一种舌鳞癌的新诊治靶标基因及其应用的制作方法
本发明涉及基因诊断及治疗领域,具体涉及一种舌鳞癌的新诊治靶标基因及其应用,更具体的涉及KLK14基因及其表达产物在诊治舌鳞癌中应用。
背景技术:
舌鳞癌(Tongue squamous cell carcinoma,TSCC)占口腔鳞癌发病首位,舌鳞癌的发生是个多因素、多步骤、多阶段的复杂过程,病因学也复杂多样,涉及理化因素、微生物感染、遗传、种族等方面。虽然有关舌鳞癌病因学及相关机制一直是研究的热点,也取得一定成果,但舌鳞癌仍然具有较高的死亡率及预后差。而预后较差的原因往往是诊断和治疗的不及时,由于舌鳞癌早期一般无任何症状,患者往往因疼痛、难以愈合溃疡、不明原因的出血、口腔或者颈部肿块等临床症状就诊,这时病情较晚,生存率较低。有报道认为早期病例及时治疗,5年生存率可达到85%,而一旦出现症状,五年生存率在50%左右。另外病灶面积大,手术切除范围广,也将严重影响患者术后生活质量。研究舌鳞癌生物学行为、发生和发展机制,有助于疾病早诊断、预防和治疗;寻找有效的分子靶基因,降低舌癌的死亡率,是迫切需要解决的问题;同时具有广阔的临床医用前景及重大的科学价值。
细胞中原癌基因激活或/和抑癌基因的失活是导致肿瘤发生的重要机制,在舌鳞状细胞癌的发生过程中同样具有重要地位,原癌基因激活或/和抑癌基因失活可以导致肿瘤细胞异常增殖、抑制凋亡,从而促进肿瘤的发生。随着人类基因组学的解密,高通量分子筛选技术如基因组学和蛋白质组学技术都已被应用到TSCC发生、发展的机制研究中。
本发明首先拟通过RNA-seq测序检测舌鳞癌、癌旁及正常口腔黏膜差异表达基因;其次通过Real-time PCR技术对测序结果进行验证,再次利用干扰技术,沉默其在舌鳞癌细胞SCC15中的表达。为KLK14基因在舌鳞癌的临床运用提供实验基础,为舌鳞癌的早期诊断和治疗提供新的靶基因和理论依据。
技术实现要素:
本发明的目的在于提供KLK14基因和/或其表达的蛋白在制备抗舌鳞癌制剂和/或舌鳞癌诊断制剂中的应用。
本发明的目的在于提供检测KLK14基因和/或其表达蛋白的制剂在制备舌鳞癌诊断制剂中的应用。
进一步,所述的舌鳞癌的诊断制剂包括用荧光定量PCR方法、基因芯片方法检测外周血中KLK14基因和/或KLK14蛋白的表达。
荧光定量PCR法是通过荧光染料或荧光标记的特异性的探针,对PCR产物进行标记跟踪,实时在线监控反应过程,结合相应的软件可以对产物进行分析,计算待测样品模板的初始浓度。荧光定量PCR的出现,极大地简化了定量检测的过程,而且真正实现了绝对定量。多种检测系统的出现,使实验的选择性更强。自动化操作提高了工作效率,反应快速、重复性好、灵敏度高、特异性强、结果清晰。
基因芯片又称为DNA微阵列(DNA microarray),可分为三种主要类型:1)固定在聚合物基片(尼龙膜,硝酸纤维膜等)表面上的核酸探针或cDNA片段,通常用同位素标记的靶基因与其杂交,通过放射显影技术进行检测。2)用点样法固定在玻璃板上的DNA探针阵列,通过与荧光标记的靶基因杂交进行检测。3)在玻璃等硬质表面上直接合成的寡核苷酸探针阵列,与荧光标记的靶基因杂交进行检测。基因芯片作为一种先进的、大规模、高通量检测技术,应用于疾病的诊断,其优点有以下几个方面:一是高度的灵敏性和准确性;二是快速简便;三是可同时检测多种疾病。
所述的用于荧光定量PCR方法检测舌鳞癌中KLK14基因的产品含有一对特异性扩增KLK14基因的引物;所述的基因芯片包括与KLK14基因的核酸序列杂交的探针。
进一步,所述的舌鳞癌的诊断制剂包括用免疫方法检测KLK14蛋白的表达。优选所述免疫检测方法检测舌鳞癌中KLK14蛋白表达的为western blot和/或ELISA和/胶体金检测方法。
