一种化合物以及在治疗结肠癌中的应用的制作方法
本发明涉及一种化合物以及在治疗结肠癌中的应用,属于生物医药领域。
背景技术:
肿瘤是目前人类健康的重大威胁,也是生命科学和医学界的巨大挑战。其中,结肠癌是欧美等发达国家最常见的恶性肿瘤,也是我国常见的九大恶性肿瘤之一。过去30多年,结肠癌在世界范围内的发病率呈现出快速上升趋势。结肠癌是导致我国男性死亡的第5大恶性肿瘤,是导致我国女性死亡的第6大恶性肿瘤。
结肠癌常见于中老年人,男性多于女性。结肠癌的发病与社会环境(尤其是空气污染和水污染等因素)及生活方式(饮食习惯及缺乏运动等)和遗传因素等息息相关。然而,结肠癌早期症状多不明显,导致确诊时往往都已是中后期。发展至中后期,多数结肠癌病人会表现出腹痛和消化道症状、腹部肿块、腹泻与便秘等,如果癌变发生于直肠则可能会出现肛门坠痛、排便不畅和里急后重等症状,给患者和家属带来巨大的痛苦和不便。
同其他恶性肿瘤一样,结肠癌也存在癌细胞转移的不利情况。结肠癌细胞可以从原发部位向淋巴管、血管或其他管路入侵,被带入其他部位生长形成与原发肿瘤类型相同的肿瘤。结肠癌可发生肝脏转移、淋巴转移、血行转移、肠腔内转移和腹腔转移等,其中肝脏是结肠癌远处转移的最主要目的地。
目前,结肠癌的治疗主要包括手术治疗、放射治疗和化学药物治疗,其中手术是根治结直肠癌最有效的办法,化学药物治疗大多采取5-氟尿嘧啶、丝裂霉素及阿霉素等通用化疗药物,目前仍缺少针对结肠癌的高效特异的临床治疗药物。
技术实现要素:
本发明的目的是提供一种化合物及其用途,本发明所提供的化合物或其药学上可接受的盐可用于预防和/或结肠癌或抑制结肠癌细胞扩散。
本发明所提供的化合物如式ⅰ所示,
式ⅰ中,r1、r2、r3和r4各自独立地选自氢原子、卤素、硝基、碳原子数为1~6的烷基和碳原子数为1~6的烷氧基;
r5选自硝基和-nr6r7,其中r6和r7各自独立地为氢原子或-ch2ar,ar表示苯基或对位被r8取代的芳基,所述芳基为苯基,r8为卤素、羟基、碳原子数为1~6的烷氧基或下述所示基团,各基团中
所述化合物的结构式具体如式ⅱ所示,
式ⅱ中,r1、r2、r3和r4的定义同式ⅰ中。
式ⅱ中,r1、r2、r3和r4各自独立地选自氢原子、f、cl、硝基、甲基和甲氧基。
式ⅱ所示化合物具体如式ⅱ-1、式ⅱ-2、式ⅱ-3、式ⅱ-4、式ⅱ-5、式ⅱ-6或式ⅱ-7所示:
本发明进一步提供了式ⅱ所示化合物的制备方法,包括如下步骤:
1)在亚硝酸钠存在的条件下,式a所示化合物与叠氮化钠经叠氮化反应得到式b所示化合物;
式a和式b中,r1、r2、r3和r4各自独立地选自氢原子、卤素、硝基、碳原子数为1~6的烷基和碳原子数为1~6的烷氧基;
2)在lewis酸的催化下,式b所示化合物与对硝基苯胺经缩合反应得到式c所示化合物;
所述lewis酸可为四氯化钛;
式c中,r1、r2、r3和r4的定义同式a中;
3)式c所示化合物在碘化亚铜的催化下经分子内环化反应即得式ⅱ所示化合物;
式ⅱ中,r1、r2、r3和r4的定义同式a中。
上述制备方法中各反应均可在常规的反应条件下进行。
所述化合物的结构式具体如式ⅲ所示,
式ⅲ中,r1、r2、r3和r4的定义同式ⅰ中。
式ⅲ中,r1、r2、r3和r4各自独立地选自氢原子和f。
式ⅲ所示化合物具体如式ⅲ-1所示,
本发明进一步提供了式ⅲ所示化合物的制备方法,包括如下步骤:
在铁粉和乙酸水溶液的还原作用下,式ⅱ所示化合物经还原反应即得式ⅲ所示化合物;
式ⅱ和式ⅲ中,r1、r2、r3和r4各自独立地选自氢原子、卤素、硝基、碳原子数为1~6的烷基和碳原子数为1~6的烷氧基。
上述制备方法中各反应均可在常规的反应条件下进行。
所述化合物的结构式具体如式ⅳ所示,
式ⅳ中,r1、r2、r3和r4的定义同式ⅰ中;
r6和r7各自独立地为氢原子或-ch2ar,但是r6和r7不同时为氢原子,
ar的定义同式ⅰ中。
式ⅳ所示化合物具体如式ⅳ-1、式ⅳ-2、式ⅳ-3、式ⅳ-4、式ⅳ-5、式ⅳ-6、式ⅳ-7、式ⅳ-8、式ⅳ-9或式ⅳ-10所示,
本发明进一步提供了式ⅳ所示化合物的制备方法,包括如下1)或2)的步骤:
1)当r6为氢原子,r7为-ch2ar,且ar为苯基时,包括如下步骤:
式ⅲ所示化合物与phch2x经取代反应即得式ⅳ所示化合物;
phch2x中,ph表示苯基,x表示卤素;
2)当r6和r7均为-ch2ar,且ar为对位被r8取代的芳基,r8为卤素、羟基、碳原子数为1~6的烷氧基或下述所示基团,各基团中
包括如下步骤:
在硼氢化钠的催化下,式ⅲ所示化合物与archo经还原胺化反应即得式ⅳ所示化合物;
式ⅳ中,r1、r2、r3和r4各自独立地选自氢原子、卤素、硝基、碳原子数为1~6的烷基和碳原子数为1~6的烷氧基。
上述制备方法中各反应均可在常规的反应条件下进行。
本发明提供的化合物或其药学上可接受的盐可用于预防和/或治疗结肠癌或抑制结肠癌细胞转移,所述结肠癌细胞具体可为结肠癌细胞dld1。
