引物可以是任何合适的长度,例如约或至少约5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、90、100个或更多个核苷酸,其任意部分或全部可互补于引物所杂交的相应靶序列(例如,约为或至少约5、10、15、20、25、30、35、40、45、50个或更多个核苷酸)。图7描述了合适的引物的三个非限制性实例。图7A显示了不使用衔接子但使用靶标特异性引物,其可以用于检测特定靶序列内的序列变体的存在与否。在一些实施方案中,在同一反应中使用针对多个靶标的多个靶标特异性引物。例如,可以在一个扩增反应中使用针对约为或至少约10、50、100、150、200、250、300、400、500、1000、2500、5000、10000、15000个或更多个不同靶序列的靶标特异性引物,以便平行地扩增相应数目的靶序列(如果存在的话)。多个靶序列可以对应于相同基因的不同部分、不同基因或非基因序列。当多个引物靶向单一基因中的多个靶序列时,可以沿着基因序列将引物间隔开(例如,间隔开约或至少约50个核苷酸,每50-150个核苷酸,或每50-100个核苷酸),以覆盖靶基因的全部或指定部分。在图7C中显示了使用与衔接子序列杂交的引物(其在一些情况下可以是衔接子寡核苷酸本身)。图7B示出了通过随机引物扩增的实例。通常,随机引物包含一个或多个随机的或接近随机的序列(例如,在一个或多个位置处从一组两个或更多个不同核苷酸中随机选择的一个或多个核苷酸,其中在一个或多个位置处选择的不同核苷酸中的每一个均在包含随机序列的衔接子的集合中被体现)。以这种方式,多核苷酸(例如,全部或基本全部的环化多核苷酸)能够以序列非特异性方式被扩增。这样的程序可被称为“全基因组扩增”(WGA);但是,典型的WGA方案(不涉及环化步骤)不能有效扩增短多核苷酸,例如本发明所涉及的多核苷酸片段。关于WGA程序的进一步阐释性讨论,参考例如Li等人(2006)JMol.Diagn.8(1):22-30。当环化多核苷酸在测序前被扩增时,可不进行富集而对扩增产物直接进行测序,或者在一个或多个富集步骤之后进行测序。富集可包括纯化一个或多个反应组分,例如通过保留扩增产物或去除一种或多种试剂。例如,扩增产物可如下纯化:与多个连接到基底上的探针杂交,随后释放捕获的多核苷酸,如通过洗涤步骤。或者,扩增产物可用结合对的一个成员进行标记,之后与连接到基底上的结合对的另一成员结合,并进行洗涤以释放扩增产物。可能的基底包括但不限于玻璃和经修饰的或功能化的玻璃、塑料(包括丙烯酸树脂,聚苯乙烯,和苯乙烯与其他材料的共聚物、聚丙烯,聚乙烯,聚丁烯、聚氨酯,TeflonTM,等等)、多糖、尼龙或硝化纤维、陶瓷、树脂、包括硅和改性硅在内的二氧化硅或硅基材料、碳、金属、无机玻璃、塑料、光纤束和多种其他聚合物。在一些实施方案中,基底为珠子或其他小的离散颗粒的形式,其可以是磁性或顺磁性珠子,以通过施加磁场促进分离。通常,“结合对”是指第一和第二部分之一,其中第一和第二部分对于彼此具有特异性结合亲和性。合适的结合对包括但不限于抗原/抗体(例如,洋地黄毒苷/抗洋地黄毒苷、二硝基苯(DNP)/抗-DNP、丹酰-X-抗-丹酰、荧光素/抗荧光素、萤光黄/抗萤光黄以及罗丹明/抗罗丹明);生物素/亲和素(或生物素/链霉亲和素);钙调蛋白结合蛋白(CBP)/钙调蛋白;激素/激素受体;凝集素/碳水化合物;肽/细胞膜受体;蛋白A/抗体;半抗原/抗半抗原;酶/辅因子;和酶/底物。在一些实施方案中,环化多核苷酸的扩增之后的富集包括一个或多个额外的扩增反应。在一些实施方案中,富集包括在扩增反应混合物中扩增包含序列A和序列B(以5’到3’方向定向)的靶序列,该扩增反应混合物包含(a)扩增的多核苷酸;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以产生扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。图10示出了相对于在单一重复情况下的靶序列(其除非为环状,否则一般不会被扩增)和包含靶序列的多个拷贝的多联体,第一和第二引物的排列实例。鉴于引物相对于靶序列单体的定向,该排列可称为“背靠背”(B2B)或“反向”引物。用B2B引物进行的扩增促进了环状和/或多联体扩增产物的富集。此外,该定向与相对较小的足印(一对引物所跨越的总距离)结合起来,使得能够扩增靶序列周围的更加多样的片段化事件,因为与在典型扩增反应中所见的引物排列(彼此面对,跨越靶序列)相比,不大可能在引物之间出现接点。在一些实施方案中,序列A的5’末端与序列B的3’末端之间的距离是大约或少于约200、150、100、75、50、40、30、25、20、15个或更少的核苷酸。在一些实施方案中,序列A是序列B的互补体。在一些实施方案中,在同一反应中使用针对多个不同靶序列的多个B2B引物对,以平行地扩增多个不同的靶序列(例如约或至少约10、50、100、150、200、250、300、400、500、1000、2500、5000、10000、15000个或更多个不同的靶序列)。引物可以具有任意合适的长度,例如在本文其他部分所描述的。扩增可包括在适当条件下的任何合适的扩增反应,例如本文所描述的扩增反应。在一些实施方案中,扩增是聚合酶链反应。在一些实施方案中,B2B引物包含至少两个序列元件:通过序列互补性与靶序列杂交的第一元件,和在第一杂交温度下的第一扩增阶段中不会与靶序列杂交的5’“尾”,在该第一扩增阶段期间第一元件发生杂交(例如,由于尾部和紧邻第一元件结合处位于其3’侧的靶序列部分之间缺乏序列互补性)。例如,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在第一杂交温度下的第一扩增阶段期间均不与多个多联体杂交。在使用了此类加尾引物的一些实施方案中,扩增可包括第一阶段和第二阶段;第一阶段包括在第一温度下的杂交步骤(其间第一和第二引物与多联体(或环化多核苷酸)杂交)以及引物延伸;而第二阶段包括在高于第一温度的第二温度下的杂交步骤(其间第一和第二引物与包含扩增的第一或第二引物或其互补体的扩增产物杂交)以及引物延伸。与通过仅引物中的第一元件与多联体内的内部靶序列之间杂交而形成较短的片段相比,较高的温度更有利于引物延伸产物中沿引物的第一元件与尾元件之间的杂交。因此,这种两阶段扩增可用于减少否则可能有利于短扩增产物的程度,从而保持了相对较高比例的具有靶序列的两个或更多个拷贝的扩增产物。例如,在进行5个循环(例如至少5、6、7、8、9、10、15、20个或更多个循环)第二温度下的杂交和引物延伸之后,反应混合物中至少5%(例如至少5%、6%、7%、8%、9%、10%、15%、20%、25%、30%或更多)的扩增多核苷酸包含靶序列的两个或更多个拷贝。根据这种两阶段的、加尾B2B引物扩增过程的实施方案的图示在图11中示出。在一些实施方案中,富集包括在倾向于增加扩增子长度的条件下从多联体的扩增。例如,可以降低引物浓度,以使得不是每一个引发位点都会与引物杂交,从而使得PCR产物更长。相似地,降低循环中的引物杂交时间会类似地使得更少的引物杂交,从而也使平均PCR扩增子大小增加。此外,增加循环的温度和/或延伸时间会类似地增加PCR扩增子的平均长度。可以使用这些技术的任意组合。在一些实施方案中,尤其是在已用B2B引物进行了扩增时,对扩增产物进行处理,以根据大小过滤所产生的扩增子,从而减少和/或去除包含多联体的混合物中的单体数目。这可以使用多种可用的技术来完成,包括但不限于,从凝胶上切下片段和凝胶过滤(例如,用于富集长度大于约300、400、500个或更多个核苷酸的片段);以及用于通过微调结合缓冲液浓度进行大小选择的SPRI珠(AgencourtAMPureXP)。例如,可以在与DNA片段混合过程中使用0.6x结合缓冲液来优先结合大于约500个碱基对(bp)的DNA片段。在一些实施方案中,当扩增产生单链多联体时,在为了测序反应而生成的测序文库形成之前或作为该形成的一部分,将单链转换为双链构建体。多种从单链核酸生成双链构建体的合适的方法是可用的。图9描述了一些可能的方法,但是也可以使用许多其他方法。如图9A所示,例如,使用随机引物、聚合酶、dNTP和连接酶会产生双链。图9B描述了当多联体包含衔接子序列时的第二链的合成,其可以在反应中作为引物使用。图9C描述了“环”的使用,其中向多联体的末端添加环衔接子的一个末端,其中环衔接子具有自杂交核酸的一个小部分。在这种情况下,环衔接子的连接产生自杂交的并且作为聚合酶引物模板的环。图9D显示了超分支引物的使用,一般多用于靶序列已知、多条链形成的情况下,特别是当使用具有强链置换功能的聚合酶时。根据一些实施方案,对环化多核苷酸(或其扩增产物,可任选地经富集)进行测序反应以生成测序读取。通过这样的方法生成的测序读取可按照本文公开的其他方法使用。多种测序方法是可用的,尤其是高通量测序方法。实例包括但不限于Illumina制造的测序系统(诸如和的测序系统)、LifeTechnologies制造的测序系统(Ion等)、Roche的454LifeSciences系统、PacificBiosciences系统等。在一些实施方案中,测序包括使用和系统来产生长度约为或多于约50、75、100、125、150、175、200、250、300个或更多个核苷酸的读取。在一些实施方案中,测序包括合成测序过程,其中随着单个核苷酸被添加至生长中的引物延伸产物上,该核苷酸被迭代地鉴定。焦磷酸测序是合成测序过程的一个实例,其通过分析所产生的合成混合物中测序反应副产物即焦磷酸的存在而鉴定核苷酸的掺入。特别是,引物/模板/聚合酶复合体与单一类型的核苷酸接触。如果该核苷酸被掺入,则聚合反应切割三磷酸链的α和β磷酸之间的三磷酸核苷,从而释放焦磷酸。然后使用化学发光酶报告系统鉴定所释放的焦磷酸的存在,该系统将含有AMP的焦磷酸转化为ATP,之后用萤光素酶测量ATP以生成可测量的光信号。检测到光时,碱基被掺入,未检测到光时,碱基未掺入。在适当的洗涤步骤后,使各种碱基循环与该复合体接触,以顺次鉴定模板序列中的后续碱基。参见,例如,美国专利号6,210,891。在相关的测序过程中,将引物/模板/聚合酶复合体固定在基底上,并且该复合体与标记的核苷酸接触。复合体的固定可通过引物序列、模板序列和/或聚合酶进行,并且可以是共价的或非共价的。例如,复合体的固定可以通过聚合酶或引物与基底表面之间的连接而实现。在备选的设置中,核苷酸具有以及不具有可去除的终止基团。在掺入后,标记物与复合体偶联,并因此是可检测的。在携带终止子的核苷酸的情况下,携带可单独鉴定的标记物的全部四种不同的核苷酸与复合体相接触。经标记的核苷酸的掺入由于终止子的存在而阻止了延伸,并且将标记物添加到复合体中,以允许鉴定掺入的核苷酸。然后将标记物和终止子从掺入的核苷酸上去除,并在适当的洗涤步骤后重复该过程。在无终止的核苷酸的情况下,如焦磷酸测序那样,将一种类型的标记的核苷酸添加到复合体中以确定其是否将会掺入。在去除核苷酸上的标记基团和适当的洗涤步骤之后,各种不同的核苷酸在同一过程中通过反应混合物进行循环。参见,例如,美国专利号6,833,246,其为了所有目的通过引用而整体并入本文。