酶联免疫吸附实验(ELISA)即将已知的抗原或抗体吸附在固相载体表面,使酶标记的抗原抗体反应在固相表面进行的技术。该技术可用于检测大分子抗原和特异性抗体等,具有快速、灵敏、简便、载体易于标准化等优点。ELISA检测试剂盒根据检测目的和操作步骤可分为间接法、双抗夹心法、竞争法、双位点一步法、捕获法测IgM抗体、应用亲和素和生物素的ELISA。ELISA检测试剂盒中生色底物可选择辣根过氧化物酶(HRP)或者碱性磷酸酶(AP)。
常用的免疫胶体金检测技术:(1)免疫胶体金光镜染色法细胞悬液涂片或组织切片,可用胶体金标记的抗体进行染色,也可在胶体金标记的基础上,以银显影液增强标记,使被还原的银原子沉积于已标记的金颗粒表面,可明显增强胶体金标记的敏感性。(2)免疫胶体金电镜染色法可用胶体金标记的抗体或抗抗体与负染病毒样本或组织超薄切片结合,然后进行负染。可用于病毒形态的观察和病毒检测。(3)斑点免疫金渗滤法应用微孔滤膜作载体,先将抗原或抗体点于膜上,封闭后加待检样本,洗涤后用胶体金标记的抗体检测相应的抗原或抗体。(4)胶体金免疫层析法将特异性的抗原或抗体以条带状固定在膜上,胶体金标记试剂(抗体或单克隆抗体)吸附在结合垫上,当待检样本加到试纸条一端的样本垫上后,通过毛细作用向前移动,溶解结合垫上的胶体金标记试剂后相互反应,当移动至固定的抗原或抗体的区域时,待检物与金标试剂的结合物又与之发生特异性结合而被截留,聚集在检测带上,可通过肉眼观察到显色结果。该法现已发展成为诊断试纸条,使用十分方便。
进一步,所述检测KLK14蛋白的ELISA法为使用ELISA检测试剂盒。所述试剂盒中的抗体可采用市售的KLK14单克隆抗体,优选abcam公司的Anti-Kallikrein 14抗体[EPR7209](ab128957)。进一步,所述的试剂盒包括:包被KLK14单克隆抗体的固相载体,酶标二抗,酶的底物,蛋白标准品,阴性对照品,稀释液,洗涤液,酶反应终止液等。
进一步,所述检测KLK14蛋白的胶体金法为使用检测试剂盒,所述的抗体可采用市售的KLK14单克隆抗体,优选abcam公司的Anti-Kallikrein 14抗体[EPR7209](ab128957)。进一步,所述的胶体金检测试剂盒采用胶体金免疫层析技术或胶体金渗滤法。进一步,所述的胶体金检测试剂盒硝酸纤维素膜上的检测区(T)喷点有抗KLK14单克隆抗体、质控区(C)喷点有免疫球蛋白IgG。
本发明的目的在于提供一种检测舌鳞癌的荧光定量PCR试剂盒,其特征在于,所述试剂盒检测基因KLK14,采用特异的上游引物和下游引物,上游引物序列为SEQ ID NO.1,下游引物序列为SEQ ID NO.2。
进一步,该PCR试剂盒适合于目前存在市场上的所有类型荧光定量基因扩增仪,灵敏度高,定量快速准确、稳定性好,具有良好的应用前景。
进一步,上述荧光定量PCR试剂盒组分包括:特异性引物、内参引物、荧光定量PCR反应液。其中所述的特异性引物包括上游引物和下游引物,上游引物序列为SEQ ID NO.1,下游引物序列为SEQ ID NO.2。所述内参引物为GAPDH内参引物,上游引物序列为SEQ ID NO.3,下游引物序列为SEQ ID NO.4。
所述的试剂盒还包含RNA抽提试剂。优选Reagent进行样本RNA提取。
本发明目的是提供了一种舌鳞癌检测试剂盒,所述的检测试剂盒检测KLK14蛋白。进一步的,所述的试剂盒还包括其他检测试剂。
本发明目的是提供了一种检测舌鳞癌的基因芯片,所述的基因芯片包括与KLK14基因的核酸序列杂交的探针。
本发明的目的在于提供KLK14基因和/或其蛋白抑制剂在制备抗舌鳞癌制剂中的应用。
为实现上述目的,本发明首先通过高通量测序结合生物信息学方法筛选到候选基因KLK14,继而通过分子生物学方法验证了KLK14与舌鳞癌的关系:KLK14与舌鳞癌具有很好的相关性,可用于制备抗舌鳞癌制剂和/或舌鳞癌诊断制剂,具有重要的临床应用价值。