本发明提供的化合物或其药学上可接受的盐还具有如下应用:
1)制备抑制结肠癌细胞生长的产品
2)制备抑制结肠癌细胞球生长的产品
3)制备抑制结肠癌细胞球形成的产品;
4)制备抑制结肠癌细胞迁移和运动能力的产品;
所述结肠癌细胞为结肠癌细胞dld1;
所述结肠癌细胞球为由结肠癌细胞dld1形成的细胞球。
活性成分为本发明提供的化合物或其药学上可接受的盐的药物也属于本发明的保护范围,该药物具有预防和/或治疗结肠癌或抑制结肠癌细胞转移的功能。
本发明通过搭建结肠癌临床病人来源的肿瘤细胞球3d培养模型,筛选并得到了结构新颖的本发明的化合物,其能特异抑制结肠癌肿瘤细胞球的形成并诱导细胞死亡的发生,但对贴壁生长的同种细胞完全没有作用(传统的化疗药物对于贴壁细胞具有显著的杀伤作用,但是对于3d的肿瘤细胞球却没有任何作用,但是,肿瘤细胞球与人体内的肿瘤更为相似,所以,这种筛选方式更为合理)。除了对结肠癌细胞系起作用,本发明化合物对结肠癌肿瘤病人来源的肿瘤细胞球也有显著的抑制效果。此外,本发明化合物还对结肠癌细胞系的迁移和运动能力具有明显的抑制作用。在本发明化合物的基础上,有望能够发展出治疗结肠癌的新型药物。
附图说明
图1为肿瘤细胞球在不同抑制剂下的形成情况。
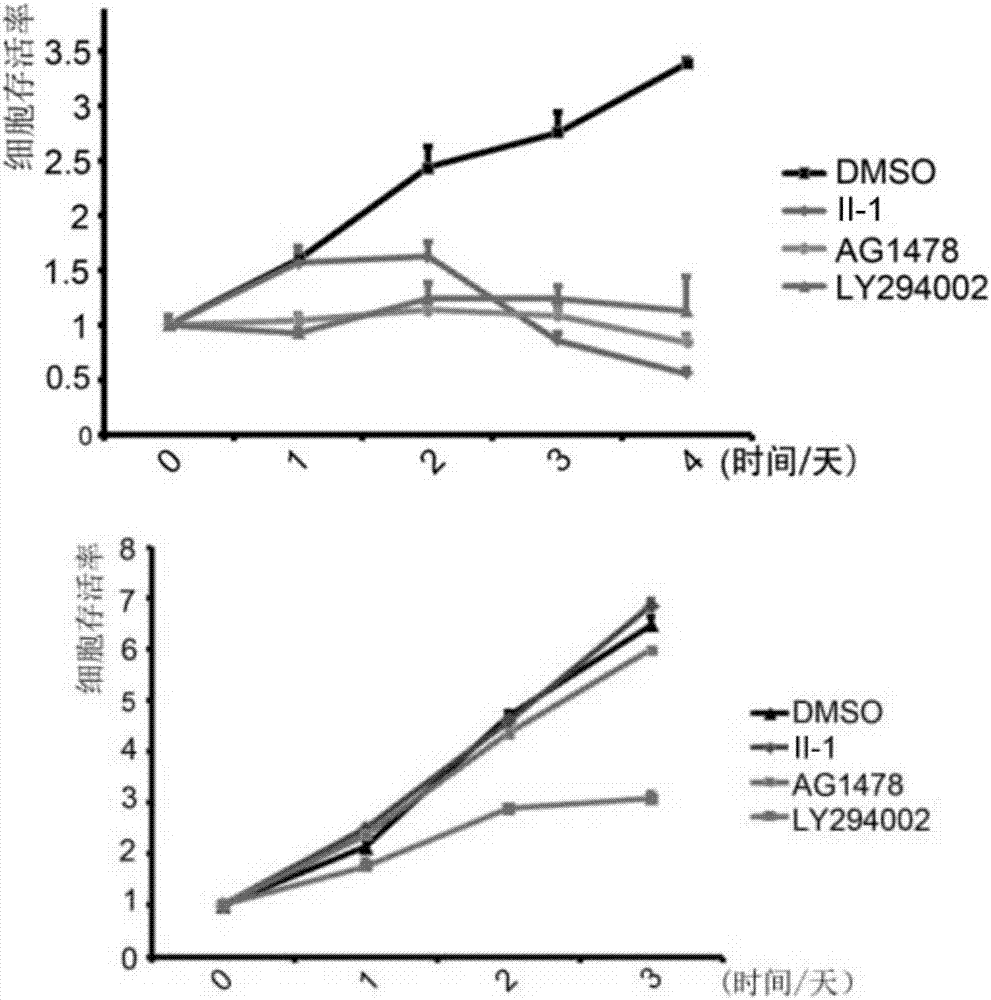
图2为在不同处理下肿瘤细胞球的生长曲线和贴壁细胞的生长曲线,其中上图为肿瘤细胞球的生长曲线,下图为贴壁细胞的生长曲线。
图3为不同浓度、不同时间本发明式ⅱ-1所示化合物处理后的肿瘤细胞球细胞状态,其中,上图为不同浓度下的状态图,下图为不同时间下的状态图。
图4为本发明式ⅱ-1所示化合物对dld1肿瘤细胞球的抑制作用随浓度的变化示意图。
图5为本发明式ⅱ-1所示化合物的transwell迁移实验结果。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、式ⅱ-1所示化合物的制备
反应方程式如下:
取1.97g2-氨基-二苯甲酮溶于20ml醋酸/水=1/1(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入1.04g亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将0.975g叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(20ml,三次),旋干溶剂后以石油醚-乙酸乙酯=70:1为洗脱剂进行硅胶柱层析纯化得到1.81g中间体a1,为油状液体,收率为81%。
取900mg中间体a1及560mg对硝基苯胺于100ml圆底烧瓶中,以氩气保护,加入15ml二氯甲烷使之溶解,小心加入329微升四氯化钛,加完后缓慢加入1.67毫升三乙胺并继续搅拌2小时至tlc检测中间体a1消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次15ml),旋干后用甲苯重结晶,得到590mg中间体a2,收率为42%。
取343mg中间体a2于50ml圆底烧瓶中,氩气保护,加入5ml无水四氢呋喃使之溶解,小心开盖加入400mg烘干除水的4a分子筛,搅拌5min后加入19mg碘化 亚铜,搅拌5min后缓慢加入129微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=20:1,得到293mg式ⅱ-1所示化合物,收率为93%。
结构征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.23(d,j=9.2hz,2h),7.78(d,j=8.8hz,1h),7.68-7.64(m,3h),7.48-7.45(m,3h),7.41-7.35(m,3h),7.15(t,j=8.4hz,2h).
经表征结构正确。
实施例2、式ⅱ-2所示化合物的制备
反应方程式如下:
取81mg2-氨基-4,4’-二氟二苯甲酮溶于5ml盐酸/水=1/4(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入48mg亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将45mg叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(10ml,三次),旋干溶剂后以石油醚-乙酸乙酯=25:1为洗脱剂进行硅胶柱层析纯化得到88g中间体a3,为油状液体,收率为98%。
