一种制备FVIII:C/FVIII:Ag比例提高的因子VIII的方法与流程
发明背景
天然因子VIII分子是在正常条件下以具有一条轻链(用于全长血浆来源的和B结构域缺失的FVIII;80kDa)和一条重链(用于200kDa血浆或重组全长来源的因子VIII和用于90kDa B结构域缺失的重组因子VIII)的复合体流通。重链和轻链关联的确切条件并未详知,但一些文章揭示了金属离子桥的参与连同疏水相互作用一起形成了复合体并使其结合在一起。已揭示,不同的金属离子参与包括钙、铜、锌、锰等的相互作用(1)。对于最近研发的B结构域缺失的重组因子VIII产品,已表明,该分子包含三种金属离子:钙、铜和锌(2)。没有金属桥的存在,仅因子VIII分子的轻链和重链不具有生物活性,但仍有抗原活性。该特点已被公开于若干出版物中(3,4),即抑制剂的形成在活体内具有风险,特别是针对因子VIII轻链。因此,在给患者注射的因子VIII产品中,应该存在尽可能少量的单独的轻链和重链,FVIII:C/FVIII:Ag活性的比例应接近1.0(一)(5),特别是对于因子VIII轻链。
一般情况下,蛋白质的聚集在纯化过程中是一个危险因素,这不仅是因为会造成所需产品的损失,还会形成潜在的抑制剂(6)。在某些生化条件下,重组因子VIII可能聚集(7,8),这将导致其生物活性显著降低。一个含有聚集的因子VIII的因子VIII产品将因此包含单体含量<100%的无活性形式的因子VIII。药物蛋白产品的单体含量应接近100%且无活性形式的因子VIII(聚集体,片段等)的含量应尽可能低。
已有许多方法描述了从血浆或重组产生因子VIII(rFVIII)的培养物中纯化因子VIII。例如WO 2009/156430公开了一系列对因子VIII的纯化层析步骤,包括基于非动物来源的Fab的亲和步骤,其中13千道尔顿的配体结合因子VIII轻链。在该申请中提到的其他层析步骤包括阴离子和阳离子混合模式树脂、阳离子交换、阴离子交换和凝胶过滤。对于除去因子VIII的无活性形式或聚集体的含量的方面的信息在该专利发明中未提供。在该文章中,描述了新重组因子VIII的纯化和表征(9),其是四步的因子VIII层析纯化方法,该方法包括用单克隆抗体作为与因子VIII的重链结合的配体的亲和步骤。其他三个步骤为混合模式层析树脂、阴离子交换树脂和凝胶过滤,对于除去因子VIII的无活性形式或聚集体的含量的方面的信息在该文章中未提供。在该文章中,使用肽配体作 为因子VIII的cGMP生产的亲和层析的建立与验证中,描述了包含五个步骤的层析方法,包括基于肽的亲和步骤,其中2.7千道尔顿的配体结合因子VIII的轻链。描述了来自于培养过程中的过量的因子VIII轻链在肽亲和树脂的洗涤过程中被除去。其他四个层析步骤为阳离子交换树脂、阴离子交换树脂、疏水相互作用树脂和凝胶过滤步骤。其阐述了该肽亲和树脂除去过量的因子VIII轻链之后的两个层析步骤,但未进一步具体限定在哪个步骤中。对于除去因子VIII轻链以外的其他无活性形式或因子VIII聚集体的含量的方面的信息在该文章中未提供。在该文章即一种用于重组因子VIII化合物的捕获和纯化的新型亲和吸附剂的应用中(11),描述了用于纯化因子VIII的基于Fab的13千道尔顿的配体亲和树脂,对于除去因子VIII的无活性形式或聚集体的含量的方面的信息在该文章中未提供。
正如该文章中描述的,市售的重组因子VIII产品含有无活性的因子VIII形式(5),(12),如用生物活性因子VIII(因子VIII:C)相对于因子VIII的总量(因子VIII:Ag)的比例所测量的,其对于患者具有潜在的免疫反应方面的副作用。
生物活性因子VIII被定义为具有因子VIII活性的因子VIII,其在活体正常条件下可以通过酶反应被激活成为因子VIIIa,这是用于止血的凝血级联的基本组成部分。生物活性因子VIII可用不同的体外分析方法进行测定(FVIII:C),例如FVIII显色测定法和/或一期凝血测定(13)。显色测定法是两阶段光度测定法,其测量因子VIII作为辅因子的生物活性。因子VIII将因子X激活为因子Xa,而这又是酶促裂解成可以通过分光光度法来定量的产品。一期凝血测定法是基于含有因子VIII的试样纠正在磷脂、接触活化剂和钙离子存在下缺乏因子VIII的血浆的凝固时间的能力。纤维蛋白凝块出现的时间是在一个步骤中测得的。
WO-A-2008/134310公开了一种稳定用于冷冻储存的重组蛋白的本体溶液的方法,其包括提供具有至少100mM的一价盐浓度的重组蛋白的部分纯化溶液,并添加足量碳水化合物至所述溶液,使得冷冻后该溶液具有-56℃或更高的玻璃化转变温度。
WO2010/115866A1公开了分子和多肽,其包含至少一个与人类因子VIII或其生物活性部分具有显著同一性(同源性)的氨基酸序列,并公开了相关分子(如编码此类多肽的核酸),包括此类多肽的组合物(例如药物制剂),以及制备和使用此类多肽的方法。
WO97/33178A1公开了一种用于检测含因子VIII的蛋白质组分的适用性的方法,对其的进一步处理包括巴氏消毒步骤,该步骤包括检测用于20-50kD范围的片段的起始材料。在此范围内的因子VIII片段会在之前用因子VIII治疗过的患者体中显著引起抑制剂的形成。甚至被这些片段污染的批次可以通过对亲水材料应用排阻层析被用于生产非常纯的无毒因子VIII。
US 4,675,385 A公开了一种用于从重组商用因子VIII:C(复合因子VIII)浓缩液中大规模纯化人、牛和猪的促凝血蛋白因子VIII的快速简单的方法,该方法首先在低盐浓度条件下以及其次在高盐浓度条件下使用有次序的高效尺寸排阻层析法。层析分离是在填充有多孔珠的高效尺寸排阻层析柱上进行,该多孔珠的粒径为约13至约35微米,孔径约500至约2000埃,孔体积每克约1.0至约1.8毫升。第一层析分离是在缓冲水溶液中进行,使用缓冲水溶液作为洗脱液。将低分子量成分(杂质)与因子VIII和高分子量成分(杂质)分离。第二层析分离可以在因子VIII在含有浓度为约0.25M至0.45M钙离子的缓冲溶液中被解离后进行。在2.5x60cm的层析柱中,4克的商用因子VIII浓缩液可以在不到两小时内被纯化。该工艺可适于规模化。
EP 0412466 A2公开了一种具有高比活性(specific activity)和稳定性的巴氏杀菌的因子VIII浓缩物的制备方法,并且包括使用Al(OH)3、阴离子交换剂或Ca3(PO4)2,优选使用选自该组的两个不同的吸附剂,通过至少两次吸附从含有因子VIII的溶液中吸附杂质。
FR 2650393 A1公开了获得具有比活性和高产率的因子VIII浓缩物,其中该浓缩物不含非人类来源的外来蛋白质。通过任意方法得到的因子VIII沉积在包含用第一缓冲液预平衡的阴离子交换凝胶的柱上。在因子VIII被装入层析柱中后,用第二缓冲液冲洗凝胶直至获得低于0.1的光密度。然后用第三缓冲液从凝胶中释放纯化的因子VIII。层析后,比活性是30/1600 IU/mg。
Cheng,Elisabeth等人在Biotechnology Letters,v.32,n.9,p.1207-1214,2010中公开了使用阴离子交换层析随后凝胶过滤来直接从血浆中纯化出人类FVIII。测试了三种Q-琼脂糖凝胶树脂,使用Q-琼脂糖凝胶XL树脂,导致40%FVIII的恢复,使用快速流Q-琼脂糖凝胶导致约80%FVIII的恢复,以及使用大珠Q-琼脂糖凝胶导致70%FVIII的恢复。维生素K依赖型凝血因子与FVIII一同从阴离子交换柱洗脱。在纯化的第二步中,当使用琼脂糖凝胶6FF时,70%的FVIII活性与维生素K依赖型因子恢复游离。
发明简述
本发明的一个目的是提供一种方法,以减少在制备因子VIII和提供高单体含量的产品的方法中单条轻链和重链的量,特别是对于重组产生的因子VIII。本发明的另一个目的是提供一种其中可以除去单条轻链和重链(片段)和凝集因子VIII形式的组合物,可以将所产生的必需的单体因子VIII溶液在冷冻和/或冷冻干燥状态储存几年,保持其较高的单体因子VIII含量。
令人惊奇的是,本发明的发明人已经发现,通过使用层析法制备在因子VIII产品中FVIII:C/FVIII:Ag比例为至少0.7并具有高单体含量的因子VIII产品的方法能够解决 本发明的目的。本发明的方法包括其中通过采用对特异性结合因子VIII有亲和性的亲和层析树脂执行至少一个层析步骤,该亲和性受到固定在亲和层析树脂上的亲和配体的影响,所述亲和配体是针对因子VIII分子的酵母衍生的13kD Fab抗体片段。
所述亲和配体结合至FVIII分子的轻链部分。令人惊奇的是,在包括含由因子VIII的轻链和重链复合形成的天然因子VIII和无任何生物FVIII活性的单条FVIII轻链的混合物的溶液中,可以通过采用特定的洗涤条件对所述亲和树脂进行处理,以从混合物中除去无任何生物凝血活性的FVIII轻链。本领域技术人员将预期到除去大量的天然因子VIII分子即重链和轻链的复合体。
所述配体特别是通过亲水间隔臂固定在亲和层析树脂上,该树脂是一种交联琼脂糖类基质,所述亲和配体是针对因子VIII分子的酵母衍生的13kD Fab抗体片段,其可从GE医疗的商品名VIIISelect下商购。
或者,该目的是通过提供FVIII:C/FVIII:Ag的比例为至少0.7并得到高因子VIII单体含量的方法来实现的,其中通过采用至少一个在阴离子交换层析树脂上的层析步骤来执行至少一个层析步骤。
在本发明的另一替代方案中,公开了提供FVIII:C/FVIII:Ag的比例为至少0.7和高因子VIII单体含量的方法,其中通过在特定层析条件和缓冲组分下采用尺寸排阻层析步骤来执行至少一个层析步骤。
在本发明的另一替代方案中,使用与用于除去FVIII:C/FVIII:Ag的相同的层析步骤,之后因子VIII产品的单体含量≥98%。
在本发明的再一替代方案中,来自前一个层析步骤的所得的高FVIII:C/FVIII:Ag比例和高FVIII:单体含量可在冷冻和/或冷冻干燥状态储存至少12个月,优选至少24个月,最优选至少36个月,保持FVIII:C/FVIII:Ag的性质和/或高因子VIII单体含量不变直至患者使用。
如果将本发明的三种方法之中的两种方法组合,则可以实现FVIII:C/FVIII:Ag的比例为至少0.8,如果三种方法组合,则比例为至少0.9成为可能。
例如,本发明的方法的代表为通过使用层析法制备因子VIII产品的方法,该产品具有FVIII:C/FVIII:Ag的比例为至少0.7且得到≥98%的高因子VIII单体含量以及基本上不含聚集的因子VIII产品,
其中,
步骤(a):通过采用对特异性结合因子VIII有亲和性的亲和层析树脂执行至少一个层析步骤,该亲和性受到固定在亲和层析树脂上的亲和配体的影响,所述亲和配体是用 本发明的方法针对因子VIII分子的酵母衍生的13kD Fab抗体片段,
其中,
步骤(b):在阴离子交换层析树脂上执行至少一个层析步骤。步骤的顺序可以是在(a)后进行(b)或在(b)后进行(a)。