例如,Illumina基因组分析系统(IlluminaGenomeAnalyzerSystem)是基于WO98/44151中描述的技术,其中DNA分子通过锚定探针结合位点(另外也称为流动池结合位点)结合到测序平台(流动池)上,并且在载玻片上原位扩增。DNA分子在其上扩增的固体表面一般包含多个第一和第二结合寡核苷酸,第一个与靠近或位于靶多核苷酸的一个末端的序列互补,而第二个与靠近或位于靶多核苷酸的另一个末端的序列互补。这种排列允许进行桥式扩增,例如US20140121116中所描述的。DNA分子然后与测序引物退火,并且使用可逆终止子方法逐个碱基地平行测序。在测序引物的杂交之前,可在锚定双链桥的结合寡核苷酸之一中的切割位点处切割双链桥多核苷酸的一条链,从而留下一条不与固体基底结合的单链,其可通过变性去除,而另一条链结合并可用来与测序引物杂交。一般来说,Illumina基因组测序分析系统使用具有8个通道的流动池,生成长度为18-36个碱基的测序读取,每次运行生成大于1.3Gbp的高质量数据(参见www.illumina.com)在另一个合成测序过程中,随着模板依赖性合成的进行而实时观察不同标记的核苷酸的掺入。具体而言,随着荧光标记的核苷酸的掺入,观察到单独的固定的引物/模板/聚合酶复合体,从而允许随着碱基的添加实时地鉴定每种添加的碱基。在该过程中,标记基团连接于在掺入过程中被切割的核苷酸的一部分上。例如,通过将标记基团连接于在掺入过程中去除的磷酸链的一部分,即,核苷多磷酸上的α、β、γ或其他末端磷酸基团上,该标记物不掺入新生链中,而是生成天然DNA。对单独的分子的观察一般涉及将复合体光学限制在非常小的照明体积内。通过对复合体进行光学限制,创造受监测的区域,其中随机分散的核苷酸在非常短的时间内存在,而掺入的核苷酸随着被掺入而在观察体积内保留更长的时间。这产生了与掺入事件相关的特征信号,其还可通过所加入的碱基的特征性信号谱被表征。在相关的方面,在聚合酶或复合体其他部分和掺入的核苷酸上提供相互作用的标记组分,例如荧光共振能量转移(FRET)染料对,从而掺入事件使得标记组分在交互距离内,并产生特征信号,这对于被掺入的碱基而言也是特征性的(参见,例如,美国专利号6,917,726、7,033,764、7,052,847、7,056,676、7,170,050、7,361,466和7,416,844;和US20070134128)。在一些实施方案中,样品中的核酸可通过连接被测序。该方法一般使用DNA连接酶来鉴定靶序列,例如,如在聚合酶克隆(polony)方法和在SOLiD技术(AppliedBiosystems,现为Invirogen)中所使用的。通常,提供固定长度的所有可能的寡核苷酸的集合,按照测序位置进行标记。将寡核苷酸退火并连接;DNA连接酶对匹配序列的优先连接会产生对应于该位置处的互补序列的信号。根据一些实施方案,如果测序读取与参考序列之间的序列差异在至少两个不同的多核苷酸(例如两个不同的环状多核苷酸,它们可以因具有不同的接点而被区分)中存在,则将其判定为真正的序列变体(例如,存在于扩增或测序前的样品中,并且不是这些过程中的任一个的结果)。因为作为扩增或测序错误而产生的序列变体不大可能在包含相同靶序列的两个不同多核苷酸上被精确地复制(例如位置和类型),所以添加这种验证参数极大地减少了错误序列变体的背景,并伴有对样品中实际序列变异的检测灵敏性和准确性的增加。在一些实施方案中,频率约为或低于约5%、4%、3%、2%、1.5%、1%、0.75%、0.5%、0.25%、0.1%、0.075%、0.05%、0.04%、0.03%、0.02%、0.01%、0.005%、0.001%或更低的序列变体充分高于背景,从而允许准确的判定。在一些实施方案中,序列变体以约为或低于约0.1%的频率发生。在一些实施方案中,当序列变体的频率在统计学上显著地高于背景误差率(例如,p值约为或小于约0.05、0.01、0.001、0.0001或更低)时,该频率充分高于背景。在一些实施方案中,当序列变体的频率约为或至少约为背景误差率的2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、25倍、50倍、100倍或更高(例如至少为5倍更高)时,该频率充分高于背景。在一些实施方案中,在准确确定给定位置的序列时的背景误差率约为或低于约1%、0.5%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%或更低。在一些实施方案中,误差率低于0.001%。在一些实施方案中,鉴定真正的序列变体(也称为“判定”或“作出判定”)包括最优地将一个或多个测序读取与参考序列进行比对以鉴定这两者之间的差异,以及鉴定接点。通常,比对包括将一个序列沿另一个序列放置,迭代地沿每一个序列引入缺口,对两个序列匹配的好坏如何进行打分,并且优选地沿着参考序列对各个位置进行重复。具有最佳得分的匹配被视为对准(alignment),并且代表了关于序列间的关系程度的推断。在一些实施方案中,与测序读取进行比较的参考序列是参考基因组,例如与受试者属于相同种的成员的基因组。参考基因组可以是完整的或不完整的。在一些实施方案中,参考基因组仅由包含靶多核苷酸的区域组成,例如该区域源自参考基因组或源自由所分析的测序读取生成的共有序列。在一些实施方案中,参考序列包含一种或多种生物体的多核苷酸序列,例如,来自一种或多种细菌、古细菌、病毒、原生生物、真菌或其他生物体的序列,或由所述序列组成。在一些实施方案中,参考序列仅由参考基因组的一部分(例如与一个或多个所分析的靶序列相对应的区域(例如,一个或多个基因,或其部分))组成。例如,为检测病原体(例如在检测污染的情况下),参考基因组是该病原体(例如HIV、HPV或有害的细菌菌株,如大肠杆菌)的整个基因组,或是可用于鉴定的其一部分,例如用于鉴定特定的菌株或血清型。在一些实施方案中,将测序读取与多个不同的参考序列进行比对,例如用以筛选多个不同的生物体或菌株。在典型的比对中,测序读取中的碱基旁边有参考序列中的不匹配碱基指示在该点发生了置换突变。类似地,当一个序列在另一序列中的碱基旁边包括缺口时,推断发生了插入或缺失突变(“indel”)。当希望指明一个序列与另一个彼此比对时,该比对有时被称为配对比对(pairwisealignment)。多序列比对通常是指两个或更多个序列的比对,包括,例如通过一系列配对比对。在一些实施方案中,给比对打分涉及为置换和插入/缺失的概然性设定数值。当单独的碱基进行比对时,匹配或不匹配导致根据置换概然性的比对得分,其可以是,例如,1为匹配而0.33为不匹配。插入/缺失从比对得分中扣除空位罚分,其可以是,例如-1。空位罚分和置换概然性可以基于经验知识或基于关于序列如何突变的先验假设。它们的值影响产生的比对。用于进行比对的算法的实例包括但不限于Smith-Waterman(SW)算法、Needleman-Wunsch(NW)算法、基于Burrows-Wheeler转换(BWT)的算法以及散列函数比对器如Novoalign(NovocraftTechnologies;可从www.novocraft.com获得)、ELAND(Illumina,SanDiego,Calif)、SOAP(可从soap.genomics.org.cn获得)和Maq(可从maq.sourceforge.net获得)。执行BWT方法的一个示例性比对程序是Burrows-Wheeler比对器(BWA),其可从Geeknet(Fairfax,Va.)维护的SourceForge网站获得。BWT一般每个核苷酸占据2比特的内存,使得用典型的台式或膝上型计算机对长达4G碱基对的核苷酸序列进行索引化(index)成为可能。预处理过程包括BWT的构建(例如,参考序列的索引化)和支持辅助数据结构。BWA包括两种不同的算法,均基于BWT。通过BWA进行的比对可使用bwa-短(bwa-short)算法进行,该算法是针对高达约200bp的短查询而设计的,具有低误差率(<3%)(LiH.和DurbinR.Bioinformatics,25:1754-60(2009))。第二个算法,BWA-SW,是针对长读取而设计的,具有更多的错误(LiH.和DurbinR.(2010).Fastandaccuratelong-readalignmentwithBurrows-WheelerTransform.Bioinformatics,Epub.)。bwa-sw比对器有时称为“bwa-长”、“bwa长算法”或类似的名称。执行Smith-Waterman算法版本的一个比对程序是MUMmer,其可从Geeknet(Fairfax,Va.)维护的SourceForge网站获得。MUMmer是一个用来快速比对完整基因组的系统,无论它是完整的形式还是草图形式(Kurtz,S.等,GenomeBiology,5:R12(2004);Delcher,A.L.等,Nucl.AcidsRes.,27:11(1999))。例如,MUMmer3.0在2.4GHzLinux台式计算机上,使用78MB的内存,能够在13.7秒内找出在一对5兆碱基基因组之间的全部20-碱基对或更长的精确比配。MUMmer也可以比对不完整的基因组;它能够容易地处理来自鸟枪法测序项目的100s或1000s的叠连群,并且使用该系统内包含的NUCmer程序将其与另一组叠连群或基因组进行比对。比对程序的其他非限制实例包括:来自KentInformatics(SantaCruz,Calif.)的BLAT(Kent,W.J.,GenomeResearch4:656-664(2002));来自北京基因组研究所(北京,Conn.)或BGIAmericasCorporation(Cambridge,Mass.)的SOAP2;Bowtie(Langmead等,GenomeBiology,10:R25(2009));高效大规模核苷酸比对数据库(ELAND)或序列及变异共同评估(CASAVA)软件的ELANDv2组件(Illumina,SanDiego,Calif.);来自RealTimeGenomics,Inc.(SanFrancisco,Calif.)的RTGInvestigator;来自Novocraft(Selangor,Malaysia)的Novoalign;Exonerate,欧洲生物信息学研究所(Hinxton,UK)(Slater,G.和Birney,E.,BMCBioinformatics6:31(2005)),ClustalOmega,来自UniversityCollegeDublin(Dublin,Ireland)(SieversF.等,MolSystBiol7,文章539(2011));来自UniversityCollegeDublin(Dublin,Ireland)的ClustalW或ClustalX(LarkinM.A.等,Bioinformatics,23,2947-2948(2007));和FASTA,欧洲生物信息学研究所(Hinxton,UK)(PearsonW.R.等,PNAS85(8):2444-8(1988);Lipman,D.J.,Science227(4693):1435-41(1985))。一般而言,测序数据从大规模平行测序反应获得。许多新一代高通量测序系统将数据输出为FASTQ文件,但是也可使用其他格式。在一些实施方案中,一般通过序列比对,分析序列以鉴定重复单元长度(例如单体长度)、通过环化形成的接点以及任何相对于参考序列的真正变异。