取88mg中间体a3及47mg对硝基苯胺于25ml圆底烧瓶中,以氩气保,加入10ml二氯甲烷使之溶解,小心加入56微升四氯化钛,加完后缓慢加入189微升三乙胺并继续搅拌2至4小时至tlc检测中间体a3消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次15ml),旋干后用甲以空气泵进行干燥,得中间体a4.取中间体a4于10ml圆底烧瓶中,氩气保护,加入2ml无水四氢呋喃使之溶解,小心加入6.5mg碘化亚铜,搅拌5min后缓慢加入24微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=20:1,得到42mg式ⅱ-2所示化合物,收率为35%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.25(d,j=8.8hz,2h),7.62(d,j=8.4hz,1h),7.58(d,j=8.0hz,1h),7.35-7.32(m,3h),7.18(t,j=8.4hz,2h),6.97(t,j=8.8hz,1h).
经表征结构正确。
实施例3、式ⅱ-3所示化合物的制备
反应方程式如下:
取2.32g2-氨基-5-氯二苯甲酮溶于20ml醋酸/水=1/1(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入1.04g亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将0.975g叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(20ml,三次),旋干溶剂后以石油醚-乙酸乙酯=70:1为洗脱剂进行硅胶柱层析纯化得到2.44g中间体a5,为油状液体,收率为95%。
取228mg中间体a5及129mg对硝基苯胺于10ml圆底烧瓶中,以氩气保,加入3ml二氯甲烷使之溶解,小心加入63微升四氯化钛,加完后缓慢加入390微升三乙胺并继续搅拌2至4小时至tlc检测中间体a5消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后旋干,得到311mg粗产品中间体a6,收率为93%。
取311mg中间体a6于一25ml圆底烧瓶中,氩气保护,加入6ml无水四氢呋喃使之溶解,小心开盖加入400mg烘干除水的4a分子筛,搅拌5min后加入18mg碘化亚铜,搅拌5min后缓慢加入129微升三乙胺并继续在室温下搅拌5小时。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干,残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=20:1,得到220mg式ⅱ-3所示化合物,收率为77%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.24(d,j=9.2hz,2h),7.21(d,j=8.8hz,1h),7.64-7.62(m,3h),7,48-7.46(m,3h),7.34-7.29(m,3h).
经表征结构正确。
实施例4、式ⅱ-4所示化合物的制备
反应方程式如下:
取126mg2-氨基-4,4’-二甲氧基二苯甲酮溶于5ml盐酸/水=1/4(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入68mg亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将64mg叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(10ml,三次),旋干溶剂后以石油醚-乙酸乙酯=10:1为洗脱剂进行硅胶柱层析纯化得到117g中间体a7,为白色固体,收率为85%。
取117mg中间体a7及57mg对硝基苯胺于25ml圆底烧瓶中,以氩气保,加入10ml二氯甲烷使之溶解,小心加入70微升四氯化钛,加完后缓慢加入230微升三乙胺并继续搅拌2至4小时至tlc检测中间体a7消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次15ml),旋干后用甲以空气泵进行干燥,得中间体a8.取中间体a8于10ml圆底烧瓶中,氩气保护,加入2ml无水四氢呋喃使之溶解,小心加入8mg碘化亚铜,搅拌5min后缓慢加入29微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=10:1,得到53mg式ⅱ-4所示化合物,收率为34%。
结构表征数据如下:
h-nmr(400mhz,cdcl3,ppm):8.21(d,j=8.4hz,2h),7.61(d,j=8.4hz,2h),7.48(d,j=9.2hz,1h),7.26(d,j=8.4hz,2h),6.97(m,3h),6.81(d,j=9.2hz,1h),3.90(s,3h),3.86(s,3h).