根据本发明,还可以执行通过使用层析法制备因子VIII产品的下列方法,该因子VIII产品具有FVIII:C/FVIII:Ag的比例为至少0.7且得到≥98%的高因子VIII单体含量以及基本上不含聚集的因子VIII产品,
其中,
步骤(a):通过采用对特异性结合因子VIII有亲和性的亲和层析树脂执行至少一个层析步骤,该亲和性受到固定在亲和层析树脂上的亲和配体的影响,所述亲和配体是用本发明的方法针对因子VIII分子的酵母衍生的13kD Fab抗体片段,
其中,
步骤(c):通过采用尺寸排阻层析步骤执行至少一个层析步骤。步骤的顺序可以是在(a)后进行(c)或在(c)后进行(a)。
根据本发明,还可以执行通过使用层析法制备因子VIII产品的下列方法,该产品具有FVIII:C/FVIII:Ag的比例为至少0.7且得到≥98%的高因子VIII单体含量以及基本上不含聚集的因子VIII产品,
其中,
步骤(b):在阴离子交换层析树脂上执行至少一个层析步骤,和
其中,
步骤(c):通过采用尺寸排阻层析步骤执行至少一个层析步骤。步骤的顺序可以是在(b)后进行(c)或在(c)后进行(b)。
更进一步的实施方案是本发明的三种方法的组合。例如,本发明的方法然后以下述方法为代表:通过使用层析法制备因子VIII产品的方法,该产品具有FVIII:C/FVIII:Ag的比例为至少0.9且得到≥99%的高因子VIII单体含量以及基本上不含聚集的因子VIII产品,
其中,
步骤(a):通过采用对特异性结合因子VIII有亲和性的亲和层析树脂执行至少一个层析步骤,该亲和性受到固定在亲和层析树脂上的亲和配体的影响,所述亲和配体是用本发明的方法针对因子VIII分子的酵母衍生的13kD Fab抗体片段,
其中,
步骤(b):在阴离子交换层析树脂上执行至少一个层析步骤;和
其中,
步骤(c):通过采用尺寸排阻层析步骤执行至少一个层析步骤。步骤的顺序可以是:(a)、(b)、(c);(a)、(c)、(b);(c)、(b)、(a);(c)、(a)、(b);(b)、(c)、(a);(b)、(a)、(c)。
根据本发明,在本发明的方法中使用的因子VIII分子是轻链和重链的复合体,提高的FVIII:C/FVIII:Ag比例是归因于因子VIII轻链、因子VIII重链和/或从复合体解离的因子VIII轻链/因子VIII重链的消耗。解离的因子VIII链可以是起源于培养生产过程由于突变、蛋白水解变性/物理变性等而存在,或是由于纯化过程中酶变性/物理变性而存在。解离的FVIII轻链和/或FVIII重链将根据环境例如缓冲液、蛋白浓度等形成因子VIII片段和/或因子VIII聚集体。因此,除去不是单体的所有形式的因子VIII和/或具有相比天然形式的因子VIII更容易形成聚集体的潜力,这是有利的。
根据本发明的另一方面,在该方法中使用的因子VIII分子可以非共价或共价结合其他物质,例如vWF、PEG、HES和/或抗体的FC片段,用于提高因子VIII产品的半衰期,得到与具有最终产品的高比例生物活性和高单体含量的本发明的相同方案。
在本发明的具体实施方案中,在洗脱因子VIII之前,在提供了用于因子VIII结合亲和树脂并通过冲洗除去解离的轻链的条件下,执行亲和层析步骤。在相当于浓度为约0.1至约0.5mol/kg氯化钠的低盐条件下发生因子VIII与亲和层析树脂的结合。然后在相当于约0.3至约0.4mol/kg范围的氯化钠的增加的盐浓度下执行亲和层析树脂的洗涤用于除去轻链,之后通过采用相当于约0.5至约4mol/kg范围的氯化钠和/或MgCl2的盐浓度与约40至约60%的醇优选乙二醇或丙二醇或其混合物的组合,任选地执行洗脱和收集步骤以获得分离级分的因子VIII。
亲和树脂已经被设计为用于结合轻链而不是因子VIII分子的其他部分。这通过使用特定尺寸的亲和配体得以实现。已知过小的亲和配体例如化学合成的分子有时由于空间位阻难以结合靶蛋白。因此,本发明所需的亲和配体的尺寸处于≥10千道尔顿的范围内。由于预期用于该尺寸的Fab片段分子(14)的强亲和性并且配体是针对FVIII轻链,因此,事实上令人惊奇的是在洗脱含有FVIII重链以及FVIII轻链的天然复合体之前,可以洗掉FVIII轻链。特别是,亲和树脂是基于平均粒径为约74μm的交联的琼脂糖基质,酵母来源的约13kD的Fab抗体片段的亲和配体通过亲水间隔臂结合到该基质,使得配体更易被利用以结合因子VIII分子。亲和配体结合生物活性因子VIII分子的因子VIII轻链。
在本发明的方法的另一实施方案中,亲和层析条件包括以下条件中的至少两个:
-至少5000IU/mL树脂的生物活性因子VIII的树脂负载,优选为至少10000IU/ml的树脂,最优选超过20000IU/mL的树脂;
-因子VIII负载期间的缓冲条件:约0.1-约0.5mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)聚山梨醇酯80,约0.5-约2%的Triton X-100,约0.1%-约1%的pH 6.2-6.8的TNBP;
-洗涤期间缓冲条件:约0.5-约4mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)pH6.2-6.8的聚山梨醇酯80;
-因子VIII洗脱期间缓冲条件:约0.5-约4mol/kg NaCl,约40%-约60%乙二醇,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)pH 6.2-6.8的聚山梨醇酯80。
在本发明的方法的另一具体实施方案中,在提供了因子VIII结合阴离子交换层析树脂的条件下执行阴离子交换层析,以及在生物活性因子VIII洗脱之前或之后从阴离子交换层析树脂除去生物无活性形式。洗脱级分中的因子VIII单体含量≥98%。根据本发明的方法,在相当于浓度为0.01至0.15mol/kg氯化钠的低盐条件下负载因子VIII用于结合因子VIII,除去因子VIII的无活性形式,在相当于浓度为0.15至0.3mol/kg氯化钠的中盐条件下清洗阴离子交换层析树脂用于除去因子VIII的无活性形式,通过采用相当于浓度为0.3-1mol/kg氯化钠的高盐条件从阴离子交换层析树脂洗脱因子VIII并将其以分离级分收集。
通过采用相当于浓度为1-2mol/kg氯化钠的高盐条件从阴离子交换层析树脂进一步洗脱无活性的因子VIII形式并将其以分离级分收集。
在本发明的阴离子交换方法的一个具体实施方案中,通过阴离子交换层析步骤除去生物无活性因子VIII,在产品洗脱级分中得到的单体含量≥98%,所述阴离子交换层析步骤包括如下层析条件中的至少两个:
-至少10000IU/mL树脂的生物活性因子VIII的树脂负载,优选至少15000IU/ml树脂,最优选超过20000IU/mL树脂;
-因子VIII负载期间缓冲条件:约0.05-约0.15mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)pH 6.0-7.5的聚山梨醇酯80;
-洗涤期间缓冲条件:约0.15-约0.3mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)pH6.0-7.5的聚山梨醇酯 80;
-因子VIII洗脱期间缓冲条件:约0.3-约0.5mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg L-组氨酸,约0.005%-约0.05%(w/w)pH 6.0-7.5的聚山梨醇酯80。
在本发明的阴离子层析方法的另一实施方案中,阴离子交换树脂是具有季铵离子的强阴离子交换剂,季铵离子作为配体耦合到球形直径为约45-约165μm的交联的6%琼脂糖基质,总离子结合能力为约0.18-约0.25mmol/mL。
在本发明的方法的另一具体实施方案中,尺寸排阻层析包括以下层析条件的至少两个:
-约4-约8%柱体积的样品负载,
-约60-约90厘米的柱高度,
-样品负载中至少10000IU/ml的生物活性因子VIII浓度,优选至少15000IU/ml,最优选超过20000IU/ml,
-用于聚集无活性形式因子VIII的柱平衡缓冲液:约0.2-约0.7mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg柠檬酸钠,约0.5%-约2%(w/w)蔗糖,约0.5%-约2%(w/w)L-精氨酸,约0.1%-约1%(w/w)pH 6.0-7.5的泊洛沙姆188,
其中生物活性因子VIII以单体形式收集,而无活性因子VIII存在于尺寸排阻层析步骤的含聚集无活性形式(可以是聚集的片段(无活性)和聚集的单体因子VIII(单体时有活性,聚集时部分无活性))的级分中和/或尺寸排阻层析步骤的含因子VIII的片段形式的级分中;和
-当在柱的出口处记录约30-约40mAU吸收峰时开始因子VIII单体的收集,当吸收峰回复至约1-约40mAU时停止因子VIII单体的收集,其涉及2-3倍点样的量。
在本发明的尺寸排阻层析的一个具体实施方案中,尺寸排阻树脂是平均直径为约34微米和最佳分离范围为10000-600000道尔顿的球形交联琼脂糖/葡聚糖介质。
在本发明的方法的另一个具体实施方案中,尺寸排阻层析洗脱液为至少12个月稳定,特别是36个月稳定,包括下列条件中的至少两个:
-缓冲液组成,包括:约0.2-约0.7mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg柠檬酸钠,约0.5%-约2%(w/w)蔗糖,约0.5%-约2%(w/w)L-精氨酸,约0.1%-约1%(w/w)pH 6.0-7.5的泊洛沙姆188,
-保存在≤-60℃的冷冻液,
-≤90分钟内完成从室温到≤-40℃的溶液冷冻,
-在≤-60℃下储存的冷冻液在≤90分钟内解冻至18-25℃,
-在用如上提及的缓冲液调整因子VIII浓度后将解冻液施加到冷冻干燥方法,并以每瓶250IU、500IU、1000IU、2000IU、3000IU、4000IU或5000IU因子VIII填充在玻璃小瓶中,
-根据上述的冷冻干燥产品,其中因子VIII单体含量≥99%,
-根据上述的冷冻干燥产品可存储至少12个月,优选24个月,最优选36个月,重构和患者使用后因子VIII单体无显著变化,
-具有以下缓冲液组成的上述重构产品:约0.2-约0.7mol/kg NaCl,约0.01-约0.05mol/kg CaCl2,约0.01-约0.05mol/kg柠檬酸钠,约0.5%-约2%(w/w)蔗糖,约0.5%-约2%(w/w)L-精氨酸,约0.1%-约1%(w/w)pH 6.0-7.5的泊洛沙姆188。
本发明的主题也是根据本发明的方法可获得的用于治疗血友病和避免抑制剂形成的因子VIII产品,该产品在冷冻和/或冷冻干燥条件下至少6个月保持稳定,优选至少12个月,更优选至少24个月,最优选高达36个月。该因子VIII是通过上述三个纯化步骤的特定处理可获得的,即以下顺序的1.亲和步骤,2.阴离子交换步骤,3.尺寸排阻层析步骤。