鉴定重复单元长度可以包括计算重复单元的区域,找出序列的参考基因座(例如,当一个或多个序列尤其以扩增、富集和/或测序为目标时)、每一个重复区域的边界和/或每次测序运行中的重复序列的数目。序列分析可以包括分析双链体的两条链的序列数据。如上文所述,在一些实施方案中,来自样品的不同多核苷酸(例如,具有不同接点的环化多核苷酸)的读取序列出现相同变体被认为是确认的变体。在一些实施方案中,如果序列变体在相同多核苷酸的多于一个重复单元中发生,则该序列变体也可被认为是确认的或真正的变体,因为相同的序列变体同样不大可能在相同多联体内的重复靶序列的相同位置上发生。序列的质量得分可在鉴定变体和确认变体时考虑,例如,可以过滤掉质量得分低于阈值的序列和碱基。可以使用其他生物信息学方法进一步提高变体判定的灵敏性和特异性。在一些实施方案中,可以使用统计学分析来确定变异(突变)及量化总DNA样品中的变异比。可使用测序数据计算特定碱基的总测量值。例如,从前面的步骤中计算的比对结果可以计算“有效读取”的数目,也即,针对每个基因座的确认读取的数目。变体的等位基因频率可用针对基因座的有效读取计数进行标准化。可计算总噪音水平,该总噪音水平是在所有基因座中观察到的变体的平均率。变体的频率和总噪音水平,结合其他因素,可用于确定变体判定的置信区间。可使用例如Poisson分布的统计模型来评估变体识别的置信区间。也可使用变体的等位基因频率作为总样品中变体的相对量的指标。在一些实施方案中,基于判定步骤鉴定微生物污染物。例如,特定的序列变体可指示潜在传染性微生物的污染。为了鉴定微生物,可在高度保守的多核苷酸内鉴定序列变体。可用于微生物的系统发生表征和鉴定的示例性高度保守多核苷酸包括在16SrRNA基因、23SrRNA基因、5SrRNA基因、5.8SrRNA基因、12SrRNA基因、18SrRNA基因、28SrRNA基因、gyrB基因、rpoB基因、fusA基因、recA基因、coxl基因和nifD基因中发现的核苷酸序列。对于真核生物,rRNA基因可以是核rRNA基因、线粒体rRNA基因或此两者。在一些实施方案中,16S-23SrRNA基因内部转录间隔区(ITS)中的序列变体可以用于区分和鉴定那些密切相关的分类群,该过程使用或不使用其他rRNA基因。由于16SrRNA的结构限制,整个基因的特定区域具有高度保守的多核苷酸序列,但非结构性区段可能具有高度的变异性。鉴定序列变体可用于鉴定操作分类单位(OTU),OTU代表了亚属、属、亚科、科、亚目、目、亚纲、纲、亚门、门、亚界或界,并且任选地确定其在群体中的频率。特定序列变体的检测可用于检测指示污染的微生物的存在以及任选地检测其量(相对的或绝对的)。示例性的应用包括对粪便或其他污染的水质检测,对动物或人类病原体的检测,定位水污染源,检测再生水或循环水,检测包括海洋排放羽流在内的污水排放流,水产养殖设施的病原体监测,监测沙滩、游泳区或其他水相关的娱乐设施,以及预测有毒藻类的繁茂情况。食物监测应用包括食品加工厂生产线的定期检测,调查屠宰场,检查饭店、医院、学校、监狱和其他机构的厨房和食品储藏区的食源性病原体,如大肠杆菌菌株O157:H7或O111:B4、单核细胞增生李斯特菌(Listeriamonocytogenes)或肠沙门氏菌肠亚种肠炎血清变种(Salmonellaentericasubsp.entericaserovarEnteritidis)。可以检测贝类和贝类所生活的水中造成麻痹性贝类中毒、神经毒性贝类中毒、腹泻型贝类中毒和遗忘型贝类中毒的藻类。此外,可以在放行之前在海关内检查进口食品以保证食品安全。植物病原体监测应用包括园艺和苗圃监测,例如监测造成栎树猝死的微生物——栎树猝死疫霉(Phytophthoraramorum),作物病原体监测及病害治理,和林业病原体监测及病害治理。也可对微生物污染是其主要安全隐患的药品、医疗器械和其他消费品或关键组件的制造环境,调查其中特定病原体如铜绿假单胞菌(Pseudomonasaeruginosa)或金黄葡萄球菌(Staphylococcusaureus)的存在,更多与人类相关的常见微生物的存在,与水的存在相关的微生物的存在,或其他代表以前在该特定环境或类似环境中鉴定的生物负荷的微生物的存在。类似地,可以对包括航天器在内的敏感设备的建造和组装区监测以前确定的、已知栖息于该环境中或最常见地引入该环境中的微生物。在一个方面,本发明提供了一种鉴定核酸样品中的序列变体的方法,该核酸样品包含少于50ng的多核苷酸,每个多核苷酸具有5’末端和3’末端。在一些实施方案中,该方法包括:(a)用连接酶环化所述样品中的单独的多核苷酸以形成多个环状多核苷酸;(b)一旦从所述环状多核苷酸中分离出所述连接酶,即扩增该环状多核苷酸以形成多联体;(c)对该多联体进行测序以生成多个测序读取;(d)鉴定该多个测序读取与参考序列之间的序列差异;和(e)将从所述少于50ng多核酸的核酸样品获得的所述多个读取中以0.05%或更高的频率发生的序列差异判定为序列变体。在样品中的多核苷酸的起始量可以很小。在一些实施方案中,起始核苷酸的量少于50ng,例如少于45ng、40ng、35ng、30ng、25ng、20ng、15ng、10ng、5ng、4ng、3ng、2ng、1ng、0.5ng、0.1ng或更少。在一些实施方案中,起始多核苷酸的量在0.1-100ng的范围内,例如1-75ng、5-50ng或10-20ng。通常,较少的起始材料提高了增加各个处理步骤的回收率的重要性。减少样品中用于参与后续反应的多核苷酸的量的过程降低了可检测出罕见突变的灵敏度。例如,Lou等(PNAS,2013,110(49))描述的方法预期仅回收起始材料的10-20%。对于大量的起始材料(例如,从实验室培养的细菌中纯化的)而言,这可能不是实质性的障碍。但是,对于起始材料显著较低的样品,在该低范围内的回收可能是检测十分罕见的变体中的实质性障碍。因此,在一些实施方案中,在本发明的方法中从一个步骤向另一步骤的样品回收率(例如,向环化步骤的输入中可用于输入到后续扩增步骤或测序步骤的质量分数)约为或大于约50%、60%、75%、80%、85%、90%、95%或更高。特定步骤的回收率可以接近100%。回收率可以是关于特定形式的回收率,例如从非环状多核苷酸输入到环状多核苷酸的回收率。所述多核苷酸可以来自任何合适的样品,例如本文针对本发明各个方面所述的样品。来自样品的多核苷酸可以是多种多核苷酸中的任意一种,包括但不限于DNA、RNA、核糖体RNA(rRNA)、转运RNA(tRNA)、微小RNA(miRNA)、信使RNA(mRNA)、以上任意一种的片段或以上任意两种或更多种的组合。在一些实施方案中,样品包含DNA。在一些实施方案中,多核苷酸是单链的,或者是直接获得的或者是通过处理(例如变性)生成的。本文描述了合适的多核苷酸的其他实例,例如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,多核苷酸不经提取步骤和/或不经纯化步骤而经历后续步骤(例如环化和扩增)。例如,流体样品可以不经提取步骤而进行处理以去除细胞,从而产生纯化的液体样品和细胞样品,随后从纯化的流体样品中分离DNA。多种用于分离多核苷酸的程序是可用的,例如通过沉淀,或与基底的非特异性结合,随后洗涤基底以释放结合的多核苷酸。当不经细胞提取步骤而从样品中分离多核苷酸时,多核苷酸大部分是细胞外的或“无细胞的”多核苷酸,其可对应于死亡或受损的细胞。对这类细胞的鉴定可用于表征它们所来源于的细胞或细胞群体,例如在微生物群落中。如果对样品进行处理以提取多核苷酸,例如从样品中的细胞中提取,有多种提取方法是可用的,其实例在本文中提供(例如,针对本发明各个方面中的任何方面)。核酸样品中的序列变体可以是多种序列变体中的任意一种。本文描述了序列变体的多个非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,序列变体是单核苷酸多态性(SNP)。在一些实施方案中,序列变体在群体中以低频率发生(也称为“罕见”序列变体)。例如,序列变体可以以约为或低于约5%、4%、3%、2%、1.5%、1%、0.75%、0.5%、0.25%、0.1%、0.075%、0.05%、0.04%、0.03%、0.02%、0.01%、0.005%、0.001%或更低的频率发生。在一些实施方案中,序列变体以约为或低于约0.1%的频率发生。根据一些实施方案,将样品的多核苷酸进行环化,例如通过使用连接酶。环化可包括将多核苷酸的5’末端连接到同一多核苷酸的3’末端,连接到样品中的另一多核苷酸的3’末端,或连接到来自不同来源的多核苷酸(例如,人工多核苷酸,如寡核苷酸衔接子)的3’末端。在一些实施方案中,多核苷酸的5’末端连接到同一多核苷酸的3’末端(也称为“自连接”)。本文提供了环化过程(使用或不使用衔接子寡核苷酸)、试剂(例如,衔接子的种类、连接酶的使用)、反应条件(例如,有利于自连接)以及任选的附加处理(例如反应后纯化)的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。通常,将多核苷酸的末端彼此连接以形成环状多核苷酸(或者是直接地,或者是使用一个或多个间插衔接子寡核苷酸)会产生具有接点序列的接点。当多核苷酸的5’末端和3’末端通过衔接子多核苷酸连接时,术语“接点”可以是指多核苷酸与衔接子之间的接点(例如5’末端接点或3’末端接点之一),或指如通过衔接子多核苷酸形成并包含衔接子多核苷酸的、多核苷酸的5’末端与3’末端之间的接点。当多核苷酸的5’末端和3’末端在不使用间插衔接子的情况下连接时(例如,单链DNA的5’末端和3’末端),术语“接点”是指这两个末端相连接的点。接点可以根据包含接点的多核苷酸的序列(也称为“接点序列”)进行鉴定。在一些实施方案中,样品包含具有通过以下过程形成的末端混合物的多核苷酸:自然降解过程(例如细胞裂解、细胞死亡和其他DNA从细胞释放到其周围环境的过程,DNA在该周围环境中可进一步降解,例如在无细胞多核苷酸中),作为样品处理的副产物的片段化(例如,固定、染色和/或存储过程),以及通过不限制于特定靶序列的切割DNA的方法进行的片段化(例如,机械片段化,如超声处理;非序列特异性核酸酶处理,如DNaseI、片段化酶(fragmentase))。当样品包含具有末端混合物的多核苷酸时,两个核苷酸具有相同5’末端或3’末端的可能性很低,并且两个核苷酸独立地具有相同的5’末端和3’末端两者的可能性极低。因此,在一些实施方案中,甚至在两个多核苷酸包含具有相同靶序列的部分时,可以使用接点来区分不同的多核苷酸。当多核苷酸末端在不使用间插衔接子的情况下连接时,接点序列可通过与参考序列比对进行鉴定。例如,当两个组分序列的顺序相对于参考序列表现出反转时,显示发生反转的点可以指示在该点具有接点。当多核苷酸末端通过一个或多个衔接子序列连接时,接点可通过与已知衔接子序列的邻近进行鉴定,或者在测序读取的长度足以从环化多核苷酸的5’和3’末端均获得序列的情况下通过上述比对进行鉴定。在一些实施方案中,特定接点的形成是十分罕见的事件,以使得其在样品的环化多核苷酸之中是独特的。环化后,反应产物可在扩增或测序之前进行纯化以提高可参与后续步骤的环化多核苷酸的相对浓度或纯度(例如,通过环状多核苷酸的分离或反应中一种或多种其他分子的去除)。