经表征结构正确。
实施例5、式ⅱ-5所示化合物的制备
反应方程式如下:
取61mg2-氨基-4-硝基二苯甲酮溶于5ml盐酸/水=1/4(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入35mg亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将33mg叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(10ml,三次),旋干溶剂后以石油醚-乙酸乙酯=10:1为洗脱剂进行硅胶柱层析纯化得到54g中间体a19,为白色固体,收率为80%。
取54mg中间体a9及28mg对硝基苯胺于25ml圆底烧瓶中,以氩气保,加入10ml二氯甲烷使之溶解,小心加入22微升四氯化钛,加完后缓慢加入84微升三乙胺并继续搅拌2至4小时至tlc检测中间体a9消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次15ml),旋干后用甲以空气泵进行干燥,得中间体a10.取中间体a10于10ml圆底烧瓶中,氩气保护,加入2ml无水四氢呋喃使之溶解,小心加入4mg碘化亚铜,搅拌5min后缓慢加入14微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=10:1,得到21mg式ⅱ-5所示化合物,收率为29%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.71(s,1h),8.29(d,j=8.8hz,2h),8.19(dd,j=9.6hz,j=9.6hz,1h),7.84(d,j=9.6hz,1h),7.68(d,j=8.8hz,2h),7.54(m,3h),7.38(m,2h).
经表征结构正确。
实施例6、式ⅱ-6所示化合物的制备
反应方程式如下:
取160mg2-氨基-4,4’-二甲基二苯甲酮溶于6ml醋酸/水=1/1(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入98mg亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将92mg叠氮化钠加入到反应液中继续搅拌1h。反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(10ml,三次),旋干溶剂后以石油醚-乙酸乙酯=20:1为洗脱剂进行硅胶柱层析纯化得到152g中间体a11,为油状液体,收率为85%。
取152mg中间体a11及83mg对硝基苯胺于25ml圆底烧瓶中,以氩气保,加入 10ml二氯甲烷使之溶解,小心加入66微升四氯化钛,加完后缓慢加入252微升三乙胺并继续搅拌2至4小时至tlc检测中间体a11消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次15ml),旋干后用甲以空气泵进行干燥,得中间体a12.取中间体a12于10ml圆底烧瓶中,氩气保护,加入3ml无水四氢呋喃使之溶解,小心加入11.5mg碘化亚铜,搅拌5min后缓慢加入42微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=30:1,得到61mg式ⅱ-6所示化合物,收率为30%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.20(d,j=8.8hz,2h),7.61(d,j=8.8hz,2h),7.52(d,j=8.8hz,1h),7.49(s,1h),7.22(dd,j=11.2hz,j=8.0hz,4h),6.96(d,j=8.8hz,1h),2.47(s,3h),2.41(s,3h).
经表征结构正确。
实施例7、式ⅱ-7所示化合物的制备
反应方程式如下:
取532mg2-氨基-2’,5-二氯二苯甲酮溶于6ml醋酸/水=1/:1(v/v)的混合溶剂中,冰水冷却至0℃,缓慢加入207mg亚硝酸钠并维持反应温度在0℃继续搅拌1h,小心缓慢地将195mg叠氮化钠加入到反应液中继续搅拌1h.反应液以碳酸氢钠缓慢中和后以二氯甲烷萃取(10ml,三次),旋干溶剂后以石油醚-乙酸乙酯=30:1为洗脱剂进行硅胶柱层析纯化得到514g中间体a13,为油状液体,收率为98%。
取53mg中间体a13及25mg对硝基苯胺于10ml圆底烧瓶中,以氩气保,加入3ml二氯甲烷使之溶解,小心加入22微升四氯化钛,加完后缓慢加入84微升三乙胺并继续搅拌2至4小时至tlc检测中间体a13消失。小心加入饱和碳酸氢钠溶液处理剩余四氯化钛,后以硅藻土过滤,滤液以二氯甲烷萃取3次(每次10ml),旋干后用甲以空气泵进行干燥,得中间体a14。取中间体a14于10ml圆底烧瓶中,氩气保护,加入3ml无水四氢呋喃使之溶解,小心加入4mg碘化亚铜,搅拌5min后缓慢加入14微升三乙胺并继续在室温下搅拌过夜。反应液以水稀释,以二氯甲烷萃取3次,合并 有机相并以无水硫酸钠干燥后减压蒸干。残余物以硅胶柱层析分离,洗脱剂为石油醚-乙酸乙酯=30:1,得到20mg式ⅱ-7所示化合物,收率为26%。
表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):8.22(d,j=8.8hz,2h),7.76(d,j=9.2hz,1h),7.62(d,j=8.8hz,2h),7.53-7.48(m,2h),7.45(s,1h),7.42(d,j=3.2hz,1h),7.33(d,j=9.2hz,1h).
经表征结构正确。
实施例8、式ⅲ-1所示化合物的制备
反应方程式如下:
取175mg化合物3于25ml圆底烧瓶中,加入10ml丙酮使之溶解,称取420mg铁粉加入上述反应液中,另取1:1(v/v)的乙酸水溶液3ml加入上述反应液中,80℃回流一小时。冷却至室温,以饱和碳酸氢钠溶液调至ph中性,反应液用硅藻土过滤,滤液以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以石油醚-乙酸乙酯=2:1为洗脱剂进行硅胶柱层析,得到130mg式ⅲ-1所示化合物,收率为82%。
结构表征数据如下:
h-nmr(400mhz,cdcl3,ppm):7.74(d,j=8.8hz,1h),7.70(d,j=1.2hz,1h),7.43-7.35(m,5h),7.29(dd,j=8.8hz,j=1.6hz,1h),7.19(d,j=8.8hz,2h),6.62(d,j=8.8hz,2h),3.87(s,2h).