通过提供该顺序的纯化方案,将所得的FVIII产品质量与使用不同的纯化方案(22)的市售的重组FVIII产品就高FVIII C/Ag比例和高单体含量(相当于低聚集体和片段)方面进行比较。本发明的因子VIII产品在高因子FVIII C/Ag比例和高单体以及低片段含量方面显示优异的结果,如可从表1看出的。在本发明的产品和市售的一种重组产品之间进行的进一步的体内比较中,如可从表2观察到的,在之前未经治疗的A型血友病患者中未注意到减量的抑制剂事件。表2中使用纯化方法A(22)的rFVIII产品不具有与本发明的纯化方法相同的高能力,来提高FVIII C/Ag比例和显示出具有高单体和低片段含量的产品,这明显地影响了之前未经治疗的A型血友病患者中免疫事件的量。
表1
表2
*已公布的柯林斯研究201421,**正在进行的研究
本发明的另一个主题是相比EP2537862A1的本发明的改进的产品性能。这特别是通过上述三个纯化步骤的特定处理而实现的,即以下顺序的1.亲和步骤,2.阴离子交换步骤,3.尺寸排阻层析步骤。此外,包括本发明的尺寸排阻步骤的特定处理条件会确保患者中所需的高(0.9)因子VIII C/Ag比例和单体含量(>99%)。当通过使用本发明的特定缓冲条件储存因子VIII时,上述步骤也在-70℃下储存12个月后实现,这种储存由通过产品的最后尺寸排阻缓冲交换步骤和冷冻干燥提供。这似乎最小化了不利的因子VIII C/Ag比例并降低了储存期间的单体含量,确保给予患者的产品对于由于注射给患者(储存的冷冻干燥产品重构之后)的溶液中的聚集体/片段所导致的不希望的免疫反应具有降低的风险。此外,使用Bradford分析法用FVIII:C/蛋白浓度测定的比活性相比EP2537862A1(8061IU/mg,n=1)中公开的值具有较高的统计显著性(10000IU/mg,n=9)。比活性指示FVIII产品的纯度(已知不同的蛋白测定方法得到不同的结果,因此可比的值需要相同的蛋白方法),表示本发明的产品相比EP2537862A1的优异性能,因为蛋白杂质为患者中抑制剂事件的风险因子之一。此外,如EP2537862A1中揭示的和在本发明的图17中发现的通过等电点聚焦的蛋白指纹分析表明各个产品的产品性质差异。
在本发明的一个具体实施方案中,因子VIII的特征在于:最后步骤之后,生物活性因子VIII(FVIII:C)相对于因子VIII的总量(FVIII:Ag)的商≥0.7,优选≥0.8,更优选≥0.9,最优选1,最后步骤之后,因子VIII单体含量≥98%,优选≥99%,最优选100%,并且基本没有检测到聚集的因子VIII。
在本发明的另一实施方案中,因子VIII保持其生物活性、高含量的单体因子VIII和低含量的聚集/片段因子VIII。
在另一个实施方案中,本发明的因子VIII的特征在于,它是血浆来源的、重组来源的和/或具有生物活性的因子VIII的缺失衍生物或截短形式,特别是B结构域缺失型FVIII,如在参考文献20中所述,其通过引用并入本文。
在本发明的又一实施方案中,因子VIII的特征在于,在它是重组来源的和/或缺失衍生物的情况下,它是在人类细胞中产生的。
在本发明的一个具体实施方案中,因子VIII的特征在于,在之前用本产品治疗或未治疗的A型血友病患者中,抑制剂的量<25%,优选<20%,更优选<10%,最优选0%。
本发明的主题也是根据本发明的方法可获得的用于治疗血友病和避免形成抑制剂的因子VIII产品。
尤其是,根据本发明的产品显示,在用本发明的产品治疗后,在之前治疗或未治疗的A型血友病患者中抑制剂的量<约25%,优选<约20%,更优选<约10%,最优选约0%。
在冷冻干燥过程和储存过程中推荐使用永冻/冻干保护剂,其通过形成围绕蛋白质的 无定形基质来保护该蛋白质。
可以包括填充剂(bulking agent)充当块状形成物以在冷冻干燥过程中给予机械支持和增加药物产品的干重。从而填充剂有助于提供冻干产品的均匀质量和外观。
此外,可以加入缓冲剂以将pH维持在适于蛋白质和产品的治疗用途的值。冻干蛋白质的适合成分例如公开于WO2010/026186A,其通过引用并入本文。
附图说明
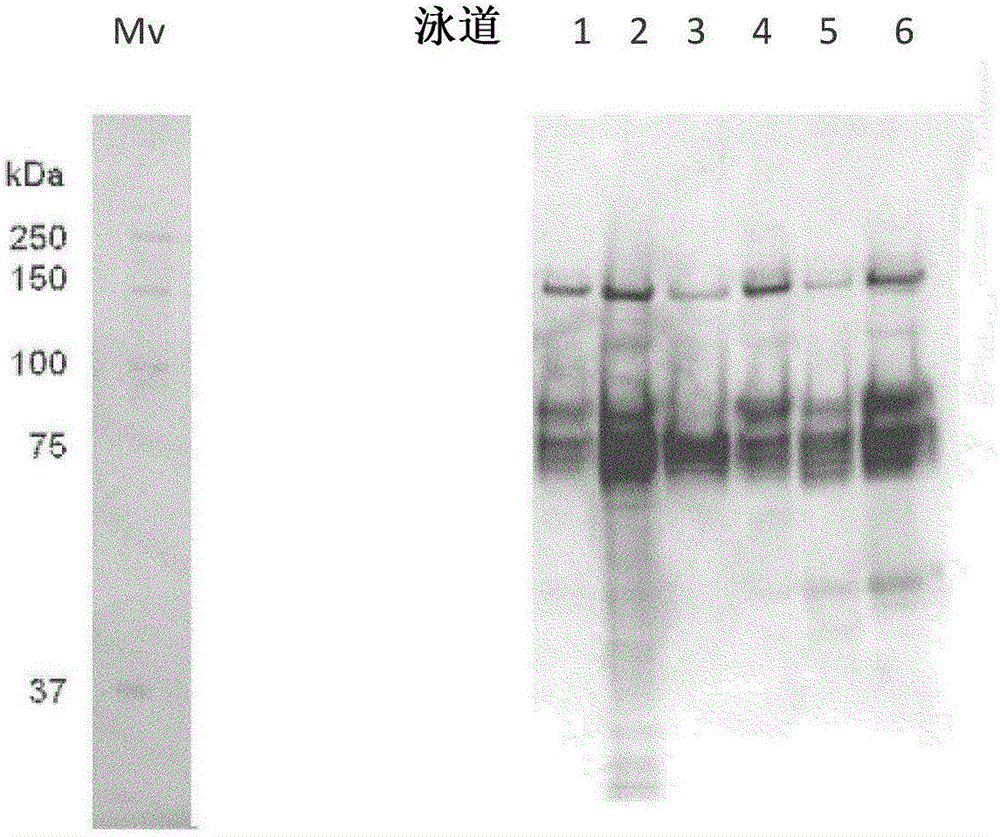
图1:使用FVIII抗体的蛋白印迹。从左边开始:分子量标准,泳道1;起始材料亲和步骤,FVIII:Ag 5.0IU/mL,泳道2;流经亲和步骤,FVIII:Ag 132IU/ml,泳道3;亲和洗涤级分,FVIII:Ag 26IU/ml,泳道4;亲和洗脱液(起始材料Q),FVIII:Ag 5.8IU/ml,泳道5;阴离子交换流经+平衡,FVIII:Ag 1.7IU/ml时,泳道6;阴离子交换洗涤,FVIII:Ag 5.0IU/ml。图1示出在亲和步骤和阴离子交换步骤的流经和洗涤级分中除去无活性FVIII:C。亲和步骤的洗涤级分几乎仅包含FVIII轻链。
图2:使用FVIII抗体的蛋白印迹。从左边开始:分子量标准,泳道1;FVIII对照,FVIII:C 5.0IU/mL,泳道2;亲和洗脱液,FVIII:C 5IU/ml,泳道3;稀释的亲和洗脱液,FVIII:C 5IU/ml,泳道4;阴离子交换洗脱液,FVIII:C 5IU/ml,泳道5;尺寸排阻洗脱液,FVIII:C 5IU/ml。图2示出了在亲和步骤后、在阴离子交换步骤后和在尺寸排阻步骤后相同的FVIII:C浓度下的FVIII蛋白印迹图案,所有均示出与FVIII对照相同的图案。
图3:来自根据实施例3的制备型尺寸排阻层析柱的层析图,示出了使用不同的树脂负载和因子VIII浓度的3个不同的实验中,聚集体和片段与单体因子VIII的分离。根据实施例3的平衡缓冲液系统和层析条件,促进了聚集,并因此促进从单体因子VIII除去聚集体(洗脱)。该层析峰反映了在280nm测定的蛋白的吸光度。
图4:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例2制备的样品(洗脱)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,聚集体<0.5%,片段<1.5%。
图5:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例3制备的样品(洗脱)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量通常>99%,无可见量的聚集体,片段<1%。
图6:来自根据实施例4的制备型尺寸排阻层析柱的层析图,示出了使用不同的树脂负载和因子VIII浓度的5个不同的实验中,聚集体和片段与单体因子VIII的分离。根据实施例4的平衡缓冲液系统和层析条件,促进了聚集,并因此促进从单体因子VIII (洗脱)除去聚集体。该层析峰反映了在280nm测定的蛋白的吸光度。
图7:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(阴离子交换洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>97%,聚集体<0.7%,片段<2.5%。
图8:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(尺寸排阻洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<2%。
图9:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(阴离子交换洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<2%。
图10:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(尺寸排阻洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<2%。
图11:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(阴离子交换洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<1.5%。
图12:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(尺寸排阻洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<1.5%。
图13:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(阴离子交换洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>99%,无可见量的聚集体,片段<1.5%。
图14:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(尺寸排阻洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<1.5%。
图15:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(阴离子交换洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>99%,无可见量的聚集体,片段<1%。
图16:来自分析型尺寸排阻层析柱(SEC-HPLC1)的层析图,示出来自实施例4制备的样品(尺寸排阻洗脱液)的层析分布,分别是聚集体、单体和片段的百分比含量。该样品的单体含量>98%,无可见量的聚集体,片段<1.5%。
图17:尺寸排阻层析后根据本发明实施例4处理的样品的电荷和尺寸分布的分析二维电泳(2D-PAGE)指纹。
图18:本发明的纯化过程中因子VIII纯度的提高。
图19:来自分析型尺寸排阻层析柱(SEC-HPLC2)的层析图,示出来自实施例7制备(根据本发明纯化的和冻干的)的样品的层析分布,分别是冻干产品(1000IU小瓶)重构后聚集体、单体和片段的百分比含量。该样品的单体含量基本上为100%(基于分子量标准保持曲线,单体峰左侧的少量肩部包括在FVIII单体峰中),在SEC-HPLC2层析中洗脱约7-10分钟,无可见的聚集体和片段。
图20:来自分析型尺寸排阻层析柱(SEC-HPLC2)的层析图,示出根据纯化方法A22和冷冻干燥制备的样品的层析分布,分别是冻干产品(1000IU小瓶)重构后聚集体、单体和片段的百分比含量。该样品的单体含量为约76%,无可见的聚集体,和约24%的片段。该rFVIII产品是具有完整B结构域的分子量约300kD的全长FVIII产品,其在SEC-HPLC2层析中大约5-8分钟时得到单体FVIII洗脱。在约8-13分钟的单体峰之后,开始直接计算片段峰。