进一步,所述抗舌鳞癌制剂是指可以抑制KLK14基因的表达的制剂。本领域人员熟知抑制基因的表达通常可以采用下述方法中的一种和/或几种:通过激活KLK14基因的抑制基因、激活KLK14基因的抑制基因表达的蛋白、采用RNA干扰技术抑制KLK14基因表达、激活促进KLK14基因mRNA降解的microRNA、导入促进KLK14基因编码蛋白降解的分子、抑制促进KLK14基因表达的因子及蛋白的表达。即通过激活KLK14基因的抑制基因、激活抑制KLK14基因表达的蛋白、导入抑制KLK14基因表达的siRNA、导入抑制KLK14基因表达的shRNA、激活促进KLK14mRNA降解的microRNA、导入促进KLK14蛋白降解的分子、抑制促进KLK14基因表达的因子及蛋白的表达。
RNA干扰(RNAi)是指外源性和内源性双链RNA在生物体内诱导同源靶基因的mRNA特异性降解,导致转录后基因沉默的现象,是一种使用小的双链RNA高效、特异地阻断体内某种特定基因的表达,促使mRNA降解,使细胞表现出特定基因缺失表型的技术。siRNA设计完成后可以采用直接合成法或者构建siRNA表达载体,制备好的siRNA可以通过磷酸钙共沉淀法、电穿孔法、DEAE-葡聚糖和polybrene法、显微注射或基因枪等机械法、阳离子脂质体试剂法等途径转染细胞。
进一步,采用shRNA慢病毒RNAi载体抑制KLK14基因或蛋白的表达。
进一步,KLK14基因和/或KLK14蛋白抑制剂在制备抗舌鳞癌细胞增殖制剂中的应用。
本发明的目的在于提供一种抗舌鳞癌制剂,所述抗舌鳞癌制剂抑制KLK14基因的表达。进一步,所述的抗舌鳞癌制剂中含有抑制KLK14基因表达的siRNA或shRNA。
附图说明
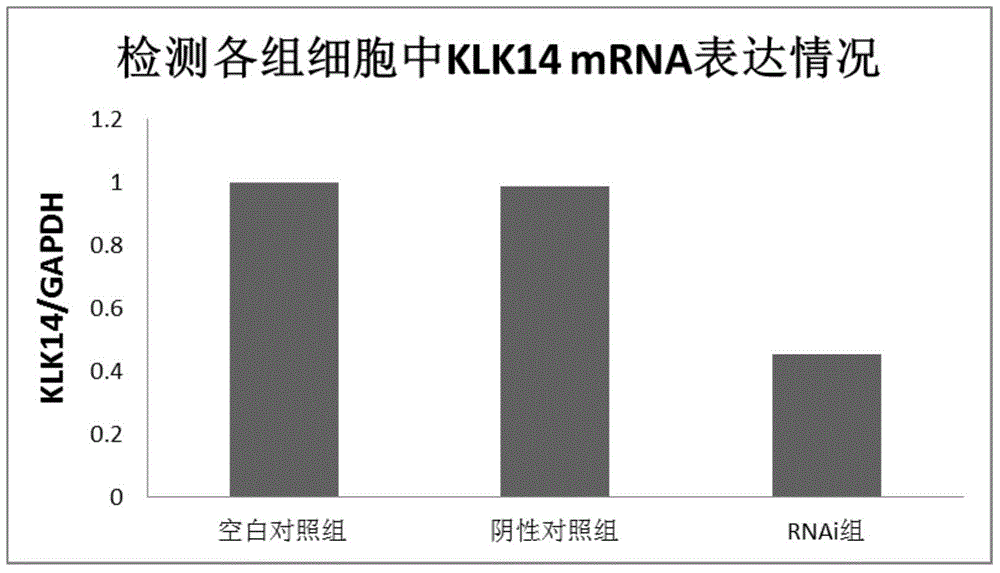
图1是RNA干扰后各组KLK14mRNA相对表达水平图
具体实施方式
下面结合具体实施例,进一步阐述本发明,仅用于解释本发明,而不能理解为对本发明的限制。本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照厂商所建议的条件实施检测。
实施例1材料的采集及准备
舌鳞癌及癌旁组织标本均取自舌鳞癌根治术患者,正常粘膜组织取自于外伤患者,所有组织取材经过病理组织学确诊,并签署自愿捐献供本次试验使用同意书。外科手术过程中标本离体30分钟内,随后在病理医生指导下采集标本。每例样本大小约为1.0cm×l.0cm×l.0cm大小,取材时癌组织为肿瘤的中心区;相应癌旁为离癌组织边缘l-2cm处组织;正常粘膜为不含黏膜下的上皮组织,标本来源于口腔外伤手术标本。所有标本分别置入1.5ml EP管放入液氮罐保存。收集所有标本患者术前未经过任何形式的抗肿瘤治疗及无其他部位肿瘤病史。根据WHO世界卫生组织1998年病理学分级:高分化鳞癌10例、中分化鳞癌(含高-中分化)8例、低分化鳞癌(含中-低分化)2例。根据国际抗癌协会(UICC)TNM(2002)分期进行临床分期,临床病理特征详见表1。
表1 20例口腔鳞癌患者临床病理资料
对上述材料进行全转录组RNA提取、纯化、RNA片段化、cDNA第一链合成、cDNA第二条链合成、末端修饰、加poly A、接头连接、选择适当长度的DNA进行扩增、纯化扩增产物、评估产量等处理,产物合格后,按照Illumina公司的HiSeq 2500高通量测序平台标准流程上机测序。
实施例2数据库及生物学分析方法
RNA-seq读段定位