例如,可处理环化反应或其组分以去除单链(未环化的)多核苷酸,例如通过外切核酸酶处理。作为进一步的实例,环化反应或其部分可进行大小排除色谱法,借此保留及丢弃小试剂(例如未反应的衔接子),或在单独的体积中保留并释放环化产物。多种用于清理连接反应的试剂盒是可用的,例如由ZymoReaserch制造的Zymo寡核苷酸纯化试剂盒所提供的试剂盒。在一些实施方案中,纯化包括用于去除或降解在环化反应中使用的连接酶和/或将环化多核苷酸从该连接酶中纯化出的处理。在一些实施方案中,用于降解连接酶的处理包括蛋白酶处理。合适的蛋白酶可从原核生物、病毒和真核生物获得。蛋白酶的实例包括蛋白酶K(来自白色念球菌(Tritirachiumalbum))、链霉蛋白酶E(来自灰色链霉菌(Streptomycesgriseus))、多粘芽孢杆菌(Bacilluspolymyxa)蛋白酶、嗜热菌蛋白酶(来自嗜热菌)、胰蛋白酶、枯草杆菌蛋白酶、弗林蛋白酶等。在一些实施方案中,该蛋白酶是蛋白酶K。蛋白酶K处理可遵循制造商的方案或使用标准条件(例如,如Sambrook和Green,MolecularCloning:ALaboratoryManual,第4版(2012)所提供的)。蛋白酶处理之后还可进行提取和沉淀。在一个实例中,环化多核苷酸如下纯化:在0.1%SDS和20mMEDTA的存在下进行蛋白酶K(Qiagen)处理,用1:1苯酚/氯仿和氯仿抽提,并用乙醇或异丙醇沉淀。在一些实施方案中,沉淀在乙醇中进行。正如针对本发明的其他方面所述,环化后可直接对环化多核苷酸进行测序。或者,测序之前可进行一个或多个扩增反应。多种扩增多核苷酸(例如,DNA和/或RNA)的方法是可用的。扩增可以是线性的,指数式的,或在多阶段扩增过程中涉及线性和指数阶段。扩增方法可包括温度的改变,例如热变性步骤,或者可以是不需要热变性的等温过程。本文描述了合适的扩增过程的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,扩增包括滚环扩增(RCA)。如本文其他部分所述,典型的RCA反应混合物包含一种或多种引物、聚合酶和dNTPs,并且生成多联体。一般来说,RCA反应中的聚合酶是具有链置换活性的聚合酶。多种这样的聚合酶是可用的,其非限制性实例包括外切核酸酶-DNA聚合酶I大(Klenow)片段、Phi29DNA聚合酶、TaqDNA聚合酶等。通常,多联体是包含来自模板核苷酸的靶序列的两个或更多个拷贝(例如靶序列的约或多于约2、3、4、5、6、7、8、9、10个或更多个拷贝;在一些实施方案中,约为或多于约2个拷贝)的多核苷酸扩增产物。扩增引物可以是任何合适的长度,例如约为或至少约5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、90、100个或更多个核苷酸,其任意部分或全部可互补于引物所杂交的相应靶序列(例如,约为或至少约5、10、15、20、25、30、35、40、45、50个或更多个核苷酸)。本文描述了多种RCA过程的实例,例如使用随机引物、靶标特异性引物和衔接子靶向引物,其中一些示于图7中。当环化多核苷酸在测序之前扩增(例如,以产生多联体)时,扩增产物可以在不进行富集的情况下直接测序,或者在一个或多个富集步骤后进行测序。本文描述了合适的富集过程的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述(例如,在第二扩增步骤中使用B2B引物)。根据一些实施方案,环化的多核苷酸(或其扩增产物,任选地可能已经富集)进行测序反应以生成测序读取。通过这样的方法生成的测序读取可按照本文公开的其他方法使用。多种测序方法是可用的,尤其是高通量测序方法。实例包括但不限于Illumina制造的测序系统(诸如和的测序系统)、LifeTechnologies制造的测序系统(Ion等)、Roche的454LifeSciences系统、PacificBiosciences系统等。在一些实施方案中,测序包括使用和系统产生长度约为或多于约50、75、100、125、150、175、200、250、300个或更多个核苷酸的读取。本文描述了扩增平台和方法的其他非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。根据一些实施方案,如果测序读取与参考序列之间的序列差异在至少两个不同的多核苷酸(例如两个不同的环状多核苷酸,它们可以因具有不同的接点而进行区分)中发生,则将其判定为真正的序列变体(例如,存在于扩增或测序前的样品中,并且不是这些过程中的任何一个过程的结果)。因为作为扩增或测序错误的结果的序列变体不大可能在包含相同靶序列的两个不同多核苷酸上准确地重复(例如位置和类型),所以添加这种验证参数极大地减少了错误序列变体的背景,同时伴有检测样品中的实际序列变异的灵敏性和准确性的增加。在一些实施方案中,频率约为或低于约5%、4%、3%、2%、1.5%、1%、0.75%、0.5%、0.25%、0.1%、0.075%、0.05%、0.04%、0.03%、0.02%、0.01%、0.005%、0.001%或更低的序列变体充分高于背景,从而允许精确的判定。在一些实施方案中,序列变体以约为或低于约0.1%的频率发生。在一些实施方案中,该方法包括将那些频率在约0.0005%至约3%的范围内,如在0.001%-2%或0.01%-1%之间的序列差异判定为真正的序列变体。在一些实施方案中,当序列变体的频率在统计学上显著地高于背景误差率(例如,p值约为或小于约0.05、0.01、0.001、0.0001或更低)时,该频率充分高于背景。在一些实施方案中,当序列变体的频率约为或至少约为背景误差率的2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、25倍、50倍、100倍或更高(例如至少高5倍)时,该频率充分高于背景。在一些实施方案中,在精确确定给定位置的序列时的背景误差率约为或低于约1%、0.5%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%或更低。在一些实施方案中,误差率低于0.001%。本文描述了确定频率和误差率的方法,如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,鉴定真正的序列变体(也称为“判定”或“作出判定”)包括最优地将一个或多个测序读取与参考序列进行比对以鉴定这两者之间的差异,以及鉴定接点。通常,比对包括将一个序列沿另一个序列放置,迭代地沿每一个序列引入缺口,对两个序列匹配的好坏如何进行打分,并且优选地沿着参考序列对各个位置进行重复。具有最佳得分的匹配被视为对准,并且代表了关于序列间的关系程度的推断。多种比对算法和执行该算法的比对器是可用的,其非限制实例在本文中描述,如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,与测序读取进行比较的参考序列是已知的参考序列,如参考基因组(例如与受试者属于同一种的成员的基因组)。参考基因组可以是完整的或不完整的。在一些实施方案中,参考基因组仅由包含靶多核苷酸的区域组成,例如该区域源自参考基因组或源自所分析的测序读取生成的共有序列。在一些实施方案中,参考序列包含一个或多个生物体的多核苷酸序列,例如,来自一个或多个细菌、古细菌、病毒、原生生物、真菌或其他生物体的序列,或由该序列组成。在一些实施方案中,参考序列仅由参考基因组的一部分,例如与一个或多个所分析的靶序列相对应的区域(例如,一个或多个基因,或其部分)组成。例如,为检测病原体(例如在检测污染的情况下),参考基因组是该病原体(例如HIV、HPV或有害的细菌菌株,如大肠杆菌)的完整基因组,或是其一部分,该部分可用于鉴定,例如鉴定特定的菌株或血清型。在一些实施方案中,测序读取与多个不同的参考序列进行比对,例如用于筛选多个不同的生物体或菌株。本文描述了可以与之对比地鉴定序列差异的参考序列的其他非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。在一个方面,本发明提供了一种在反应混合物中扩增多个不同多联体的方法,该多联体包含靶序列的两或更多个拷贝,其中该靶序列包含以5’到3’方向定向的序列A和序列B。在一些实施方案中,该方法包括将反应混合物进行核酸扩增反应,其中该反应混合物包含:(a)多个多联体,其中该多个多联体中的单独的多联体包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过序列B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以生成扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。在一个相关方面中,本发明提供了一种在反应混合物中扩增多个包含靶序列的不同环状多核苷酸的方法,其中该靶序列包含以5’到3’方向定向的序列A和序列B。在一些实施方案中,该方法包括将反应混合物进行核酸扩增反应,其中该反应混合物包含:(a)多个环状多核苷酸,其中该多个环状多核苷酸中的单独的环状多核苷酸包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过序列B与B’之间的序列互补性特异性地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以生成扩增的多核苷酸;其中序列A和序列B为内源性序列,并且靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。无论是扩增环状多核苷酸还是扩增多联体,此类多核苷酸都可来自任何合适的样品来源(或者是直接地,或者是间接地,例如通过扩增)。本文描述了多种合适的样品来源、任选的提取方法、多核苷酸的类型以及序列变体的类型,例如针对本发明各个方面中的任何方面进行了描述。环状多核苷酸可由非环状多核苷酸的环化产生。本文提供了环化过程(例如,使用及不使用衔接子寡核苷酸)、试剂(例如,衔接子的类型、连接酶的使用)、反应条件(例如,有利于自连接)、任选的附加处理(例如反应后纯化)以及由此形成的接点的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。多联体可由环状多核苷酸的扩增产生。多种扩增多核苷酸(例如,DNA和/或RNA)的方法是可用的,其非限制实例也在本文中描述。在一些实施方案中,多联体通过环状多核苷酸的滚环扩增产生。图10示出了第一和第二引物相对于在单一重复情况下的靶序列(其除非为环状,否则一般不会扩增)和包含靶序列的多个拷贝的多联体的排列实例。如针对本文描述的其他方面所指出的,该引物排列可称为“背靠背”(B2B)或“反向”引物。用B2B引物进行的扩增促进了环状和/或多联体模板的富集。此外,该定向与相对较小的足印(一对引物所跨越的总距离)结合起来,使得能够扩增靶序列周围的更加多样的片段化事件,因为与在典型扩增反应中所见的引物排列(彼此面对,跨越靶序列)相比,不大可能在引物之间出现接点。