图21:来自分析型尺寸排阻层析柱(SEC-HPLC2)的层析图,示出根据纯化方法B22和冷冻干燥制备的样品的层析分布,分别是冻干产品(1000IU小瓶)重构后聚集体、单体和片段的百分比含量。该样品的单体含量为约99%(基于分子量标准保持曲线(图23),单体峰左侧的少量肩部包括在FVIII单体峰中),无可见的聚集体,和约1%的片段。该rFVIII产品是分子量约170kD的B结构域缺失的FVIII产品,其在SEC-HPLC2层析中大约7-10.5分钟时得到单体FVIII洗脱。在约10.5-13分钟的单体峰之后,开始直接计算片段峰。
图22:来自分析型尺寸排阻层析柱(SEC-HPLC2)的层析图,示出根据纯化方法C22和冷冻干燥制备的样品的层析分布,分别是冻干产品(1000IU小瓶)重构后聚集体、单体和片段的百分比含量。该样品的单体含量为约80%,无可见的聚集体,和约20%的片段。该rFVIII产品是具有完整B结构域的分子量约300kD的全长FVIII产品,其在SEC-HPLC2层析中大约5-8分钟时得到单体FVIII洗脱。在约8-13分钟的单体峰之后,开始直接计算片段峰。
图23:来自分析型尺寸排阻层析柱(SEC-HPLC2)的层析图,示出来自分子量标准样品的层析分布(15-600kD,69385,Sigma-Aldrich Chemie GmbH)。
本发明的术语“无生物活性形式的因子VIII”是指由于化学、生物化学或酶的原因导致其经失去了生物活性的因子VIII形式。无生物活性因子VIII可为例如单一因子VIII的轻链或重链,因子VIII的截短形式或因子VIII的活性形式(如FVIIIa)(但不稳定, 如通过在凝血周期中的激活所定义的)和/或呈片段的因子VIII的聚集形式或其它形式。
本申请中使用的mol/kg的定义是:加入1000克水中的摩尔量,摩尔的定义是:加入水中至1000ml的摩尔量。
一份样品中有生物活性和无生物活性的因子VIII的总量用基于抗原的(FVIII:Ag)的ELISA分析方法来测定。在有完全生物活性的样品(如在体内)中生物活性因子FVIII(FVIII:C)与因子VIII(FVIII:Ag)抗原含量的比例应等于1.0。如果该比例低于1,则表示样品中存在无活性形式的因子VIII。无活性的形式可以为聚集的因子VIII和/或被解离成单一因子VIII轻链和重链的因子VIII分子。
在生物活性因子VIII分子中,FVIII轻链(因子VIII的A3、C1和C2结构域,分子量约为80kD)是具有FVIII重链(因子VIII的A1和A2结构域,分子量为90-210kD)的金属的/疏水的复合体。这种复合体是体内天然的因子VIII分子,其在正常条件下循环结合到维勒布兰因子(vWF),可防止其变性。当凝血系统被激活时,该天然因子VIII分子从vWF释放并结合到活化的血小板并经蛋白水解转化成FVIIIa(活化的因子VIII),FVIIIa为天然FVIII分子的活化形式,是凝血系统中的一个重要部分。FVIIIa分子通过凝血级联被迅速消耗,然后通过不同的蛋白酶抑制剂被酶促失活(15)。如果该复合体被解离(16),则该轻链或重链不具有或具有很少的生物活性,且FVIII:C/FVIII:Ag的比例接近于0。如果该天然复合体被蛋白水解而失活,那么该轻链和重链的分子量都会减少,该因子VIII降解产物具有最初余留的生物活性(例如FVIIIa),但是它们不稳定并且在体内会较快地失活(1)。因子VIII的降解产物(蛋白水解降解和非蛋白水解解离的)可用因子VIII蛋白印迹分析法检测到,该方法会根据尺寸具体显示有生物活性和无活性的因子VIII分子。该天然的因子VIII分子的分子量约为170kD或290kD,取决于该分子的B结构域是否缺失。该B结构域在因子VIII分子中不具有生物活性功能,因此,具有生物活性的因子VIII分子可以是具有B结构域的或者不具有B结构域的(或者不具有它的一部分)。
因子VIII的单体产品在本文中被定义为具有相同分子量质量的生物活性的因子VIII分子,该分子量质量由天然缓冲条件下进行的分析型HPLC尺寸排阻层析法所定义。片段形式的因子VIII主要是无活性的,但聚集形式的因子VIII具有降低的生物活性。两种形式都会在体内使理论上的抑制剂形成的风险增加,且其在治疗A型血友病的因子VIII产品中应尽可能低。该单体因子VIII产品在纯化过程结束时应尽可能高,并且也应在因子VIII产品的制药处理之前(冷冻状态)和实际制药处理期间(冷冻干燥或填充液体溶液于小瓶中)稳定直到重构和被患者使用。该时间段常常可为几个月到1-3年。US 8187799 B2提供了一个因子VIII在冷冻溶液中的稳定性的示例,其中保护了特定的缓冲组合物,但是在该文献中并未讨论因子VIII聚集体/单体的含量。
本发明的重组因子VIII尤其是完全或部分缺失B结构域的缺失衍生物,因此提供了在最终的纯化产品中比活性极大地超过5000IU/mg。EP-A-1136553和EP-A-1739179中公开了完全或部分缺失B结构域的这种缺失衍生物以及由人类细胞系中制备这样的缺失衍生物的示例。应认识到,如以下部分中描述的本发明的组合物特别适用于因子VIII的这种缺失衍生物或其他相似高纯度的因子VIII产品。
具体实施方式
因子VIII亲和层析步骤的详细描述
因子VIII在平衡缓冲条件(0.3mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 6.4-6.6,导电率30-36mS/cm)下结合因子VIII亲和层析柱(VIIISelect),该条件中具有或没有溶剂洗涤剂(1%Triton X-100+0.3%三正丁基磷酸酯),在病毒(脂质包膜)灭活目的的亲和步骤之前可将其应用到因子VIII溶液。来自上游因子VIII纯化的其他化学物质也可以存在于因子VIII负载溶液,如可添加用于因子VIII稳定目的的0.2mol/kg山梨醇和0.045mol/kg精氨酸。典型地,因子VIII负载为约5000-约25000IU/ml亲和树脂,可将基本上任何纯度的因子VIII溶液(1-15000IU因子VIII/mg蛋白)应用到亲和柱。该方法在环境温度例如室温下进行。
在已通过柱处理含有因子VIII的溶液后,将柱用15个柱体积的平衡缓冲液(0.3mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)的聚山梨醇酯80,pH 6.4-6.6,导电率30-36mS/cm)冲洗,以除去杂质(产品和相关方法)。随后用增加的盐浓度(1mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)的聚山梨醇酯80,pH 6.4-6.6,导电率83-89mS/cm)清洗5个柱体积,以专门除去单条FVIII轻链。此后使用具有增加的盐浓度和乙二醇的4柱体积缓冲液(1.5mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,50%(w/w)乙二醇,0.02%(w/w)的聚山梨醇酯80,pH 6.4-6.6,导电率36-42mS/cm)从柱洗脱生物活性因子VIII。
此后通过酸性洗涤(pH为2)除去结合到亲和树脂的剩余蛋白,其中平衡后的树脂用于另一个纯化循环。
阴离子交换层析步骤的详细描述
在平衡缓冲条件(0.1mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)pH 7.4-7.6的聚山梨醇酯80,导电率12-16mS/cm)下因子VIII结合强阴离子交换柱(Q琼脂糖FF)。来自上游因子VIII纯化的其他化学物质也可以存在于因子VIII负载溶液,如5%乙二醇。典型地,因子VIII负载为10000-100000IU/ml亲和树脂,起始材料的纯度是>5000IU因子VIII/mg蛋白。该方法在环境温度例如室温下进行。
在已通过柱处理含有因子VIII的溶液后,将柱用15个柱体积的平衡缓冲液(0.1 mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 7.4-7.6,导电率12-16mS/cm)冲洗,以除去杂质(产品和相关方法)。随后用增加的盐浓度(0.30mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)的聚山梨醇酯80,pH 7.4-7.6,导电率30-35mS/cm)清洗5个柱体积,以除去无活性形式的因子VIII。此后使用1个柱体积的具有增加的盐浓度的缓冲液(0.39mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 5.9-6.1,导电率38-42mS/cm)从柱洗脱生物活性单体因子VIII。
此后通过增加盐浓度至2mol/kg NaCl除去通过离子相互作用结合于阴离子交换树脂的任何剩余蛋白,之后使用高pH(14)对树脂消毒,平衡后该柱用于另一纯化循环。
尺寸排阻层析步骤的详细描述
因子VIII以4-8%的柱体积被装载到尺寸排阻层析柱(Superdex 200p.g.),其具有70厘米的床高,并用以下溶液来平衡:30.7g/kg NaCl,0.5g/kg氯化钙,2.0g/kg柠檬酸钠,9.2g/kg L-精氨酸盐酸盐,9.2g/kg蔗糖,2.0g/kg泊洛沙姆188,pH值6.9-7.1,导电率47-51mS/cm。典型地,起始材料中因子VIII的浓度为>10000IU/ml,缓冲液的组成:0.4mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH值6.0-6.5,导电率35-42mS/cm。起始材料的纯度是>8000IU因子VIII/mg蛋白。该方法是在室温下进行;18-25℃。
在已经将含有因子VIII的溶液施加到尺寸排阻柱后,将柱用平衡缓冲液处理,直到在该柱的出口处280nm下测定的吸光度升高到40mAU,当收集单体生物活性因子VIII产品直到吸光度返回到它的起点(40-1mAU)。在单体生物活性因子VIII达到峰值前(<40mAU)除去生物无活性的聚集因子VIII,在单体生物活性因子VIII达到峰值后(<40mAU)除去无活性因子VIII片段。尺寸排阻步骤后单体生物活性因子VIII溶液通常是起始材料的2-3倍体积。
本文引用的所有参考文献都通过引用以其并入不与本文所表达的教导矛盾的程度并入。本文引用的所有参考文献,包括出版物、专利申请和专利,在此通过引用以其整体并入并以每个参考文献被单独并具体地引用并入的相同程度并入,并在本文中(以法律允许的最大程度)以其整体进行详细说明,而不管在本文别处以任何单独提供的特定文件并入。
在描述本发明的上下文中使用的术语“一”、“一个”、“该”和类似的指代被解释为包括单数和复数,除非本文另有说明或与上下文明显矛盾。除非另有说明,本文提供的所有精确值代表相应近似值(例如,对于特定因子或测量提供的所有精确示例值可以被视为也提供了相应的近似测量值,适当时由“约”修饰)。