经表征结构正确。
实施例9、式ⅳ-1和式ⅳ-2所示化合物的制备
反应方程式如下:
取32mg式iii-1所示化合物于10ml圆底烧瓶中,加入13微升苄溴,17mg碘化钾,及14mg碳酸钾,向反应瓶中加入2ml乙腈,将反应瓶置于60摄氏度油浴锅中加 热搅拌24h。反应液减压蒸干后以石油醚:乙酸乙酯=5:1过硅胶柱,得到25mg式ⅳ-1所示化合物,收率为61%;得到15mg式ⅳ-2所示化合物,收率为30%。
式ⅳ-1的表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.71(d,j=9.2hz,1h),7.68(s,1h),7.43-7.35(m,9h),7.27(d,j=9.6hz,2h),7.20(d,j=8.4hz,2h),6.58(d,j=8.4hz,2h),4.33(s,2h),4.25(s,1h).
式ⅳ-2的表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.76-7.69(m,3h),7.56(m,1h),7.43-7.34(m,10h),7.31-7.21(m,10h),6.71(d,j=8.8hz,2h),4.69(s,4h).
经表征结构正确。
实施例10、式ⅳ-3所示化合物的制备
反应方程式如下:
取32mg式iii-1所示化合物与25mg对羟基苯甲醛于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另取2.4微升乙酸加入到上述溶液中,室温搅拌过夜。将反应液置于冰水浴中搅拌5min,称取57mg硼氢化钠缓慢加入到上述反应液中,继续搅拌1小时。以水稀释反应液并以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以石油醚-乙酸乙酯=3:1为洗脱剂进行硅胶柱层析,得到25mg式ⅳ-3所示化合物,收率为59%。
表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.71(d,j=8.8hz,1h),7.67(s,1h),7.43-7.35(m,6),7.19(d,j=8.4hz,4h),7.78(d,j=8.4hz,2h),6.57(d,j=8.0hz,2h),4.23(s,2h).
经表征结构正确。
实施例11、式ⅳ-4所示化合物的制备
反应方程式如下:
取378微升3-丁炔-1-醇于50ml圆底烧瓶中,加入20ml二氯甲烷使之溶解,另取774微升甲磺酰氯在冰水浴下加入到上述溶液中,缓慢滴加1.39ml三乙胺,继续搅拌1小时。反应液以乙酸乙酯稀释并用水洗涤3次,分取乙酸乙酯层并以无水硫酸钠干燥后旋干,得到540mg中间体a15,收率为73%。
取540mg中间体a15与445mg4-羟基苯甲醛于50ml圆底烧瓶中,称取1.0g碳酸钾加入反应瓶中,加入20ml乙腈,85℃搅拌3.5小时。冷却至室温,减压蒸干除去溶剂后加水稀释并以二氯甲烷萃取3次(每次15ml)。合并有机相并以无水硫酸钠1干燥后减压蒸干。残余物以石油醚-乙酸乙酯=5:1为洗脱剂进行硅胶柱层析,得到200mg中间体a16,收率为32%。
取32mg式iii-1所示化合物与35mg中间体a16于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另取2.4微升乙酸加入到上述溶液中,室温搅拌过夜。将反应液置于冰水浴中搅拌5min,称取100mg硼氢化钠缓慢加入到上述反应液中,继续搅拌1小时。以水稀释反应液并以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以石油醚-乙酸乙酯=3:1为洗脱剂进行硅胶柱层析,得到30mg式ⅳ-4所示化合物,收率为63%。
表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.70(d,j=8.8hz,1h),7.67(m,1h),7.42-7.33(m,7h),7.28-7.25(m,2h),7.17(d,j=6.8hz,2h),6.55(d,j=6.8hz,2h),4.24(s,2h),4.15(s,1h),4.08(t,j=7.0hz,2h),2.68(t,j=7.0hz,2h),2.04(s,1h).
经表征结构正确。
实施例12、式ⅳ-5所示化合物的制备
反应方程式如下:
取32mg式iii-1所示化合物与30mg对溴苯甲醛于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另取1.8微升乙酸加入到上述溶液中,室温搅拌过夜。将反应液置于冰水浴中搅拌5min,称取57mg硼氢化钠缓慢加入到上述反应液中,继续搅拌1小时。以水稀释反应液并以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以石油醚-乙酸乙酯=5:1为洗脱剂进行硅胶柱层析,得到30mg式ⅳ-5所示化合物,收率为61%。
表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.69(d,j=9.28hz,1h),7.66(d,j=1.44hz,1h),7.44(d,j=8.36hz,2h),7.39-7.32(m,5h),7.27-7.24(m,1h),7.20-7.15(m,4h),6.53(d,j=11.76hz,2h),4.25(s,2h),
经表征结构正确。
实施例13、式ⅳ-6所示化合物的制备
反应方程式如下:
取1.22g4-羟基苯甲醛与1.87g2-溴乙醇于50ml圆底烧瓶中,加入15mln,n-二甲基甲酰胺,称取2.76g碳酸钾加入到上述反应液中,80℃搅拌2小时。冷却至室温,以乙酸乙酯稀释并用水洗涤4次,分取有机相并以无水硫酸钠干燥后减压蒸干。残余物依次以石油醚-乙酸乙酯=4:1,石油醚-乙酸乙酯=1:1为洗脱剂进行硅胶柱层析,得到1.35g中间体a17,为黄色油状液体,收率为81%。
取64mg式iii-1所示化合物与50mg中间体a17于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另加入3.6微升乙酸,反应液在室温下搅拌24小时。将反应瓶置于冰水浴中搅拌5min,缓慢加入189mg硼氢化钠,继续搅拌2小时。反应液用水稀释, 以二氯甲烷萃取3次(每次15ml),合并有机相并以无水硫酸钠干燥后减压蒸干。剩余物依次用石油醚-乙酸乙酯=2:1,二氯甲烷-丙酮=10:1进行硅胶柱层析,得到50mg式ⅳ-6所示化合物,为一白色固体,收率为53%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.65(m,2h),7.43-7.40(m,3h),7.38-7.30(m,3h),7.27(d,j=8.4hz,2h),7.08(d,j=8.8hz,2h),6.90(d,j=8.8hz,2h),6.61(d,j=8.8hz,2h),4.25(s,2h),4.02(t,j=4.6hz,2h),3.85(t,j=4.6hz,2h).