在一些实施方案中,序列A的5’末端与序列B的3’末端之间的距离约为或少于约200、150、100、75、50、40、30、25、20、15个或更少的核苷酸。在一些实施方案中,序列A是序列B的互补体。在一些实施方案中,在同一反应中使用针对多个不同靶序列的多个B2B引物对,以平行地扩增多个不同的靶序列(例如约或至少约10、50、100、150、200、250、300、400、500、1000、2500、5000、10000、15000个或更多个不同的靶序列)。引物可以具有任意合适的长度,例如在本文其他部分所描述的。扩增可包括在适当条件下的任意合适的扩增反应,例如本文所描述的扩增反应。在一些实施方案中,扩增是聚合酶链反应。在一些实施方案中,B2B引物包含至少两个序列元件:通过序列互补性与靶序列杂交的第一元件,和在第一杂交温度下的第一扩增阶段中不会与靶序列杂交的5’“尾”,在该扩增阶段期间第一元件发生杂交(例如,由于在尾部和紧邻第一元件结合处位于其3’侧的靶序列部分之间缺乏序列互补性)。例如,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在第一杂交温度下的第一扩增阶段期间均不与多个多联体(或环状多核苷酸)杂交。在使用了此类加尾引物的一些实施方案中,扩增可包括第一阶段和第二阶段;第一阶段包括在第一温度下的杂交步骤,其间第一和第二引物与多联体(或环状多核苷酸)杂交,以及引物延伸;而第二阶段包括在高于第一温度的第二温度下的杂交步骤,其间第一和第二引物与包含延伸的第一或第二引物或其互补体的扩增产物杂交,以及引物延伸。在两个温度中的每个温度下的扩增循环数可基于所需的产物进行调整。一般来说,第一温度将用于数目相对较少的循环,例如约或少于约15、10、9、8、7、6、5个或更少的循环。在较高温度下的循环数可独立于第一温度下的循环数进行选择,但一般是同样多的或更多的循环,例如约或至少约5、6、7、8、9、10、15、20、25个或更多的循环。与通过仅引物中的第一元件与多联体内的内部靶序列之间杂交而形成较短的片段相比,较高的温度更有利于引物延伸产物中沿引物的第一元件与尾元件之间的杂交。因此,这种两阶段扩增可用于减少可能有利于短扩增产物的程度,从而保持了相对较高比例的扩增产物具有靶序列的两个或更多个拷贝。例如,在5个循环(例如至少5、6、7、8、9、10、15、20个或更多个循环)的第二温度下的杂交和引物延伸之后,反应混合物中至少5%(例如至少5%、6%、7%、8%、9%、10%、15%、20%、25%、30%或更多)的扩增多核苷酸包含靶序列的两个或更多个拷贝。根据这种两阶段、加尾B2B引物扩增过程的实施方案的图示在图11中示出。在一些实施方案中,扩增的条件设置为偏向于增加自多联体的扩增子长度。例如,可以降低引物浓度,以使得不是每一个引发位点都会与引物杂交,从而使得PCR产物更长。类似地,减少循环中的引物杂交时间会类似地使得更少的引物能够杂交,从而也使平均PCR扩增子大小增加。此外,增加循环的温度和/或延伸时间同样会增加PCR扩增子的平均长度。可以使用这些技术的任意组合。在一些实施方案中,尤其是在已用B2B引物进行了扩增时,对扩增产物进行处理,以根据大小过滤所得到的扩增子,从而减少和/或去除包含多联体的混合物中的单体数目。这可以使用多种可用的技术来完成,包括但不限于,从凝胶上切下片段和凝胶过滤(例如,用于富集长度大于约300、400、500个或更多个核苷酸的片段);以及用于通过微调结合缓冲液浓度进行大小选择的SPRI珠(AgencourtAMPureXP)。例如,可以在与DNA片段混合过程中使用0.6x结合缓冲液来优先结合大于约500个碱基对(bp)的DNA片段。在一些实施方案中,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在第一杂交温度下的第一扩增阶段期间均不与多个环状多核苷酸杂交。扩增可包括第一阶段和第二阶段;其中第一阶段包括在第一温度下的杂交步骤,其间第一和第二引物在引物延伸前与环状多核苷酸或其扩增产物杂交;而第二阶段包括在高于第一温度的第二温度下的杂交步骤,其间第一和第二引物与包含延伸的第一或第二引物或其互补体的扩增产物杂交。例如,第一温度可以选择为约为或高于大约序列A’、序列B的Tm或其平均值,或比这些Tm之一高1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃或更高的温度。在这种实例中,第二温度可以选择为约为或高于约组合序列(A’+C)、组合序列(B+D)的Tm或其平均值,或比这些Tm之一高1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃或更高的温度。术语“Tm”也被称为“解链温度”,并且通常代表50%的由参考序列(其实际上可能是较大多核苷酸内的子序列)及其互补序列组成的寡核苷酸发生杂交(或分离)时的温度。通常,Tm随长度的增加而升高,因此,序列A’的Tm预期低于组合序列(A’+C)的Tm。在一个方面,本发明提供了一种用于按照本发明的方法实施方法的反应混合物。该反应混合物可以包含如本文对于任意多种方法所述的各种组分中的一种或多种,包括在本文所述的方法中描述的反应混合物。在一些实施方案中,该反应混合物是用于扩增多个包含靶序列的两个或更多个拷贝的不同多联体或包含靶序列的一个或多个拷贝的环状多核苷酸(例如环状单体)的混合物,其中该靶序列包含以5’到3’方向定向的序列A和序列B,该反应混合物包含:(a)多个多联体(或环状多核苷酸),其中该多个多联体(或环状多核苷酸)中的单独的多联体(或环状多核苷酸)包含通过环化具有5’末端和3’末端的单独多核苷酸而形成的不同接点;(b)包含序列A’的第一引物,其中该第一引物与靶序列的序列A通过序列A与序列A’之间的序列互补性特异性地杂交;(c)包含序列B的第二引物,其中该第二引物与存在于包含靶序列互补体的互补多核苷酸中的序列B’通过B与B’之间的序列互补性特异地杂交;以及(d)聚合酶,其延伸第一引物和第二引物以生成扩增的多核苷酸;其中靶序列的序列A的5’末端与序列B的3’末端之间的距离为75nt或更短。样品、多核苷酸、引物、聚合酶、其他试剂和反应条件可以是任何本文所述的那些,例如针对各个方面中的任何方面所述的那些,其可以以任意合适的组合包括在反应混合物中。在一些实施方案中,第一引物包含相对于序列A’位于5’侧的序列C,第二引物包含相对于序列B位于5’侧的序列D,并且序列C和序列D在扩增反应的第一扩增步骤期间均不与所述两个或更多个多联体杂交。在一个方面,本发明提供了在本文所述的方法中有用的或由其产生的组合物,例如在本发明的各个其他方面的任意方面中。在一些实施方案中,该组合物包含多个为单链的且基本上不含连接酶的环化多核苷酸。在一些实施方案中,该组合物包含多个多联体,其中该多个多联体对应于一组10000个或更少的靶多核苷酸,并且进一步地,其中该多个多联体中的单独多联体的特征在于:(a)它们包含序列重复的两个或更多个拷贝,其中所有的所述拷贝均对应于相同的靶多核苷酸;且(b)一个单独的多联体中序列重复的两个或更多个拷贝之间的接点与所述组合物中的另一个单独多联体中的不同。样品、多核苷酸、引物、聚合酶和其他试剂可以是任何本文所述的那些,例如针对各个方面中的任何方面所述的那些,其可以以任意合适的组合包含在组合物中。该组合物可包括一对或多对引物,例如本文所述的B2B引物,其设计用于扩增一个或多个靶序列。组合物可以以试剂盒的形式提供。试剂盒中的试剂和其他材料可以容纳在任何合适的容器中,并且可以是直接可用的形式,或者需要与试剂盒中的其他试剂或使用者提供的试剂进行组合(例如,浓缩的组合物的稀释或冻干组合物的重建)。试剂盒可提供缓冲液,其非限制性实例包括碳酸钠缓冲液、碳酸氢钠缓冲液、硼酸盐缓冲液、Tris缓冲液、MOPS缓冲液、HEPES缓冲液及其组合。试剂盒可进一步包含关于实施本文中针对各个方面中的任何方面所描述的一种或多种方法的说明。说明可以用一种或多种语言(例如2、3、4、5种或更多种语言)提供。在一个方面,本文公开提供了一种用于检测序列变体的系统。在一些实施方案中,该系统包含(a)计算机,其配置为接收对样品进行检测反应的用户请求;(b)扩增系统,其响应于用户请求对样品或其一部分进行核酸扩增反应,其中该扩增反应包括以下步骤:(i)将单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸都在5’末端与3’末端之间具有接点;和(ii)扩增该环状多核苷酸;(c)测序系统,其针对由该扩增系统扩增的多核苷酸生成测序读取,鉴定测序读取与参考序列之间的序列差异,并且将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体;和(d)向接收方发送报告的报告生成器,其中该报告包含用于序列变体检测的结果。在一些实施方案中,该接收方为用户。图32示出了在本发明方法中有用的系统的非限制性实例。用于在所述系统中使用的计算机可包含一个或多个处理器。处理器可以与一个或多个控制器、计算单元和/或计算机系统的其他单元相关联,或者根据需要植入固件中。如果在软件中实现,则例程可存储在任何计算机可读存储器中,诸如存储在RAM、ROM、闪速存储器、磁盘、激光盘或其他合适的存储介质中。同样地,该软件可经由任何已知的传送方法而传送至计算设备,所述传送方法例如包括通过诸如电话线、因特网、无线连接等通信信道,或者经由诸如计算机可读盘、闪存驱动器等可移动介质。各个步骤可实现为各个块、操作、工具、模块和技术,而所述各个块、操作、工具、模块和技术转而可在硬件、固件、软件或者硬件、固件和/或软件的任何组合中实现。当在硬件中实现时,所述块、操作、技术等之中的一些或全部块、操作、技术可例如在定制集成电路(IC)、专用集成电路(ASIC)、现场可编程逻辑阵列(FPGA)、可编程逻辑阵列(PLA)等中实现。在系统的实施方案中可以使用客户端-服务器关系数据库架构。客户端-服务器架构是这样的网络架构:在其中网络上的每个计算机或过程是客户端或服务器。服务器计算机通常是专用于管理磁盘驱动器(文件服务器)、打印机(打印服务器)或网络流量(网络服务器)的强大的计算机。客户端计算机包括用户在其上运行应用程序的PC(个人计算机)或工作站,以及本文所公开的示例输出设备。客户端计算机依靠服务器计算机获得资源,诸如文件、设备甚至处理能力。在一些实施方案中,服务器计算机处理所有的数据库功能。客户端计算机可具有处理所有前端数据管理的软件,并且还可接收来自用户的数据输入。该系统可被配置用于接收对样品进行检测反应的用户请求。该用户请求可以是直接的或间接的。直接请求的实例包括通过诸如键盘、鼠标或触摸屏等输入设备传输的请求。间接请求的实例包括经由通信介质,例如通过因特网(有线或无线)的传输。该系统可进一步包含响应于用户请求对样品或其部分进行核酸扩增反应的扩增系统。多种扩增多核苷酸(例如,DNA和/或RNA)的方法是可用的。扩增可以是线性的,指数式的,或在多阶段扩增过程中涉及线性和指数阶段。扩增方法可包括温度的改变,例如热变性步骤,或者可以是不需要热变性的等温过程。本文描述了合适的扩增过程的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。在一些实施方案中,扩增包括滚环扩增(RCA)。