在本发明的任何方面或实施方案的描述中的参考一个或多个要素所使用的术语如“包括”、“具有”、“包含”或“含有”旨在为本发明的类似方面或实施方案提供支持,该类似方面或实施方案“由该特定要素组成”、“基本上由该特定要素组成”或“基本上包括该特定要素”,除非另有说明或与上下文明显矛盾(例如,本文所述的包含特定要素的组合物应理解为也描述了由该要素组成的组合物,除非另有说明或与上下文明显矛盾)。
本文中所用的所有标题和小标题仅为方便起见且不应该解释为以任何方式限制本发明。除非另有声明,本文所提供的任何和所有实施例或示例性语言(例如,“如”)的使用仅仅是为了更好地阐明本发明,并不造成对本发明的范围的限制。说明书中的任何语言都不应被解释为表示任何非要求保护的要素对于本发明的实施是必要的。
本文中专利文献的引用和并入仅为方便起见,并不反映这类专利文件的有效性、可专利性和/或可执行性。本发明包括由适用法律允许的本文所包括的权利要求书和/或方面中的主题的所有修改和等同方案。
通过以下实施例进一步描述本发明,但其并不限制本发明。
实施例1:
用耦合在树脂上的酵母来源的因子VIII亲和配体来纯化。
以下方法示出了在酵母来源的因子亲和配体层析步骤(VIIISelect)中去除无VIII:C活性的因子VIII形式。
柱和树脂
对BPG140柱填充VIIISelect树脂至床高11厘米,得到1.7升的柱体积。VIIISelect树脂获自GE医疗(目录No.17-5450)。
起始材料:
含因子VIII的材料用作起始材料,其具有:1614IU因子VIII/mg蛋白的纯度,0.34mol/kg NaCl,0035mol/kg氯化钙,0.01mol/kg L-组氨酸,0.045mol/kg L-精氨酸,0.2mol/kg山梨醇,0.02%(w/w)聚山梨醇酯80,1%(w/w)Triton X-100,0.3%三正丁基磷酸酯(TNBP,w/w),pH 6.5。在WO2009156430A1中进一步描述了起始材料的生产,其通过引用并入本文。FVIII:C在树脂上的负载为15529IU/mL树脂。
缓冲液组成:
平衡缓冲液(含Triton X-100和TNBP)
0.3mol/kg NaCl,0.02mol/kg氯化钙(2x H2O),0.02mol/kg L-组氨酸,1%w/w的Triton X-100,0.3%w/w的TNBP,pH:6.5±0.1,导电率:在+25℃为31±3mS/cm。
洗涤缓冲液1(不含Triton X-100和TNBP的平衡缓冲液)
0.3mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.5±0.1,导电率:+25℃下为31±3mS/cm。
洗涤缓冲液2
1.0mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.5±0.1,导电率:+25℃为85±3mS/cm。
洗脱缓冲液
1.5mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,50%(w/w)乙二醇(EG),pH:6.5±0.1,导电率:+25℃为39±3mS/cm。
平衡、洗涤和洗脱缓冲液不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
用平衡缓冲液平衡该柱后,装载起始材料。此后使树脂经历如表1所述的洗涤缓冲液1和洗涤缓冲液2和其后的洗脱缓冲液。分别从级分取出样品(起始材料负载期间的流经+洗涤1、洗涤2和洗脱),并就FVIII:C、FVIII:Ag和因子VIII蛋白印迹方面进行分析。
表3,来自VIIISelect处理的结果
表3示出了从起始材料去除无活性形式因子VIII。起始材料中,用FVIII:C测定的生物活性因子VIII与通过FVIII:Ag测定的可用因子VIII的总量的比例是0.61。还测定流经和洗涤级分中的C/Ag比例,其非常低(<0.05)。在洗脱级分中,C/Ag比例从起始材料中的0.61增加至0.87。这清楚地表明了经过VIIISelect亲和步骤去除了无活性因子VIII形式。这可通过观察图1中的因子VIII蛋白印迹分析泳道1-4得到进一步证实。泳 道1示出亲和柱之前的“起始材料”。泳道2示出许多因子VIII相关的条带,显示出在“流经+洗涤1级分”中除去了因子VIII变性的产品。泳道3显示在“洗涤2级分”中有与单条(离解的)因子VIII轻链(80kD)具有相同分子量的一条明显的主要的因子VIII条带。该因子VIII级分也示出没有或很少的因子VIII:C活性(见表3)。泳道4示出亲和步骤的洗脱级分,包括因子VIII轻链(80kD),因子VIII重链(90kD)和未裂解的因子VIII分子(170kD)。
实施例1的结论
如在表3和图1(泳道1-4)可观察到的,VIIISelect步骤除去了在流经和洗涤级分中具有减少的FVIII:C活性和/或没有FVIII:C活性的因子VIII分子。
实施例2,阴离子交换层析步骤(Q-琼脂糖FF)
以下方法示出了用阴离子交换树脂(Q琼脂糖FF)除去无因子VIII:C活性的因子VIII形式,得到较高的因子VIII单体产品。
柱和树脂
对BPG140柱填充Q琼脂糖FF树脂至床高8厘米,得到1.23升的柱体积。Q琼脂糖FF树脂获自GE医疗(目录No.17-0510)。
起始材料:
含因子VIII的材料用作起始材料,其具有:9470IU因子VIII/mg蛋白的纯度,0.1mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 6.5。按实施例1中进一步描述的来生产起始材料(产品洗脱级分)。FVIII:C在树脂上的负载为15383IU/mL树脂。
缓冲液组成:
平衡缓冲液
0.1mol/kg NaCl,0.02mol/kg氯化钙(2x H2O),0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:7.5±0.1,导电率:在+25℃为15±1mS/cm。
洗涤缓冲液
0.32mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:7.5±0.1,导电率:+25℃为32.5±2.5mS/cm。
洗脱缓冲液
0.39mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.0±0.1,导电率:+25℃为40±2mS/cm。
平衡、洗涤和洗脱缓冲液不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
用平衡缓冲液平衡该柱后,装载起始材料。此后使树脂再次经历平衡缓冲液,以使得因子VIII结合,之后应用洗涤缓冲液,接着是洗脱缓冲液,如表3所述。分别从级分取出样品(起始材料负载期间的流经+洗涤1、洗涤2和洗脱),并就FVIII:C、FVIII:Ag、SEC-HPLC1、FVIII蛋白印迹、N聚糖指纹图谱和胰蛋白酶肽指纹图谱方面进行分析。
表4,阴离子交换方法的结果
表4示出了通过阴离子交换步骤从起始材料除去无活性因子VIII形式。起始材料中,用FVIII:C测定的生物活性因子VIII与通过FVIII:Ag测定的可用因子VIII的总量的比例是0.78。还测定流经和洗涤级分中的C/Ag比例,其非常低(<0.35)。在洗脱级分中,C/Ag比例从起始材料中的0.78增加至0.91。这清楚地表明了经过阴离子交换步骤去除了无活性因子VIII形式。这可通过观察图1中的泳道5-6得到进一步证实,这两个泳道均显示在流经+平衡级分和在洗涤级分中除去了因子VIII变性的产品。图2中泳道4显示了因子VIII主级分(洗脱液)的FVIII蛋白印迹分布,没有可见的FVIII变性产品。如用SEC-HPLC1分析的,图4中示出洗脱液中的高单体含量(>98%)。
实施例2的结论
如在表4和图1(泳道5-6)可观察到的,阴离子交换步骤除去了在流经和洗涤级分中具有减少的FVIII:C活性和/或没有FVIII:C活性的因子VIII分子。如可在图4中观察到的,生物活性产品级分(洗脱)含有较高的单体因子VIII。
实施例3,尺寸排阻层析法
以下方法示出了用尺寸排阻层析柱(Superdex 200P.g.)除去无因子VIII:C活性的因子VIII形式。
柱和树脂
对BPG100柱填充Superdex 200p.g.树脂至床高69厘米,得到5.4升的柱体积。Superdex 200p.g.树脂获自GE医疗(目录No.17-1043)。
起始材料:
含因子VIII的材料用作起始材料,其具有:10200IU因子VIII/mg蛋白的纯度,0.4mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 6.2。如实施例2中描述地生产起始材料(产品洗脱级分)。样品在树脂上的负载为5.5%的柱体积。
缓冲液组成:
平衡缓冲液
30.7g/kg NaCl,0.5g/kg氯化钙,2.0g/kg柠檬酸钠,9.2g/kg精氨酸,9.2g/kg蔗糖,0.02%(w/w)聚山梨醇酯80,pH:7.0±0.1,导电率:+25℃为49±2mS/cm。
平衡不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
用平衡缓冲液平衡该柱后,装载起始材料。此后使树脂再次经历平衡缓冲液,以使得因子VIII溶液在尺寸排阻柱上分离。当在柱的出口处的在280nm下的吸光度升高至超过0.035AU时,开始收集洗脱液,当吸光度降低到0.05,停止收集洗脱液。从负载和洗脱液中取出样品,并就FVIII:C、FVIII:Ag、SEC-HPLC1和FVIII蛋白印迹方面进行分析。
表5,尺寸排阻方法的结果
表5示出了通过尺寸排阻步骤从起始材料中除去无活性因子VIII形式。用FVIII:C测定的生物活性因子VIII与通过FVIII:Ag测定的可用因子VIII的总量的比例在起始材料中是0.83,在洗脱级分中是0.94。这表明通过尺寸排阻柱除去了由于聚集和/或片段化导致的无活性因子VIII形式的除去。图2中,泳道5显示了洗脱级分与对照相同的因子 VIII的条带图案分布。图5显示了天然条件下使用SEC-HPLC1的洗脱液的高因子VIII单体含量(>99%)和低聚集体含量(<1%)。图3示出了聚集体和片段从单体因子VIII产品的实际除去,如生产尺寸排阻层析图所描述的。
实施例3的结论
尺寸排阻层析步骤通过分离不同尺寸的分子除去了具有减少的FVIII:C活性和/或没有FVIII:C活性的因子VIII分子。层析环境和工艺参数有利于该过程中这些分子的聚集和除去,得到具有高生物活性(C/Ag>0.9)的高(>99%)单体因子VIII产品,如图5和表5中可见。
实施例4,依次使用亲和、阴离子交换和尺寸排阻层析法
以下方法示出了无因子VIII:C活性的因子VIII形式的除去,执行由三种不同的层析技术组成的纯化序列,依次为:
1.亲和层析(VIIISelect)
2.阴离子交换层析(Q琼脂糖FF)
3.尺寸排阻层析(Superdex 200p.g.)