经表征结构正确。
实施例14、式ⅳ-7所示化合物的制备
反应方程式如下:
取366mg4-羟基苯甲醛与540mg4-(3-氯丙基)-吗啉于50ml圆底烧瓶中,称取1.24g碳酸钾加入到上述混合物中,另加入15mln,n-二甲基甲酰胺,于85℃搅拌2小时。冷却至室温,加乙酸乙酯稀释并以水洗涤四次,分取有机相以无水硫酸钠干燥后减压蒸干。残余物以二氯甲烷-甲醇=10:1为洗脱剂进行硅胶柱层析,得到400mg中间体a18,为一淡黄色油状液体,收率为54%。
取65mg式iii-1所示化合物与75mg中间体a18于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另加入3.6微升乙酸,反应液在室温下搅拌4小时。将反应瓶置于冰水浴中搅拌5min,缓慢加入189mg硼氢化钠,继续搅拌2小时。反应液用水稀释,以二氯甲烷萃取3次(每次15ml),合并有机相并以无水硫酸钠干燥后减压蒸干。剩余物依次用石油醚-乙酸乙酯=3:1,纯乙酸乙酯,二氯甲烷-甲醇=10:1进行硅胶柱层析,得到53mg式ⅳ-7所示化合物,为一白色固体,收率为47%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.71(d,j=9.2hz,1h),7.67(s,1h),7.66-7.34(m,5h),7.28-7.24(m,3h),7.19(d,j=8.8hz,2h),6.87(d,j=8.4hz,2h),6.58(d,j=8.8hz,2h),4.25(s,2h),4.02(t,j=6.3hz,2h),3.72(t,j=4.6hz,4h),2.54-2.47(m,6h),1.97(m,2h).
经表征结构正确。
实施例14、式ⅳ-8所示化合物的制备
反应方程式如下:
取870微升吗啉于50ml圆底烧瓶中,加入20ml二氯甲烷使之溶解,冰水浴下慢慢滴加820微升氯乙酰氯,另慢慢滴加1.4ml三乙胺,继续搅拌1小时。反应液以水稀释,并以二氯甲烷萃取3次(每次15ml)。合并有机相并以无水硫酸钠干燥后减压蒸干,得到827mg中间体a19,收率为50%。
取244mg4-羟基苯甲醛及344mg中间体a19于50ml圆底烧瓶中,加入10mln,n-二甲基甲酰胺,另加入828mg碳酸钾,将此反应液于80℃油浴锅中搅拌1小时,冷却至室温。将反应液以乙酸乙酯稀释,用水洗涤4次,分取乙酸乙酯层以无水硫酸钠干燥,减压蒸干。剩余物用二氯甲烷-甲醇=10:1为洗脱剂进行硅胶柱层析,得到433mg中间体.a20,收率为85%。
取32mg式iii-1所示化合物与50mg中间体a20于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另加入1.2微升乙酸,反应在室温下搅拌6小时。将反应瓶置于冰水浴中,缓慢加入100mg硼氢化钠,继续搅拌2小时。反应液用水稀释并以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残余物依次用石油醚-乙酸乙酯=1:1,纯乙酸乙酯,二氯甲烷-甲醇=10:1为洗脱剂进行硅胶柱层析,得到56mg式ⅳ-8所示化合物,收率为87%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.70(d,j=9.2hz,1h),7.69(s,1h),7.66-7.34(m,5h),7.28-7.25(m,3h),7.19(d,j=8.8hz,2h),6.91(d,j=8.8hz,2h),6.57(d,j=8.8hz,2h),4.67(s,2h),4.26(s,2h),4.20(s,1h),3.66-3.59(m,8h).