多种用于扩增多核苷酸的系统均是可用的,并且可基于将要进行的扩增反应的类型而不同。例如,对于包括温度变化循环的扩增方法,扩增系统可包含热循环仪。扩增系统可包含实时扩增和检测仪器,例如由AppliedBiosystems、Roche和Strategene制造的系统。在一些实施方案中,扩增反应包括以下步骤:(i)将单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸都在5’末端与3’末端之间具有接点;和(ii)扩增该环状多核苷酸。样品、多核苷酸、引物、聚合酶和其他试剂可以是任何本文所述的那些,例如针对各个方面中的任何方面所述的那些。本文提供了环化过程(例如,使用及不使用衔接子寡核苷酸)、试剂(例如,衔接子的类型、连接酶的使用)、反应条件(例如,有利于自连接)、任选的附加处理(例如反应后纯化)以及由此形成的接点的非限制性实例,例如针对本发明各个方面中的任何方面进行了描述。可将系统选择和/或设计为执行任何这样的方法。系统可进一步包含测序系统,该测序系统针对由扩增系统扩增的多核苷酸生成测序读取,鉴定该测序读取与参考序列之间的序列差异,并且将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体。测序系统和扩增系统可以是相同的,或包含重叠的设备。例如,扩增系统和测序系统都可使用相同的热循环仪。供该系统使用的多种测序平台是可用的,并且基于所选定的测序方法进行选择。本文描述了测序方法的实例。扩增和测序可以包括液体处理器的使用。几种市售的液体处理系统可用于运行这些过程的自动化操作(参见,例如,来自Perkin-Elmer、BeckmanCoulter、CaliperLifeSciences、Tecan、Eppendorf、ApricotDesign、Velocity11的液体处理器作为示例)。多种自动测序仪是可商购获得的,并且包括由LifeTechnologies(SOLiD平台,和基于pH的检测)、Roche(454平台)、Illumina(例如,基于流动池的系统,如GenomeAnalyzer装置)制造的测序仪。在2、3、4、5台或更多的自动化设备之间(例如,在液体处理器和测序装置中的一个或多个之间)的转移可以是手动的或自动化的。本文描述了相对于参考序列鉴定序列差异和判定序列变体的方法,例如针对本发明各个方面中的任何方面进行了描述。测序系统通常包括软件,该软件用于响应于测序数据的输入和所需参数的输入(例如参考基因组的选择)而执行这些步骤。比对算法和执行这些算法的比对器的实例在本文中描述,并且可构成测序系统的一部分。所述系统可进一步包含向接收方发送报告的报告生成器,其中该报告包含用于序列变体的检测的结果。报告可实时生成,例如在测序读取期间或在分析测序数据时,并且随着过程的进展定期更新。此外,或可替代地,报告可在分析结束时生成。该报告可自动生成,例如当测序系统完成判定所有序列变体的步骤时。在一些实施方案中,该报告响应于用户的指令而生成。除了测序变体的检测结果,报告还可包含基于一个或多个序列变体的分析。例如,当一个或多个序列变体与特定污染物或表型相关时,该报告可包括关于此种相关性的信息,例如该污染物或表型存在的可能性、处于什么水平,以及任选的基于此信息的建议(例如额外的测试、监控或补救措施)。该报告可以采取多种形式中的任何形式。可以预想到,与本公开内容相关的数据可通过这样的网络或连接(或任何其他合适的传送信息的手段,包括但不限于邮寄体检报告,诸如打印输出)传送,以供接收和/或由接收者查阅。接收者可以是但不限于个人或电子系统(例如,一个或多个计算机,和/或一个或多个服务器)。在一个方面,本发明提供了一种包含代码的计算机可读介质,该代码一旦由一个或多个处理器执行,即实施检测序列变体的方法。在一些实施方案中,该实施的方法包括:(a)接收对样品进行检测反应的客户请求;(b)响应于客户请求对样品或其一部分进行核酸扩增反应,其中该扩增反应包括以下步骤:(i)将单独的多核苷酸进行环化以形成多个环状多核苷酸,其中每一个环状多核苷酸都在5’末端与3’末端之间具有接点;和(ii)扩增该环状多核苷酸;(c)进行测序分析,其包括以下步骤:(i)针对在扩增反应中扩增的多核苷酸生成测序读取;(ii)鉴定测序读取与参考序列之间的序列差异;以及(iii)将存在于至少两个具有不同接点的环状多核苷酸中的序列差异判定为序列变体;和(d)生成包含用于序列变体检测的结果的报告。包含计算机可执行代码的机器可读介质可以采取许多形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质例如包括光盘或磁盘(诸如任何计算机中的任何存储设备)等,诸如可用于实现数据库的存储介质等。易失性存储介质包括动态存储器,例如这样的计算机平台的主存储器。有形传输介质包括同轴线缆、铜线和光纤,包括构成计算机系统内的总线的导线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(RF)和红外(IR)数据通信期间所生成的电信号或电磁信号或者声波或光波。因此,计算机可读介质的常见形式包括,例如:软盘、柔性盘、硬盘、磁带、任何其他磁介质、CD-ROM、DVD或DVD-ROM、任何其他光学介质、穿孔卡片纸带、任何具有孔洞图案的其他物理存储介质、RAM、ROM、PROM和EPROM、FLASH-EPROM、任何其他存储器芯片或盒、载波传输数据或指令、传输此类载波的缆线或链路,或者任何可让计算机从中读取编程代码和/或数据的其他介质。这些计算机可读介质的形式中的许多形式可参与向处理器传送一个或多个序列的一个或多个指令以供执行。本发明的计算机可执行代码可以在包括服务器、PC或者诸如智能电话或平板计算机等移动设备在内的任何包含处理器的合适的设备上执行。任何控制器或计算机任选地包括监视器,该监视器可以是阴极射线管(“CRT”)显示器、平板显示器(例如,有源矩阵液晶显示器、液晶显示器等)或其他显示器。计算机电路通常被置于盒中,该盒包含许多集成电路芯片,诸如微处理器、存储器、接口电路及其他芯片。该盒任选地还包含硬盘驱动器、软盘驱动器、高容量可移动驱动器诸如可写CD-ROM,以及其他常见的外设元件。诸如键盘、鼠标或触摸屏等输入设备任选地提供来自用户的输入。计算机可包括用于接收用户指令的适当的软件,所述用户指令的形式为向一组参数字段中的用户输入(例如,在GUI中),或者形式为预编程指令(例如,预编程用于多种不同的具体操作)。实施例下列实施例是为了说明本发明的各种实施方案而给出的,并非意在以任何方式限制本发明。这些实施例以及其中描述的方法目前均代表优选的实施方案,是示例性的,并且不应作为对本发明范围的限制。本领域技术人员将会想到由权利要求范围限定的本发明范围内所包含的其变化和其他用途。实施例1:制备用于突变检测的串联重复测序文库由在12μL水或10mMTris-HClpH8.0中的>10ng约150bpDNA片段起始,加入2μL10XCircLigase缓冲液混合物,并加热到95℃2分钟,在冰上冷却5分钟。加入4μL5M甜菜碱、1μL50mMMnCl2和1μLCircLigaseII。在60℃孵育至少12小时。加入2μLRCA引物混合物(各50nM,至5nM终浓度),并混合。加热至95℃2分钟,并冷却至42℃2小时。用ZYMO寡核苷酸纯化试剂盒纯化CirLigation产物。根据制造商的说明,将28μL水加至22μLCircLigation产物至50μL的总体积。与100μL寡核苷酸结合缓冲液和400μL乙醇混合。以>10,000×g离心30秒,然后弃去流出液。加入750μLDNA洗涤缓冲液,以>10,000×g离心30秒,弃去流出液,并以最高速度再离心1分钟。将柱子转移至新的Eppendorf管中,并用17μL水洗脱(最终洗脱体积大约为15μL)。在约50μL的体积中进行滚环扩增。向15μL洗脱样品中加入5μL10XRepliPHI缓冲液(Epicentre)、1μL25mMdNTP、2μL100mMDTT、1μL100U/μLRepliPHIPhi29和26μL水。在30℃孵育反应混合物1小时。通过加入80μLAmpure珠子纯化RCA产物,而剩余的洗涤步骤遵循制造商的说明。对于洗脱,加入22.5μL洗脱缓冲液并将珠子在65℃孵育5分钟。将管返回到磁体之前短暂离心。将大约20μL来自RCA反应的洗脱产物与25μL2XPhusion主混合物、2.5μLDMSO和0.5μL10μM的各种B2B引物混合物混合。运行以下PCR程序:95℃1分钟,5个循环的延伸(95℃15秒、55℃15秒、72℃1分钟),13-18个循环的复制(95℃15秒、68℃15秒、72℃1分钟)以及72℃7分钟的最终延伸。运行E-凝胶以检查PCR产物的大小。如果范围为100-500bp,则进行0.6XAmpure珠纯化,以富集300-500bp,并取1-2ng进行另一轮采用小RNA文库衔接子引物的PCR。如果产物大小范围>1000bp,则用1.6XAmpure珠进行纯化,并取2-3ng用于NexteraXT扩增子文库制备,以通过0.6XAmpure珠纯化来富集在400-1000bp范围内的大小。为了对测序数据进行生物信息学分析,从MiSeq运行中获得FASTQ文件。使用BWA将FASTQ文件中的序列与包含靶向序列(例如KRAS和EGFR)的参考基因组序列进行比对。使用比对结果找到每个序列(两个读取)的重复单元的区域和长度及其参考位置。使用每个序列的比对结果和重复单元的信息,找到所有基因座中的变体。将来自两个读取的结果合并。计算变体的标准化频率和噪声水平。在从确认的变体得到的变体判定中应用多个附加标准,包括qscore>30和p值<0.0001。通过这些标准的确认的变体被报告为真正的变体(突变)。该过程可以由计算机语言(例如Python)自动化。实施例2:制备用于序列变体检测的串联重复测序文库将12μL体积中的10ng平均长度为150bp的DNA片段用于串联重复测序文库构建。DNA预先用T4多核苷酸激酶(NewEnglandBiolabs)处理,以在5'末端添加磷酸基团以及在3'末端留下羟基。对于由DNaseI或酶促片段化生成的或从血清或血浆中提取的DNA片段,跳过末端处理步骤。将DNA与2μL10XCircLigase缓冲液(EpicentreCL9021K)混合。将混合物加热至95℃2分钟,并在冰上冷却5分钟,然后加入4μL甜菜碱、1μL50mMMnCl2和1μLCircLigaseII(EpicentreCL9021K)。在60℃进行连接反应至少12小时。将1μL200nm的各种RCA引物混合物(至最终10nM的终浓度)加入到连接产物中并混合,加热至96℃保持1分钟,冷却到42℃,并在42℃孵育2小时。将杂交有RCA引物的环化连接(CircLigation)产物用ZYMO寡核苷酸纯化试剂盒(ZYMOResearch,D4060)纯化。为此,用28μL水和1μL载体RNA(Sigma-Aldrich,R5636,用1XTE缓冲液稀释至200ng/μL)将21μL产物稀释到50μL。将稀释的样品与100μL寡核苷酸结合缓冲液和400μL100%乙醇混合。将混合物加载到柱上,在>10,000×g下离心30秒。弃去流出液。用750μLDNA洗涤缓冲液对通过在>10,000×g下离心30秒而洗涤该柱,弃去流出液,并以最高速度再离心1分钟。