柱和树脂
1.亲和层析(VIIISelect)
对BPG140柱填充VIIISelect树脂至床高10厘米,得到1.5升的柱体积。VIIISelect树脂获自GE医疗(目录No.17-5450)。
2.阴离子交换层析(Q琼脂糖FF)
对BPG100柱填充Q琼脂糖FF树脂至床高7厘米,得到0.55升的柱体积。Q琼脂糖FF树脂获自GE医疗(目录No.17-0510)。
3.尺寸排阻层析(Superdex 200p.g.)
对BPG100柱填充Superdex 200p.g.树脂至床高69厘米,得到5.4升的柱体积。Superdex 200p.g.树脂获自GE医疗(目录No.17-1043)。
起始材料、缓冲液组成、亲和步骤:
以下用作起始材料:0.34mol/kg NaCl,0.035mol/kg氯化钙,0.01mol/kg L-组氨酸,0.045mol/kg L-精氨酸,0.2mol/kg山梨醇,0.02%(w/w)聚山梨醇酯80,1%(w/w)Triton X-100,0.3%三正丁基磷酸酯(TNBP,w/w),pH 6.5。如WO2009/156430A1中进一步描述地生产起始材料,其通过引用并入本文。
起始材料、缓冲液组成、阴离子交换步骤:
以下用作起始材料:0.15mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 7.5。
起始材料、缓冲液组成、尺寸排阻步骤:
以下用作起始材料:0.39mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 6.2。
缓冲液组成、亲和层析:
平衡缓冲液(含Triton X-100和TNBP)
0.3mol/kg NaCl,0.02mol/kg氯化钙(2x H2O),0.02mol/kg L-组氨酸,1%w/w的Triton X-100,0.3%w/w的TNBP,pH:6.5±0.1,导电率:+25℃为31±3mS/cm。
洗涤缓冲液1(不含Triton X-100和TNBP的平衡缓冲液)
0.3mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.5±0.1,导电率:+25℃下为31±3mS/cm。
洗涤缓冲液2
1.0mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.5±0.1,导电率:+25℃下为85±3mS/cm。
洗脱缓冲液
1.5mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,50%(w/w)乙二醇(EG),pH:6.5±0.1,导电率:+25℃下为39±3mS/cm。
平衡、洗涤和洗脱缓冲液不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
缓冲液组成、阴离子交换层析:
平衡缓冲液
0.1mol/kg NaCl,0.02mol/kg氯化钙(2x H2O),0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:7.5±0.1,导电率:+25℃下为15±1mS/cm。
洗涤缓冲液
0.32mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:7.5±0.1,导电率:+25℃下为32.5±2.5mS/cm。
洗脱缓冲液
0.39mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH:6.0±0.1,导电率:+25℃下为40±2mS/cm。
平衡、洗涤和洗脱缓冲液不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
缓冲液组成、尺寸排阻层析:
平衡缓冲液
30.7g/kg NaCl,0.5g/kg氯化钙,2.0g/kg柠檬酸钠,9.2g/kg精氨酸,9.2g/kg蔗糖,0.02%(w/w)聚山梨醇酯80,pH:7.0±0.1,导电率:+25℃下为49±2mS/cm。
平衡不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
用如上定义的平衡缓冲液平衡各个柱(亲和、阴离子交换和尺寸排阻层析)。
首先处理亲和层析树脂,通过装载如上定义的和表6中的起始材料,随后用如上定义的洗涤缓冲液1和洗涤缓冲液2清洗柱。此后,通过洗脱缓冲液洗脱生物活性因子VIII产品。洗脱液是采样后(FVIII:C,FVIII:Ag)用于下列阴离子交换层析步骤的起始材料。
将来自亲和层析步骤的洗脱液稀释10倍,以实现如上定义的起始材料的条件。将稀释的起始材料用阴离子交换柱处理,接着用如上定义的洗涤1和洗涤2处理。此后,通过洗脱缓冲液洗脱生物活性因子VIII产品。洗脱液是采样后(FVIII:C,FVIII:Ag)用于下列尺寸排阻层析步骤的起始材料。
如果冷冻则将来自阴离子交换步骤的洗脱液解冻,否则通过如上定义的和表6中的尺寸排阻步骤直接处理。在将产品施加到柱上后,通过柱处理平衡缓冲液直至洗脱单体因子VIII级分。当在280nm处测定的吸光度升高至0.04AU以上时,开始单体因子VIII级分的收集。并且,当在280nm处测定的吸光度回到0.01AU时,停止单体因子VIII级分的收集。对洗脱液采样并就FVIII:C、FVIII:Ag、FVIII蛋白印迹、SEC_HPLC和氨基酸组成方面进行分析。
在特定条件下对5个不同批次(表6)重复上述程序以研究可再现性,结果可在表7中看出。
表6,5个不同批次柱负载性质
*尺寸排阻柱进行2个循环,以不超过特定的最高柱负载的8%,之后将接收的两个洗脱液混合到一个池中,从其中取出分析样品。
表7,来自连续纯化过程的FVIII C/Ag结果
表7示出通过该过程的第一纯化步骤(亲和层析),接着是该过程的第二纯化步骤(阴离子交换层析),最后是该过程的最后一个纯化步骤(尺寸排阻层析),从来自5个不同批次的起始材料中除去无活性因子VIII形式。
图6示出在尺寸排阻层析步骤进行的5个生产批次(根据表6-7)的层析覆盖图。收集尺寸排阻洗脱级分的开始和停止表示在图中。
用FVIII:C测定的生物活性因子VIII与通过FVIII:Ag测定的可用因子VIII的总量的比例是0.70,其作为用于亲和步骤之前起始材料的平均值。FVIII:C/Ag比例然后增加至0.80,其作为亲和步骤后的平均值,并且进一步增加至0.85,其作为阴离子交换步骤后的平均值。最终在最后的尺寸排阻纯化步骤之后,该比例终止在0.95,接近于最佳的FVIII:C/Ag比例1.0。这可在表7中进行进一步研究,表7示出对于执行的所有5个批次的各个步骤的所有的FVIII:C和FVIII:Ag的浓度,以及FVIII:C/FVIII:Ag比例。
图7、9、11、13和15示出对表6-7定义的批次1-5的阴离子交换洗脱液的SEC-HPLC1分析之后的结果。所有阴离子交换洗脱液样品的SEC-HPLC1结果显示没有(4个批次)或非常低(<1%)的聚集体含量(5个批次的平均值为0.1%),>97%的FVIII单体含量(5个批次的平均值为98.5%),以及<2%的片段含量(5个批次的平均值为1.4%)。执行亲和步骤以及随后的阴离子交换步骤后,阴离子交换洗脱液显示高含量的因子VIII单体和低含量的可能的免疫原性聚集体/片段FVIII部分。
图8、10、12、14和16示出对表6-7定义的批次1-5的尺寸排阻洗脱液的SEC-HPLC1分析之后的结果。所有尺寸排阻洗脱液样品的SEC-HPLC1结果显示没有可见的或可检测量的聚集体,>98%的FVIII单体含量(5个批次的平均值为98.6%),和<2%的片段含量(5个批次的平均值为1.4%)。在执行亲和步骤以及随后的阴离子交换步骤和尺寸排阻步骤之后,尺寸排阻洗脱液显示高含量的因子VIII单体,没有可检测的或可见的聚集体迹象,以及低含量的FVIII片段。
实施例4的结论
根据本发明,根据实施例4中按顺序1-3执行的所有三个层析步骤有助于除去无活性因子VIII分子。这表明通过不同步骤除去的无活性因子VIII分子在其生物物理学性质上有所区别,并且执行按顺序彼此结合的步骤提供了最终纯化的因子VIII产品,其具有最低程度的无活性因子VIII含量和最高程度的因子VIII单体含量,这使得患者的免疫原性反应的风险最低。
实施例5,尺寸排阻层析参数(柱高度,FVIII:C浓度,负载)
下面的实施例说明尺寸排阻参数(FVIII:C浓度和柱负载)对生物活性(FVIII:C/Ag)的重要性。
柱和树脂
对BPG200柱填充Superdex 200p.g.树脂至床高62厘米,得到19.5升的柱体积。Superdex 200p.g.树脂获自GE医疗(目录No.17-1043)。
起始材料:
含因子VIII的材料用作起始材料,具有以下组成:0.4mol/kg NaCl,0.02mol/kg氯化钙,0.02mol/kg L-组氨酸,0.02%(w/w)聚山梨醇酯80,pH 6.2。如实施例2中所描述地生产起始材料(产品洗脱级分)。
缓冲液组成:
平衡缓冲液
30.7g/kg NaCl,0.5g/kg氯化钙,2.0g/kg柠檬酸钠,9.2g/kg精氨酸,9.2g/kg蔗糖,0.02%(w/w)聚山梨醇酯80,pH:7.0±0.1,导电率:+25℃下为49±2mS/cm。
平衡不限于规定的pH值、浓度和缓冲液、盐或洗涤剂类型。
按照下表8执行七个不同批次(A-G)。用平衡缓冲液平衡该柱,随后负载特定的起始材料。此后使树脂经历平衡缓冲液以使因子VIII溶液通过尺寸排阻柱分离。当在柱的出口处在280nm下的吸光度升高至超过0.035AU时,开始收集洗脱液,当吸光度降低到0.005AU,停止收集洗脱液。从起始材料和洗脱液中取出样品,并就FVIII:C、FVIII:Ag和SEC-HPLC1方面进行分析。
表8,试验条件和所得的生物活性FVIII:C/Ag
表8表明通过尺寸排阻步骤从起始材料除去无活性因子VIII形式取决于不同的参数。相比批次A-D(A-0.90,B-0.81,C-0.99和D-1.01),在各个洗脱液(E-0.81,F-0.79,G-0.82)中,如批次E(15925IU/ml,4.3%)、批次F(14844IU/ml,1.9%)和批次G(9149IU/ml,3.9%)中相对低的因子VIII:C的浓度与相对低的柱负载的结合似乎对于用FVIII:C/FVIII:Ag比例测定的生物活性是起负作用的。
实施例5的结论
FVIII:C浓度与柱负载的组合是尺寸排阻步骤的结果在生物活性方面的重要性的参数。根据实施例3和实施例4中描述的数据,两者都使用69厘米的床高,似乎具有略低床高(62厘米)的实施例5中所述的结果与两个其它重要性因子(FVIII:C浓度和柱负载)的组合可能也影响尺寸排阻步骤的结果。这表明具有高因子VIII:C浓度、高柱床高度和≥4%的柱负载的尺寸排阻步骤得到的效果更好。
实施例6,处于冷冻状态的生物活性单体因子VIII的稳定性。
下面的实施例说明了根据实施例4处理的生物活性单体因子VIII溶液在冷冻状态下的的稳定性。
起始材料:根据实施例4制备的因子VIII溶液,具有>99%的因子VIII单体含量,<1%的聚集体含量,和>0.9的无活性因子VIII的量,如用生物活性因子VIII相对于因子VIII的总量的比例(FVIII:C/FVIII:Ag)测定的。
因子VIII缓冲环境:
30.7g/kg NaCl,0.5g/kg氯化钙,2.0g/kg柠檬酸钠,9.2g/kg精氨酸,9.2g/kg蔗糖,0.02%(w/w)聚山梨醇酯80,pH:7.0±0.1,导电率:+25℃下为49±2mS/cm。
冷冻和储存条件:
将因子VIII溶液填充在塑料低密度聚乙烯容器中,通过快速冷冻法在最多60分钟期间冷冻至-40℃。此后将冷冻溶液储存在-60℃至-80℃之间的温度36个月。储存0、6、9、12、18、24和36个月后取出样品,并对生物活性和单体、聚集体、片段含量进行分析。
表9,在-60℃至-80℃之间的温度下冷冻储存的因子VIII溶液的稳定性
如从表9中可以看出,因子VIII溶液在-60℃至-80℃之间的温度下储存至少36个月,其在生物活性和因子VIII单体含量方面(聚集体和片段的形成)是完全稳定的。
实施例7,冷冻干燥的产品中生物活性单体因子VIII的稳定性。
下面的实施例说明了根据实施例4和实施例6处理及此后冷冻干燥的生物活性单体因子VIII溶液的稳定性。
起始材料:根据实施例4和实施例6制备的冷冻因子VIII溶液,具有>99%的因子VIII单体含量,<1%的聚集体含量,和>0.9的无活性因子VIII的量,如用生物活性因子VIII相对于因子VIII的总量的比例(FVIII:C/FVIII:Ag)测定的。
冷冻干燥后一小瓶因子VIII的含量:
冷冻干燥程序:
将2.5毫升因子VIII起始材料填充在8毫升模压玻璃小瓶(I型)中至总量为250IU因子VIII。如表10所述,使小瓶进行冷冻干燥过程。
表10,冷冻干燥过程
在冷冻干燥过程后,将小瓶用溴化丁基塞子封闭并用铝盖封装。将小瓶在+5℃下储存24个月。通过在2.5毫升注射用水中重构冻干产品,在储存0、6、9、12、18和24个月后进行取样,然后对生物活性和单体、聚集体、片段含量进行分析,参见表11。
表11,在+5℃下储存的冷冻干燥的因子VIII溶液的稳定性
如从表11可以看出,冷冻干燥的因子VIII产品在+5℃的温度下储存至少24个月,其在生物活性和因子VIII单体含量方面(聚集体和片段的形成)是完全稳定的。
实施例8,测定根据本发明的最终冻干产品中FVIII C/Ag比例以及聚集体、单体和片段,并将其与使用不同纯化方法的重组产品进行比较。
下面的实施例说明了根据本发明的产品的优异性能并将其与使用不同纯化方法的其它市售重组产品进行比较。
起始材料:将根据本发明的实施例7制备的冻干因子VIII产品重构并与根据3种不同的纯化方法(A-C)纯化并用不同的制剂缓冲液稳定的3个不同的竞争rFVIII产品进行比较。就稳定剂/FVIII方面分析至少1瓶的强度(250IU,1000IU或3000IU),以在不同制剂组合物之间进行良好比较。根据SEC-HPLC2分析方法在天然电泳条件下对所有样品的FVIII C/Ag和聚集体、单体和片段进行分析。
表1
实施例8的结论:
表1示出了根据本发明生产的重组FVIII的纯化和冷冻干燥产品的优良质量,即100%的单体含量和>0.9的FVIII C/Ag。这表明,相比在不同的条件下纯化和稳定的所有竞争产品,在最终产品中没有无生物活性因子VIII或有非常少量的无生物活性因子VIII。
实施例9,比较之前未用根据本发明纯化、配制、冷冻干燥和储存的rFVIII产品治疗的A型血友病患者中的抑制剂形成,并将其与对一种竞争的重组FVIII产品的公布数据21进行比较。并且,还比较两个产品之间的FVIII C/Ag比例和聚集体、单体和片段。
下面的实施例说明了根据本发明的产品的优异性能,并将其与一种在市场上可获得的市售重组FVIII产品进行比较。
起始材料:将根据本发明的实施例7制备的冻干因子VIII产品与一种根据实施例8所述的方法A纯化的市场上可获得的竞争rFVIII产品进行比较。将这两种产品给予至患者,并根据之前未治疗的A型血友病患者的标准临床协议21随访抑制剂形成的发展。对于各种产品发现抑制剂的患者量以及因子FVIII C/Ag比例和聚集体/单体/片段分布可从表2中看出。
表2
*发表的柯林斯研究201421,**正在进行的研究的未发表数据
实施例9的结论:
该表示出了与市场上市售的一种rFVIII产品相比,使用本发明的产品在先前未治疗的A型血友病患者中检测到显著较低量的抑制剂。此外,表10示出了对于3个不同小瓶强度(250IU,1000IU和3000IU)中的2个(250IU和1000IU),各个产品的实施例中因子FVIII C/Ag比例和聚集体/单体/片段分布。可假设产品中较低量的生物无活性FVIII会减少在患者中形成的抑制剂的量。因此,暗示了重组FVIII产品的纯化、稳定、冷冻干燥和储存对于最小化患者在免疫反应方面的风险的重要性。
实施例10,根据本发明纯化的重组FVIII产品的比活性(纯度)。
以下实施例说明了根据本发明纯化的重组FVIII产品的优异纯度。
起始材料:根据实施例4和实施例6制备的冷冻因子VIII溶液,其具有>99%的因子VIII单体含量,<1%的聚集体含量,和>0.9的无活性因子VIII的量,如用生物活性因子VIII相对于因子VIII的总量的比例(FVIII:C/FVIII:Ag)测定的,分析该溶液的FVIII:C并根据Bradford分析总蛋白含量。
实施例10的结论:
图18显示了通过FVIII:C/mg蛋白测定(如用Bradford测定)的在生产规模上根据本发明纯化(最后3个步骤:亲和洗脱液,阴离子交换洗脱液和药物(=尺寸排阻层析洗脱液))的9个批次的纯度的增加,结束纯度为10000IU/mg蛋白的范围,其是基本上纯的rFVIII。
分析说明
基于测定作为辅因子的因子VIII的生物活性的两阶段光度测定法(17),用显色法(COATEST SP FVIII试剂盒,82 4086 63,Chromogenix/仪器实验室(US))测定因子VIII的生物活性(FVIII:C)。
如进一步描述的,用用于样品稀释的Tris-NaCl缓冲液+1%牛血清白蛋白替换提供的试剂盒缓冲液(18),用ELISA试剂盒(VIII:Ag,用于因子VIII的 酶免疫测定,试剂盒,Diagnostica Stago(法国))测定因子VIII抗原含量(FVIII:Ag)。
使用两个不同的尺寸排阻层析(SEC-HPLC)分析柱(SEC-HPLC 1,Superdex 200,10/300GL,GE医疗和SEC-HPLC2,BioSEC-3,安捷伦科技)测定因子VIII单体、聚集体和片段。
天然缓冲条件(25mM HEPES,0.5M氯化钠,0.3M精氨酸,50mM氯化钙,0.02%聚山梨酯醇80,pH 7.5)下进行SEC-HPLC1方法(Superdex 200)。样品负载是约1%的尺寸排阻柱,因子VIII:C的浓度是约1000IU/ml。单体被定义为主要的FVIII层析图谱峰,聚集体为FVIII单体峰之前洗脱的峰,片段为FVIII单体峰之后洗脱的层析峰。
SEC-HPLC2法(BiSEC-3):使用尺寸排阻层析(BioSEC-3,30×4.6毫米SEC柱,安捷伦科技)在非变性缓冲条件(25mM Tris-HCl,50mM CaCl2,500mM NaCl,pH 7.0)下以0.4mL/分钟的流速在Shimadzu HPLC系统上,对重组表达FVIII的样品针对FVIII聚集体、单体和片段的组成进行分析。根据确定的FVIII活性(FVIII:C)值,样品负载为约0.2-0.4μg。单体被定义为主要的FVIII层析图谱峰,聚集体为FVIII单体峰之前洗脱的峰,片段为FVIII单体峰之后洗脱的层析图谱峰。同样还将用于分析的rFVIII样品的保留时间与分子量样品(15-600kD,69385,Sigma-Aldrich Chemie GmbH)的保留时间进行比较。
使用FVIII蛋白印迹基于尺寸测定因子VIII变性产品。根据分子量在还原条件下通过十二烷基硫酸钠(SDS)聚丙烯酰胺凝胶电泳(PAGE)分离因子VIII制剂中的FVIII分子质量分布的蛋白和肽。
此后,通过电泳将蛋白从凝胶基质转移至硝酸纤维素膜,其随后用封闭剂孵育。然后将针对整个人因子VIII分子的市售多克隆羊抗体加入,之后加入酶标记的次级抗体作为探针。作为第三步骤,加入化学发光底物并且当其与酶相结合,产生作为副产物的光。使用冷却电荷耦合装置相机将光输出捕获为实时图像。信号强度与印迹膜上抗原(FVIII)的丰度相关。
为了研究因子VIII蛋白链的电泳带图案,实施银染色的2D-电泳。使用pH为3-10的线性pH梯度执行等电点聚焦,作为第一维跑胶(run)。使用Tris-醋酸酯(3-8%)凝胶使第二维SDS-PAGE跑胶。第二维跑胶后用银染料对凝胶进行染色。
通过在蛋白的酸水解后对组成氨基酸的分析来进行氨基酸组成分析。在110℃下将蛋白质在6M盐酸中水解24小时,此后通过阳离子交换层析在磺化聚苯乙烯树脂上分离氨基酸,并在洗脱液中进行连续检测。该检测是基于茚三酮柱后衍生化法,其使用在用于脯氨酸的440nm处和用于所有其他氨基酸的570nm处同时测定的双光度计。由于这种方法不能适当地测量半胱氨酸或色氨酸的残基,因此没有测得来自半胱氨酸或色氨酸 的值。由于缓冲制剂中存在赖氨酸和精氨酸的干扰,因此还省略了赖氨酸和精氨酸的值。
通过高效阴离子交换层析与脉冲安培检测法(HPAEC-PAD)执行N-聚糖指纹法,以确定N-连接的聚糖(19)。通过酶促裂解(N-聚糖酶Plus,产品编号GKE-5010B,来自Prozyme)使N-连接的聚糖链从蛋白释放,随后在高pH(pH为13)下结合到阴离子交换层析柱,接着经过离子强度增加和pH降低的梯度。由于电荷、尺寸和结构的差异导致实现了层析系统中各种聚糖链的分离。阴离子交换柱是来自Dionex的CarboPacTM PA200层析柱(250毫米×3毫米ID,粒径5.5微米,产品编号062896)和来自Dionex的CarboPacTM PA 200保护柱(50×3mm的ID,产品编号062895)。所用的检测系统是具有3mm金膜的ICS-3000CD电化学检测器(ICS-3000Au工作电极,3毫米,产品编号063723),其由软件Chromeleon控制。
按级分收集对应于不同层析峰的样品,每个级分对应于1分钟的保留时间。在多孔石墨碳柱上对聚糖脱盐,然后浓缩各级分。使用PGC-柱在毛细管液相层析系统上进行层析分离,在线将聚糖电喷雾进入飞行时间质谱仪。通过大量输入并搜索可能的匹配来使用来自功能糖组学[www.functionalglycomics.org]的数据库。由于高质量准确度(约30ppm),各聚糖的鉴定被确定是准确的。
执行包括两个主要步骤的胰蛋白酶肽指纹映射。在第一步骤中,使用胰蛋白酶消化待被分析成多肽的蛋白,在第二步骤中将其分离并用HPLC技术记录。
通过这种方式产生了特定的图案(“指纹”)。可以通过荧光(主要为色氨酸)检测肽,相比UV检测得到更为简化的图。随时间记录的荧光图案表示用于所分析的样品的肽图。
使用Bradford测定法23测定蛋白浓度。
VIIISelect是设计用于纯化重组B-结构域贫乏的因子VIII的亲和层析介质(树脂)。
根据数据文件28-9662-37 AB,VIIISelect的GE医疗关键特性包括:
·重组B-结构域缺失的因子VIII的高效纯化,产率高并保留特定活性
·高选择性
·优良的可扩展性
·不含动物成分的生产
治疗血友病患者需要重组凝血因子的高效纯化处理。VIIISelect是设计用于纯化重组B-结构域贫乏的因子VIII的亲和层析介质,重组B-结构域贫乏的因子VIII是用于治疗A型血友病的关键重组血液因子。由于因子VIII分子的敏感性质,重要的是限制下游处理中步骤的数量。使用VIIISelect得到的高选择性和高产率允许获得具有在一个步骤 中得到的优良纯度的稳健有效的纯化方法。不含动物成分的生产和低配体泄漏是附加属性,使得该介质高度适合于大规模生产重组B-结构域贫乏的因子VIII。VIIISelect是GE医疗客户定制媒体计划的一部分。