经表征结构正确。
实施例15、式ⅳ-9所示化合物的制备
反应方程式如下:
取1.04g化合物1于100ml圆底烧瓶中,加入30ml丙酮使之溶解,称取1.85g铁粉加入上述反应液中,另取1:1(v/v)的乙酸水溶液6ml加入上述反应液中,80℃回流一小时。冷却至室温,以饱和碳酸氢钠溶液调至ph中性,反应液用硅藻土过滤,滤液以二氯甲烷萃取3次,合并有机相并以无水硫酸钠干燥后减压蒸干。残余物以石油醚-乙酸乙酯=2:1为洗脱剂进行硅胶柱层析,得到778mg中间体a21,收率为83%。
取50mg中间体a21与87mg中间体a18于10ml圆底烧瓶中,加入2ml甲醇使之溶解,另加入3.6微升乙酸,反应液在室温下搅拌5小时。将反应瓶置于冰水浴中搅拌5min,缓慢加入189mg硼氢化钠,继续搅拌2小时。反应液用水稀释,以二氯甲烷萃取3次(每次15ml),合并有机相并以无水硫酸钠干燥后减压蒸干。剩余物依次用石油醚-丙酮=10:1,石油醚-丙酮=5:1为洗脱剂进行硅胶柱层析,得到55mg式ⅳ-9所示化合物,为一白色固体,收率为61%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.79(d,j=8.8hz,1h),7.72(d,j=8.4hz,1h),7.41-7.33(m,6h),7.28-7.21(m,4h),7.15-7.11(m,1h),6.88(d,j=8.4hz,2h),6.58(d,j=8.8hz,2h),4.26(d,j=5.2hz,2h),4.16-4.13(m,1h),4.03(d,j=6.4hz,2h),3.74-3.72(m,4h),2.53(t,j=6.8hz,2h),2.48(m,4h),2.01-1.95(m,2h)
经表征结构正确。
实施例19、式ⅳ-10所示化合物的制备
反应方程式如下:
取哌嗪乙醇3.0g于100ml圆底烧瓶中,另取5.5g二叔丁基二碳酸酯溶于15ml四氢呋喃中,将此溶液慢慢滴加于圆底烧瓶中,室温搅拌4小时,直至tlc检测哌嗪乙醇消耗完。旋蒸除去大部分溶剂,剩余物用水稀释,并以二氯甲烷萃取三次(每次50ml)。合并有机相并以无水硫酸钠干燥后旋干得到油状液体。将此油状液体溶于300ml二氯甲烷中,室温下慢慢加入3.34ml二氯亚砜,室温搅拌过夜。反应液加水稀释并以碳酸氢钠固体调至ph中性,分取有机相,以无水硫酸钠干燥后旋干得到3.16g中间体a22。两步收率55%。
取4-羟基苯甲醛1.86g及3.72g中间体a22于50ml圆底烧瓶中,加入15mln,n-二甲基甲酰胺,另加入4.2g碳酸钾,将此反应液于85℃油浴锅中搅拌3小时,冷却至室温。将反应液以乙酸乙酯稀释,用水洗涤4次,分取乙酸乙酯层以无水硫酸钠干燥,减压蒸干。剩余物依次用石油醚-乙酸乙酯=4:1,石油醚-乙酸乙酯=1:1,石油醚-乙酸乙酯=1:1并加入1%体积三乙胺为洗脱剂进行硅胶柱层析,得到3.35g中间体a23,收率为66%。
取57mg中间体a21及65mg中间体a23于10ml圆底烧瓶中,加入3ml甲醇使之溶解,另加入3.6微升乙酸,反应在室温下搅拌过夜。将反应瓶置于冰水浴中,缓慢加入189mg硼氢化钠,继续搅拌2小时。反应液用水稀释并以二氯甲烷萃取3次(每次10ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残余物依次用石油醚-乙酸乙酯=2:1,1:1,纯乙酸乙酯为洗脱剂进行硅胶柱层析,得到85mg中间体a24,收率为70%。
取85mg中间体a24于10ml圆底烧瓶中,加入2ml二氯甲烷使之溶解,室温下加入600微升三氟乙酸,室温搅拌过夜。反应液以饱和碳酸氢钠溶液调至ph中性,以二氯甲烷萃取3次(每次15ml),合并有机相并以无水硫酸钠干燥后减压蒸干。残 余物依次用石油醚-乙酸乙酯=1:1,纯乙酸乙酯,二氯甲烷-甲醇=10:1,二氯甲烷-甲醇=5:1,二氯甲烷-甲醇=10:1另加2%体积三乙胺为洗脱剂进行柱层析,得到50mg式ⅳ-10所示化合物,收率为71%。
结构表征数据如下:
1h-nmr(400mhz,cdcl3,ppm):7.79(d,j=8.8hz,1h),7.70(d,j=8.8hz,1h),7.39-7.33(m,6h),7.26(s,2h),7.20(d,j=8.8hz,2h),7.12(t,j=7.6hz,1h),6.87(d,j=7.6hz,2h),6.58(d,j=8.8hz,2h),4.26(d,j=8.8hz,2h),4.15(s,1h),4.10(d,j=5.2hz,2h),3.10(s,4h),2.85(t,j=5.2hz,2h),2.78(s,4h).