将该柱转移至新的1.5mLEppendorf管中并使用17μL洗脱缓冲液(10mMTris-CLpH8.0,最终洗脱体积约为15μL)洗脱DNA。将5μL10XRepliPHI缓冲液、2μL25mMdNTP、2μL100mMDTT、1μL100U/μLRepliPHIPhi29和25μL水(Epicentre,RH040210)加入到15μL从柱上洗脱的样品中,总反应体积为50μL。将反应混合物在30℃下孵育2小时。通过加入80μLAmpureXP珠(BeckmanCoulter,A63881)而纯化RCA产物。洗涤步骤依照制造商的说明。在22.5μL洗脱缓冲液中65℃孵育5分钟之后洗脱RCA产物。在返回到磁体前将管短暂离心。将约20μL来自RCA反应的洗脱产物与25μL2XPhusion主混合物(NewEnglandBiolabsM0531S)、2.5μL水、2.5μLDMSO和0.5μLB2B引物混合物(各10μM)混合。使用以下热循环程序进行扩增:95℃2分钟,5个循环的延伸(95℃30秒、55℃15秒、72℃1分钟),18个循环的复制(95℃15秒、68℃15秒、72℃1分钟)以及72℃7分钟的最终延伸。通过电泳检查PCR产物大小。一旦长PCR产物通过电泳得到确认,PCR产物即与30μLAmpure珠(0.6X体积)混合以供纯化,以富集>500bp的PCR产物。使用Qubit2.0QuantificationPlatform(Invitorgen)对纯化的产物进行定量。大约1ng纯化的DNA用于NexteraXT扩增子文库制备(IlluminaFC-131-1024)。通过用0.6XAmpure珠进行纯化来富集插入片段大小>500bp的文库元件。使用适用于2100Bioanalyzer(AgilentTechnologiesInc.,SantaClara,CA)的AgilentDNA高灵敏度试剂盒分析扩增的文库的浓度和大小分布。使用IlluminaMiSeq和2-250bpMiSeq测序试剂盒进行测序。根据MiSeq手册,将12pM变性的文库加载于测序运行上。在此过程的变化形式中,将Illumina衔接子用于文库制备而不是Nextera制备。为此,将约1ng类似地纯化的DNA用于PCR扩增,该扩增使用一对包含B2B引物的通用部分和Illumina衔接子序列的引物(P5和P7;5'CAAGCAGAAGACGGCATACGA3'和5'ACACTCTTTCCCTACACGACGCTCTTCCGATCT3')。使用Phusion主混合物,进行12个循环的复制步骤(95℃30秒、55℃15秒、72℃60秒)。该扩增步骤的目的是加入Illumina衔接子以供扩增子测序。用0.6XAmpure珠富集长度>500bp的扩增子。使用适用于2100Bioanalyzer(AgilentTechnologiesInc.,SantaClara,CA)的AgilentDNA高灵敏度试剂盒分析扩增子文库的浓度和大小分布。使用IlluminaMiSeq和2x250bpMiSeq测序试剂盒进行测序。B2B引物的通用部分还充当测序引物序列,并且如果引物不包含在Illumina试剂盒中则加入定制的测序引物。将12pM变性的文库加载于测序运行上。在图33中示出了在一个实例分析中的靶区域覆盖。以下表3描述了靶向区域的分析结果。表1提供了在本发明的方法中有用的RCA引物的实例。表2提供了在本发明的方法中有用的B2B引物的实例。表1表2表3结果读取1.5M%靶碱基,1x97.8%%靶标命中63.4%%复制18.2%平均覆盖度74.5x覆盖度的标准差0.21实施例3:用于测序文库构建的基因组DNA的片段化使用NEBNextdsDNA片段化酶试剂盒(NewEnglandBiolabs)按照生产商的方案对1μL基因组DNA进行处理。孵育时间延长至45分钟,37℃。通过加入5μL0.5MEDTApH8.0而终止片段化反应,并通过添加2X体积的AmpureXP珠(BeckmanCoulter,A63881)根据制造商的方案进行纯化。使用高灵敏度DNA试剂盒在Bioanalyzer(Agilent)上分析片段化的DNA。片段化的DNA的大小范围通常是约100bp至约200bp,并具有约150bp的峰。实施例4:文库制备程序在本实施例中,为了说明的目的,使用KAPA文库制备试剂盒(KK8230)。对于包括珠纯化的步骤,将AMPureXP珠(目录号A63881)平衡至室温,并且在与样品混合之前充分重悬。在涡旋混合器上与样品充分混合后,将其在室温下孵育15分钟,以使DNA结合到珠子上。然后将珠子置于磁架上直到液体澄清。然后用200μL80%的乙醇将珠子洗涤两次,并在室温下干燥15分钟。为了进行末端修复反应,至多50μL(2-10ng)无细胞DNA与20μL末端修复主混合物(8μL水、7μL10XKAPA末端修复缓冲液和5μLKAPA末端修复酶混合物)混合,并在20℃孵育30分钟。然后将120μLAMPureXP珠加入到70μL末端修复反应液中。然后如上所述对样品进行纯化。为了进行A-加尾反应,将包含末端修复的DNA片段的干燥的珠子与A-加尾主混合物(42μL水、5μL10XKAPAA加尾缓冲液和KAPAA-加尾酶)混合。将反应于30℃孵育30分钟。在加入90μLPEG溶液(20%PEG8000,2.5MNaCl)之后,将混合物按上述珠纯化方案洗涤。对于平端连接反应跳过这一A-加尾步骤。对于连接体连接,具有下述序列(5'至3')的两种寡核苷酸用于形成衔接子多核苷酸双链体:/5Phos/CCATTTCATTACCTCTTTCTCCGCACCCGACATAGAT*T和/5Phos/ATCTATGTCGGGTGCGGAGAAAGAGGTAATGAAATGG*T。包含末端修复的(对于平端连接)或A-加尾(对于基于连接体的连接)的干燥珠子与45μL连接主混合物(30μL水、10μL5xKAPA连接缓冲液和5μLKAPAT4DNA连接酶)以及5μL水(对于平端连接)或5μL连接体寡核苷酸的等摩尔混合物(对于基于连接体的连接)混合。将珠子充分重悬,并在20℃孵育15分钟。加入50μLPEG溶液(见上)之后,按上述珠纯化方案洗涤该混合物。使用IllustraGenomiphiV2DNA扩增试剂盒进行多重置换扩增(MDA)。将包含连接片段链的干燥的珠子重悬于9μL包含随机六聚体的缓冲液中,并在95℃加热3分钟,接着在冰上迅速冷却。加入1μL酶混合物之后,将冷却的样品在30℃孵育90分钟。然后通过在65℃加热10分钟停止反应。加入30μLPEG溶液(见上)后,将混合物按照上述纯化方案进行洗涤,并重悬于200μLTE中(并在65℃孵育5分钟)。如有需要,可通过定量PCR、数字微滴PCR(ddPCR)或通过新一代测序(NGS)对纯化的产物进行定量。MDA之后,使用CovarisS220将连接的长片段链(例如>2kb)在130μL总体积中超声处理至约300bp。制造商的方案标明140W峰值功率、10%的占空系数、每脉冲(burst)200个循环和80秒的处理时间。选择大约300bp的片段长度以增加保持完整的原始无细胞DNA片段的几率。在需要时可使用标准文库制备方案将衔接子置于超声处理的DNA片段上以供测序。从Illumina测序仪(HiSeq或MiSeq)上的配对末端测序运行中返回多种读取组合。接点(或者是自连接接点,或者在连接步骤中包括衔接子的情况下是衔接子接点)处于读取内部(其5'和3'侧翼为非衔接子序列)的那些读取用来对感兴趣的序列进行条形码编码。实施例5:环化和扩增本实施例提供环化和扩增程序(也被称为“Nebula”程序)的示例性描述。该程序使用以下器材:PCR仪(例如MJresearchPTC-200Peltier热循环仪);CircligaseII、ssDNA连接酶Epicentre目录号CL9025K;外切核酸酶(例如Exol,NEBBiolabs目录号M0293S;Exolll,NEBBiolabs目录号M0206S);T4多核苷酸激酶(NEBBiolab目录号M0201S);全基因组扩增试剂盒(例如GEHealthcare,Illustra,Ready-To-Go,Genomiphi,V3DNA扩增试剂盒);GlycoBlue(例如Ambion目录号AM9515);微型离心机(例如Eppendrof5415D);DNA纯化珠(例如Agencourt,AMpureXP,BeckmanCoulter目录号A63881);磁架(例如MagnaRackTMInvitrogen目录号CS15000);2.0荧光计(Invitrogen,目录号Q32866);分子探针dsDNAHS测定试剂盒(LifeTechnology目录号032854);和Bioanalyzer(Agilent2100),以及高灵敏度DNA试剂(目录号5067-4626)。为了扩增缺少5'末端磷酸的DNA片段(例如无细胞DNA),第一步是末端修复和单链的形成。使DNA在96℃下变性30秒(例如在PCR仪中)。通过将40μLDNA与5μL10XPNK反应缓冲液混合,接着在37℃孵育30分钟,从而准备多核苷酸激酶(PNK)反应。将1mMATP和PNK酶加入该反应中,并在37℃孵育45分钟。通过沉淀和重悬DNA进行缓冲液更换。将50μL来自PNK反应的DNA与5μL0.5M乙酸钠pH5.2、1μLGlycoBlue、1μL寡核苷酸(100ng/μL)和150μL100%乙醇混合。在-80℃孵育30分钟,并以16Krpm离心5分钟以沉淀DNA。用500μL70%乙醇洗涤DNA沉淀物,在室温下风干5分钟,并将DNA悬浮于12μL10mMTris-ClpH8.0中。然后通过连接将重悬的DNA环化。使DNA在96℃下变性30秒,样品在冰上冷却2分钟,并加入连接酶混合物(2μL10XCircLigase缓冲液、4μL5M甜菜碱、1μL50mMMnCl2、1μLCircLigaseII)。连接反应在PCR仪上60℃孵育16小时。通过外切核酸酶消化来降解未连接的多核苷酸。为此,使DNA在80℃下变性45秒,并向各管中加入1μL外切核酸酶混合物(ExoI20U/μL:ExoIII100U/μL=1:2)。通过移液器上下吹吸5次进行混合,并短暂离心。消化混合物在37℃下孵育45分钟。加入30μL水使体积达到50μL,并通过如上所述的沉淀和重悬进行进一步的缓冲液更换。为了进行全基因组扩增(WGA),通过使纯化的DNA在65℃下变性5分钟而起始。将10μL来自GEWGA试剂盒的变性缓冲液加入到10μL纯化的DNA中。在冷却块或冰上冷却DNA2分钟。将20μLDNA加至Ready-To-GoGenomiPhiV3饼(WGA)。WGA反应在30℃下孵育1.5小时。在65℃下热灭活10分钟。使用AmpureXP磁珠(1.6X)纯化样品。将珠子涡旋振荡,分装80μL于1.5mL管中。将30μL水、20μL扩增的DNA和80μL珠子混合。在室温下孵育3分钟。将管置于磁架上2分钟,并吸出澄清溶液。用80%乙醇将珠子洗涤两次。通过加入200μL10mMTris-ClpH8.0而洗脱DNA。DNA珠子混合物在65℃下孵育5分钟。将管放回磁架上2分钟。将195μLDNA转移到新的管中。取1μL使用Qubit进行定量。最后,使用CovarisS220超声处理130μLWGA产物以达到约400bp的大小。