介质特征
VIIISelect是基于高度交联的琼脂糖基质,其能够实现快速处理大样本容量。将配体即13kD的重组蛋白通过亲水间隔臂连接至多孔基质,使其容易用于结合到重组结构域贫乏的因子VIII(图1)。表1概括了VIIISelect的主要特征。
功能原理
亲和层析利用在适合的结合条件下吸附特定分子或特定分子组并在适合的洗脱条件下对它们解吸附的固定化配体。这些条件取决于靶分子、进料组合物和层析介质,并且必须与其它层析参数(例如样品负载、流速、床高、
VIIISelect的部分结构、再生和就地清洁)一起研究,以建立将以最高的产品回收率结合靶分子的条件。
重组因子VIII可以直接从澄清的细胞裂解物或上清液施加到VIIISelect柱。
表1.VIIISelect的主要特征
为了促进因子VIII的活性构象的形成,缓冲液应总是包含Ca2+离子。需要表面活性剂的存在来抑制表面诱导的变性/吸附。中性pH缓冲液和组氨酸应总是用于结合、洗 涤和洗脱,以保持特定的因子VIII活性。根据施加至VIIISelect的材料的性质,通常在每个循环后需要再生,接着是在平衡/负载缓冲液中的重新平衡。
稳定性
将该配体通过稳定的酰胺键连接到高度交联的基质。图2示出了VIIISelect在不同pH值于室温下储存1周的研究。该图表明稳定性在pH为3和10之间较高。GE建议在pH为3和10之间进行长期储存,在pH为2和12之间进行短期储存。
储存
建议储存条件是在4℃-8℃下的20%乙醇。在20%的乙醇溶液中供应预溶胀的VIIISelect。
就地清洁
用于VIIISelect的清洁方案可以由0.1M柠檬酸或0.5M磷酸组成。然而,由于琼脂糖基质的分解,应避免长时间暴露于pH<2。可将氢氧化钠(0.01M)单独使用或与硫酸钠/氯化钠组合使用作为稳定剂。
参考文献
1.Wang etal.,Coagulation Factor VIII;structure and stability(结构和稳定性),International Journal of pharmaceutics 259(2003)1-15.
2.Svensson etal.,Evaluation of the metal binding site in a recombinant coagulation factor VIII identifies two sites with unique metal binding sites(重组凝血因子VIII中金属结合位点的评价鉴定具有独特的金属结合位点的两个位点),Biological Chemistry,DOI:10.1515/hsz-2012-0298
3.Peerlinck etal.,Factor VIII inhibitors in previous treated Haemophilia A patientse with a double virus inactivated plasma derived Factor VIII concentrate(先前用双重病毒灭活的血浆来源的因子VIII浓缩液治疗的A型血友病患者中的因子VIII抑制剂),Thrombosis and Haemostasis 77(1)80-86(1997).
4.Fang etal.,The protein structure and effect of Factor VIII(因子VIII的蛋白结构和作用),Thrombosis Research(2007)119,1-13.
5.Lin etal.,Relationship between Factor VIII:Ag and Factor VIII in recombinant and plasma derived Factor VIII concentrate(重组和血浆来源的因子VIII浓缩液中因子 VIII:Ag和因子VIII之间的关系),Haemophilia(2004),10,459-469.
6.Mire-Sluis etal.,Analysis and immunogenic potential of aggregates and particles(聚集体和颗粒的分析和致免疫潜力),Bioprocess International 9(11)December 201138-43.
7.Grillo etal.,Conformational origin of the aggregation of recombinant human Factor VIII(重组人因子VIII的聚集的构象起源),Biochemistry 2001,40,586-595.
8.Wang etal.,Correlation with rFVIII inactivation with aggregation in solution(rFVIII失活与溶液中的聚集的关系),Pharmaceutical Research,Vol.20,No.4,April 2003.
9.Thim etal.,Purification and characterization of new recombinant Factor VIII(新重组因子VIII的纯化和表征)(N8),Haemophilia(2010),16,349-359.
10.Kelley etal.,Development and validation of ab affinity chromatography step using a peptide ligand for cGMP production of Factor VIII(使用用于cGMP生产因子VIII的肽配体建立和验证ab亲和层析步骤),Biotechnology and Bioengineering,Vol.87,No.3,August5,2004.
11.McCue etal.,Application of a novel affinity adsorbent for the capture and purification of recombinant Factor VIII compounds(用于捕获和纯化重组因子VIII化合物的新亲和吸附剂的应用),Journal of Chromatography A,1216(2009)7824-7830.
12.Kusch etal.,Factor VIII assay mimicking in vivo coagulation conditions(模仿体内凝血条件的因子VIII试验),Haemophilia(2013),1-7.
13.Sommer etal.,Comparative field study evaluating activity of recombinant FVIII Fc fusion protein in plasma samples at clinical haemostatis laboratories(临床止血实验室评价血浆样品中重组FVIII Fc融合蛋白的活性的比较现场研究),Heamophilia(2013)1-7.
14.Muyldermans,Single domain camel antibodies:current status(骆驼单域抗体:现状),Reviews in Moleculer Biotechnology 74,(2001),277-302.
15.Fay,Factor VIII:Function and structure(因子VIII:功能和结构),International Journal of Hematology 83(2006)103-108.
16.Metal ion-independent association of Factor VIII subunits and the roles of calcium and copper ions for cofactor activity and inter-sub-unit affinity(因子VIII亚基与钙和铜离子对辅因子活性和亚基间亲和性的作用的金属离子非依赖性联系),Biochemistry 2001,40,10293-10300.
17.Rosen,Assay of Factor VIII:C with a chromogenic substrate(使用显色底物的因子VIII:C的试验),Scand J Haemetol-Suppl 40,Vol.33,1984,139-145.
18.Girma etal.,Assay of Factor VIII antigen(FVIII:CAg)in 294Haemophilia A patients by a new commercial ELISA using monoclonal antibodies(使用单克隆抗体通过新商用ELISA对294名A型血友病患者中因子VIII抗原(FVIII:CAg)的测定),(Haemophilia 1998),4,98-103.
19.Cataldi etal.,Carbohydrate analysis by high performance anion-exchange chromatography with pulsed amperometric detection:the potential is still growing(通过高效阴离子交换层析与脉冲安培检测进行的糖类分析:可能性仍在增加),Fresenius J Anal Chem 2000;368:739-58.
20.Casademunt et al.,The first recombinant human coagulation factor VIII of human origin:human cell line and manufacturing characteristics(人来源:人细胞系的第一重组人凝血因子VIII和制备特征).Eur J Haematol.2012;89(2):165-76.
21.Collins etal.,Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe haemophilia A(在先前未治疗的患有严重A型血友病的UK儿童中因子VIII印记和因子VIII抑制剂的发生),2000-2011,Bloodjournal.org,DO1 10.1182/blood-2014-07-580498
22.Boedeker,Production processes of licensed recombinant factor VIII preparations(经许可的重组因子VIII制剂的生产方法),Seminars in thrombosis and hemostasis volume 27,number 4,2001.
23.Bradford MM.A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding(利用蛋白-染料结合的原理定量微克量的蛋白的快速且灵敏的方法).Anal Biochem.1976;72:248-54.
- 还没有人留言评论。精彩留言会获得点赞!