经表征结构正确。
实施例20、本发明化合物对肿瘤细胞球的抑制作用
一、实验原理
将贴壁培养的结肠癌细胞接种于超低吸附培养板进行悬浮培养,能够形成肿瘤细胞球。因此可以通过观察肿瘤细胞球的形成能力和细胞增殖能力来评价本发明化合物的抑制效率。
二、实验材料的制备
(一)肿瘤细胞球的培养
将各种结肠癌细胞接种于超低吸附培养板以使其悬浮生长,单细胞悬液接种培养后,显微镜下观察,这些结肠癌细胞依然具备较强的运动能力,相邻的细胞迅速粘附在一起,形成团状。结肠癌细胞系dld1和hct116都能形成较为致密的肿瘤细胞球。
具体步骤如下:
1.配制相应的无血清培养基dmem/f12,并加入对应的添加剂n2、b27以及10ng/mlbfgf和20ng/mlegf。
2.利用胰酶将贴壁培养的结肠癌肿瘤细胞系dld1消化为单细胞。
3.将消化为单细胞的dld1细胞接种在超低吸附的96孔板中,进行悬浮培养,每孔接种200个细胞,培养5天后即有明显的肿瘤细胞球形成。
(二)其它实验材料
dmem/f12:购自gibco,产品目录号为10565-018。
n2:购自gibco,产品目录号为17502048。
b27:购自gibco,产品目录号为12587010。
egf:购自gibco,产品目录号为phg0314。
bfgf:购自gibco,产品目录号为phg0264。
三、不同化合物对肿瘤细胞球形成能力的抑制作用
1、显微镜观察肿瘤细胞球的大小和数目
筛选系统:dld1细胞形成的肿瘤细胞球。
筛选方法:利用96孔板的筛选体系,检测本发明化合物对dld1肿瘤细胞球形成数量和大小的影响。
评价标准:接种细胞时(每孔200个细胞),每个孔中分别加入一种浓度为10μm的本发明化合物(dmso溶解)处理5天,比较本发明化合物处理和dmso对照形成的肿瘤细胞球的差异,利用显微镜观察肿瘤细胞球的大小和数目。
结果如图1所示,在众多不同新型化合物中,式ⅱ-1所示化合物抑制肿瘤细胞球形成的效果最佳,能够明显抑制肿瘤细胞球形成的数量,在式ⅱ-1所示化合物处理的第5天,基本无明显的完整肿瘤细胞球的形成。egfr抑制剂ag1478和pi3k-akt抑制剂ly294002作为对照,可以看出,二者并不能明显抑制肿瘤细胞球的形成。
2、检测不同化合物诱导肿瘤细胞球死亡和贴壁肿瘤细胞死亡的能力
检测细胞生存能力的实验步骤:
1)96孔板每孔接种1000个细胞(不同细胞系的具体接种数目应视细胞生长速度做预实验而定),每组样品设置3个重复。同时,加入dmso及各种小分子处理。
2)从第二天开始,每天收取对照组和实验组的数据:每孔加入20μlmts溶液,在细胞培养箱中避光培养4个小时。设置3个只加培养基和mts溶液的空白对照。剩余继续培养的细胞每天补充新鲜的培养基。
3)4个小时后,将已经完成反应的细胞液全部转移至新的96孔板中,用酶标仪检。
4)利用商业化的egfr抑制剂和pi3k-akt抑制剂作为阳性对照,同时,对比化合物与这两个抑制剂的效果。
检测细胞死亡的实验步骤:
钙黄绿素-am是一种可以穿透活细胞膜并对活细胞进行荧光标记的细胞染色剂。在活细胞内的酯酶作用下,钙黄绿素-am会被水解产生钙黄绿素而发出绿色荧光。碘化丙啶仅能穿过死细胞膜到达细胞核内,并嵌入dna双螺旋结构中而发出红色荧光。因此,可以同时使用钙黄绿素-am和碘化丙啶双重染色来区别活细胞和死细胞。
具体实验步骤如下:
1)准备细胞样品:培养细胞,根据实验需求用不同小分子处理。
2)染色:离心收集悬浮生长的肿瘤细胞球细胞,pbs清洗细胞一次,加入用pbs稀释的钙黄绿素-am和碘化丙啶,37℃孵育15-20分钟。其中,钙黄绿素-am和碘化丙啶的浓度根据细胞类型不同而变化,本实验中肿瘤细胞球使用的两者浓度分别为0.2μg/ml和0.75μg/ml。
3)观察:钙黄绿素发出的绿色荧光可以由488nm激发光检测,碘化丙啶发出的 红色荧光可以由535nm激发光检测,拍照记录。
结果如图2和图3所示,式ⅱ-1所示化合物诱导肿瘤细胞球死亡的能力较强。但是式ⅱ-1所示化合物基本不影响贴壁肿瘤细胞的增殖能力。另外,试验结果表明,式ⅳ-7所示化合物和式ⅳ-10所示化合物诱导肿瘤细胞球死亡的能力最强,并且发挥作用所需的时间最短,仅需要24小时。
3、测定本发明化合物杀死肿瘤细胞球的ec50
测定方法:使用不同浓度的化合物处理dld1形成的肿瘤细胞球,检测相应浓度下肿瘤细胞球的存活能力。
测定公式:根据不同浓度下测出的细胞存活能力,根据四参数logistic模型计算ic50的数值:
结果如图4所示,可以看出,式ⅱ-1所示化合物抑制dld1肿瘤细胞球细胞增殖的ec50浓度为1.353μm,另外,效果更好的式ⅳ-4所示化合物的ec50浓度为0.76μm,式ⅳ-7所示化合物的ec50浓度为0.58μm,式ⅳ-10所示化合物的ec50浓度为0.53μm。
4、检测本发明化合物对肿瘤细胞dld1的迁移能力的影响
检测方法:transwell小室实验能够检测细胞迁移能力,对于研究肿瘤细胞迁移能力,通常在上室加肿瘤细胞,下室加血清或者其他趋化因子以吸引细胞向下迁移,最终计数转移至下室的细胞数目从而反应肿瘤细胞的迁移能力。具体实验步骤如下:
1)接种细胞:消化细胞,用无血清培养基或pbs重悬细胞,细胞计数板计数。24孔板中每孔上室加入100l无血清培养基并接种2×104个dld1细胞,下室加入500l含2%血清的培养基。同时加入本发明式ⅱ-1、式ⅳ-4、式ⅳ-7和式ⅳ-10所示化合物处理。每组样品设置3个重复。
2)固定:培养12小时后,吸弃上下培养基,4%多聚甲醛室温固定30分钟。
3)染色:结晶紫染液室温染细胞20分钟。
4)清洗并拍照:pbs清洗3次,用棉签轻轻擦去上室内膜的细胞。显微镜下明场拍照记录,每个孔至少拍6张照片,计数并计算迁移细胞平均值。
结果如图5所示,可以看出,式ⅱ-1所示化合物可以明显在体外实验中抑制肿瘤细胞dld1的迁移能力,并且试验结果表明式ⅳ-4所示化合物、式ⅳ-7所示化合物和式ⅳ-10所示化合物同样具有抑制肿瘤细胞dld1迁移的能力。
- 还没有人留言评论。精彩留言会获得点赞!