实施例6:带有额外的纯化的环化和扩增本实施例提供带有苯酚氯仿抽提步骤的环化和扩增程序(也被称为“Nebula”程序)的示例性描述。步骤1是去除竞争性RNA(从提取中的RNA载体中)和天然RNA(共纯化的)以用于Circligase反应。通过将1μLRNaseA(10mg/mL)(Qiagen1007885)加至50μLcfDNA(2-10ng)而去除RNA,并在PCR仪(MJresearchPTC-200Peltier热循环仪)上在37℃下孵育30分钟。步骤2是缓冲液更换与盐和乙醇沉淀。该步骤对于清理和浓缩输入物以用于连接是非常有用的,具有接近100%的回收率(而柱通常仅回收30%)。乙醇共沉淀混合物(50μL来自RNase处理的DNA、5μL0.5M乙酸钠pH5.2、1μLGlycoBlue(AmbionAM9515)、1μL载体寡核苷酸(100ng/μL)、150μL100%乙醇)在-80℃下孵育30分钟,并以16Krpm(Eppendorf5415D)离心5分钟以沉淀DNA。使用20-mer非特异性载体寡核苷酸(我们使用PCR引物)略微增加了沉淀回收的产率和稳定性。将DNA沉淀物用500μL70%乙醇洗涤。将DNA沉淀物在室温下风干5分钟,并用13μL10mMTris-ClpH8.0重悬。步骤3是环化。使12μLcfDNA在96℃下变性30秒,并在冰块上冷却2分钟。在冷却块上进行连接混合物(12μLcfDNA、2μL10XCircligase缓冲液、4μL5M甜菜碱、1μL50mMMnCl2、1μLCircligaseII(Epicentre#CL9025K)的添加,并在60℃下进行连接16小时。步骤4是外切核酸酶消化。连接DNA混合物在PCR仪上于80℃下孵育45秒,随后进行外切核酸酶处理。向各管中加入1μL外切核酸酶混合物(ExoI20U/μL:ExoIII100U/μL=1:2),并且反应在37℃下孵育30分钟。出于质量控制的目的,没有必要去除线性模板。步骤5是苯酚氯仿抽提和缓冲液更换与盐和乙醇沉淀。苯酚/乙醇有助于实现超过80%的连接效率(环化产物的量约等于输入多核苷酸的量)。将180μL10mMTris加入到20μL来自外切核酸酶处理的DNA中以达到200μL的体积,并使用200μL苯酚抽提DNA。收集水层,并且通过乙醇沉淀回收DNA。乙醇共沉淀混合物(200μL苯酚抽提后的DNA溶液、20μL0.5M乙酸钠pH5.2、1μLGlycoBlue、1μL载体寡核苷酸(100ng/μL)、600μL100%乙醇)于-80℃孵育30分钟,并以16Krpm离心5分钟以沉淀DNA。将DNA沉淀物用500μL的70%乙醇洗涤。DNA沉淀物在室温下风干5分钟,并用11μL10mMTris-ClpH8.0重悬。步骤6是全基因组扩增。将10μL纯化的DNA在加热块上65℃孵育5分钟,并加入10μL变性缓冲液(来自GEHealthcare,Ready-To-Go,Genomiphi,V3DNA扩增试剂盒)。DNA在室温下冷却5分钟后,将20μLDNA加至Ready-To-GoGenomiPhiV3饼(WGA)。扩增反应在30℃孵育1.5小时,并通过在65℃下热灭活10分钟终止反应。步骤7是使用AmpureXP磁珠(1.6X)的珠纯化。这按照前面的实施例进行。步骤8是如同前面的实施例中的超声处理。然后将DNA准备好用于定量PCR、ddPCR或测序文库构建。实施例7:连接效率与靶标命中率的分析如上述实施例一样环化并且进行全基因组应用的cfDNA通过定量PCR(qPCR)进行分析。样品靶标的qPCR扩增曲线结果(使用KRAS引物)示于图18中。如图18A中所示,1/10的输入cfDNA的qPCR扩增得到31.75的平均Ct值(循环阈值),而1/10的同一样品的连接产物得到31.927的平均Ct值,显示出约88%的高连接效率。连接效率可以在大约或高于约70%、80%、90%、95%的范围内,或更高,例如约100%。在一些实例中去除未环化的线性DNA,以使得大致所有的DNA均可以从环状形式扩增。每个样品运行三次,每次两个重复。如图18B所示,10ngWGA产物和参考基因组DNA(gDNA)(12878,10ng)的扩增曲线几乎彼此重叠。WGA样品的平均Ct值为26.655,而gDNA样品的平均Ct值为26.605,表明超过96%的高的靶标命中率。给定量的扩增的DNA中KRAS的数目与未扩增的gDNA具有可比性,显示出无偏倚的扩增过程。每个样品测试三次,每次两个重复。作为对比的点,还测试了Lou等人(PNAS,2013,110(49))提供的环化方案。使用Lou的方法(该方法缺少上述实施例中的沉淀和纯化步骤),仅有10-30%的线性输入DNA转化为环状DNA。如此低的回收率显示出对下游测序和变体检测的挑战。实施例8:通过ddPCR对扩增的环化DNA的分析用微滴数字PCR(ddPCR)评估由环化的多核苷酸产生的全基因组扩增产物中的等位基因频率保持和偏倚。通常,ddPCR是指通过对核酸分子进行计数而测量绝对量的数字PCR分析,该核酸分子封装在支持PCR扩增的离散的、容积限定的、油包水型微滴分区中(Hinson等,2011,Anal.Chem.83:8604-8610;Pinheiro等,2012,Anal.Chem.84:1003-1011)。单个ddPCR反应可以由每孔至少20,000个分区的微滴构成。微滴数字PCR可以使用能够进行数字PCR分析的任何平台进行,该数字PCR分析通过对核酸分子进行计数而测量绝对量,所述核酸分子封装在支持PCR扩增的离散的、容积限定的、油包水型微滴分区中。微滴数字PCR的典型策略可归纳如下:将样品稀释并分配到数千至数百万个单独的反应室(油包水微滴)中,使得每个反应室包含目标核酸分子的一个拷贝或不包含其拷贝。检测到的“阳性”微滴(其含有靶扩增子(即,目标核酸分子))的数目,相对于“阴性”微滴(其不包含靶扩增子(目标核酸分子))的数目,可以用于确定原始样品中目标核酸分子的拷贝数。微滴数字PCR系统的实例包括Bio-Rad的QX100TM微滴数字PCR系统,其将含有核酸模板的样品分配到20,000纳升大小的微滴中;以及RainDance的RainDropTM数字PCR系统,其将包含核酸模板的样品分配到1,000,000至10,000,000皮升大小的微滴中。在WO2013181276A1中提供了用于ddPCR的方法的其他例子。在本实施例中,来自黑色素瘤细胞系的BRAFV600E基因组DNA(gDNA)与参考基因组DNA12878以特定比例(0%、0.67%、2.0%、6.67%、20%或100%)混合,并进行片段化以产生与在cfDNA中所见的大小相似(在该情况下,大约150bp)的片段。将混合的DNA样品(10ng)根据实施例2进行环化和扩增。对于BRAFV600E和野生型,将40ng扩增的DNA进行ddPCR。所观察到的突变等位基因频率在图19中以图形示出并制成表格。如图所示,所观察到的进行扩增时的突变等位基因频率(图19表格的中间行)反映了输入突变等位基因频率(最上行),以及100ng基因组DNA在不进行扩增时的ddPCR结果(最下行)。根据ddPCR输出的等位基因频率被计算为含有BRAF突变的微滴的数目除以含有突变体和含有野生型的微滴的总和。进行扩增的DNA被表示为空心圆,而不进行扩增的DNA被表示为缩小的实心圆。除了在0.67%处有小的偏差外,这两个数据集完全重叠。这表明了突变等位基因频率的真实表现的保持,基本没有偏倚。实施例9:高于背景的序列变体的检测10ng经超声处理的gDNA(150bp,多基因多重参考DNA,Horizon)如实施例2所述进行环化和扩增,随后进行超声处理。片段化的DNA随后进行Rubicon测序文库构建。捕获测序后,标绘距离参考热点50bp内的变体。图20中显示了变体检测的结果,其中判定变体需要在根据不同接点区分的两个不同多核苷酸中均可检测到。将七个预期的参考热点(KITD816V、EGFRG719S、EGFRT790M、EGFRL858R、KRASG13D、KRASG12D、NRASQ61K)绘制在位置0。也确认了另外两个变体,在图20中表示为空心三角形和菱形。根据类似方法检测不同浓度的变体序列的其他结果示于图17中。为了比较,如上所述对gDNA进行超声处理,但根据常规实践将经超声处理的10nggDNA直接进行Rubicon测序文库构建,而不经过环化并且不需要确认在两个不同多核苷酸上的序列变体。捕获测序后,再次绘制距离参考热点50bp内的变体,其结果示于图21中。将7个预期的参考热点(KITD816V、EGFRG719S、EGFRT790M、EGFRL858R、KRASG13D、KRASG12D、NRASQ61K)绘制在位置0。其他位置的变体不是预料中的,并且最有可能是由于测序错误。与生成图20所用的方法的结果相比,图21中的结果表明标准测序方法具有高得多的随机误差率,当等位基因频率低(例如低于5%)时其可掩盖真正的突变信号。这一观点的另一例证由绘制于图16中的相似的结果加以阐明。实施例10:GC组成和大小分布的分析10ng经超声处理的gDNA(150bp,多基因多重参考DNA,Horizon)如实施例2所述进行环化和扩增,测序,并用变体判定双多核苷酸验证过滤器(左)进行分析。将具有一系列CG百分比的序列的数目制成表格并绘制图形,如图22所示。如最左侧的图所示,按照实施例2制备的样品的序列除中央峰(对应于基础基因组的总GC含量)以外基本上类似于理论分布。与之相比,当使用Rubicon测序文库构建试剂盒直接使用相同量的gDNA不经扩增而构建测序文库时,测序结果与理论分布之间的差异非常明显(见中间的图)。这种直接Rubicon测序的中央峰比理论分布更高。Newman等人(2014;NatureMedicine,(20):548-54)报道,当使用32ngcfDNA时,cfDNA测序GC含量分布类似于理论分布。这在最右侧的图中示出。对如实施例2所述环化、扩增并测序的cfDNA进行DNA大小分布的评估。如图23所示,测序结果指出的片段长度分布的峰位于约150-180bp处,其类似于cfDNA的典型分布模式。实施例11:扩增均一性的评估将按照实施例2环化并扩增的10个产物的qPCR结果与未扩增的参考DNA(来自12878细胞系的gDNA,CoriellInstitute)进行比较。10ng基因组参考DNA或扩增产物用于每个实时qPCR反应,并通过扩增产物相对于基因组参考的相对定量生成比值。如图24所示,每个PCR的比值在2倍的变化内,这表明在扩增的DNA库中这些靶标的拷贝数非常类似于未扩增的参考DNA。设计并预先验证了来自6个基因(BRAF、cKIT、EGFR、KRAS、NRAS、PI3KCA)的10对PCR引物。尽管本发明的优选实施方案已经在本文中显示和描述,但对于本领域技术人员显而易见的是,这些实施方案仅以示例的方式提供。在不背离本发明的情况下,本领域技术人员现在将会想到许多更改、改变和替代。应当理解,本文所述的本发明实施方案的各种替代方案可以在本发明的实践中使用。意图以下列权利要求限定本发明的范围,并由此涵盖在这些权利要求的范围内的方法和结构及其等同物。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1