首先将低质量的读段去除得到清洁读段然后利用TopHat v1.3.11将清洁片段与UCSC H.sapiens参考基因组(hg19)进行匹配,人类参考基因组序列(hgl9,2009年3月20日更新)从UCSC数据库中下载获得。Hgl9基因组中包括25个参考序列(reference assemblies)和68个可变序列(alternative assemblies)。H.sapiens UCSC hg19版的预先构建的索引从TopHat主页下载,并作为参考基因组,利用TopHat与基因组匹配时,允许每个读段(默认到20)有多个匹配位点,最多2次错配。TopHat根据外显子区域和GT-AG剪切信号建立可能的剪切位点库,根据这些剪切位点库将没有定位到基因组的读段定位到基因组上。我们使用的TopHat方法的系统默认参数。
转录丰度评估
匹配上的读段文件通过Cufflinks v1.0.32处理,Cufflinks v1.0.3将RNA-seq片段数目进行标准化计算转录本的相对丰度。FPKM值指的是每一百万测序片段中匹配到特定基因1kb长的外显子区域的片段数目。通过贝叶斯推理方法3计算FPKM估计值的置信区间。Cufflinks使用的参考的GTF注释文件从Ensembl数据库下载(Homo_sapiens.GRCh37.63.gtf)。
差异表达基因的检测
将下载的Ensembl GTF文件和通过TopHat匹配的原始文件传输到Cuffdiff,Cuffdiff使用原始的匹配文件重新估算GTF文件中列出的转录本的表达丰度,检测差异表达。在Cuffidff输出中只有q值<0.01,测试显示成功的比较才被认为是差异表达。
差异表达基因的分析
为了计算基因的表达从而鉴定出不同样品间的差异表达基因,我们利用Cuffdiff方法估计基因的表达,从而鉴定表达失调的基因。每个基因的标准化的表达水平按照每一百万测序片段中匹配到特定基因1kb长的外显子区域的片段数目(FPKM)计算。FPKM是tophat和cuffdiff算出来的,基于106*103*C/NL,其中C是map至该基因的外显子上的片断数,N是所有map至基因组的测序reads的碱基数,L就是该基因外显子碱基全长。将FPKM设计定为>1,分别在癌变、癌旁及正常组织,我们检测到14854–15168个表达基因,其中包括大部分注释的人类参考基因。我们进一步分析不同样品之间基因表达的相关性。总体基因表达普遍与Pearson关联系数高度相关。两个样本表达量的相关性计算的是pearson相关系数,先把FPKM取log2(FPKM+1),然后用R里的cor.test函数计算的相关性。判断检测的样本是否合理选择以及检验结果是否可靠的主要指标是样品间差异基因表达水平的相关性。相关系数越接近1,表明样品之间表达模式的相似度越高。我们发现癌变组与癌旁组的相关系数为0.93,癌变组与正常口腔黏膜组的相关系数也为0.93,说明样品之间表达模式的相似度高。
我们检测到癌变与癌旁组织之间有252个差异表达基因,其中表达升高者117个,表达降低者135个;癌变与正常组织之间有234个差异表达基因,其中表达升高者67个,表达降低者167个。其中,癌变与癌旁组织之间差异表达明显的KLK14基因进入我们的研究范围。
实施例3舌鳞癌组织及癌旁组织中KLK14基因表达情况
一、材料和方法
1、材料
采集标准见实施例1,采集36例舌鳞癌组织及癌旁组织,对其进行分组编号。
2、方法
2.1RNA的提取
(1)从液氮罐中取出样本后,取50-100mg(黄豆大小)组织研磨后,加1ml Trizol试剂。把Trizol-组织裂解混合液转移到1.5ml离心管中,之后按Trizol试剂的标准操作抽提总RNA;
(2)分离阶段每1mlTrizol中加0.2ml氯仿,用力摇晃试管10秒,混匀,室温静置5分钟。然后12000rmp离心15分钟;
(3)RNA的沉淀吸出上层水相样液体,置入另一个新的1.5mlEP管中,加入等体积的异丙醇,轻轻摇晃后置放于-20℃,20分钟,然后12000rmp离心10分钟;
(4)RNA的洗脱轻轻去掉上清,保留沉淀。加入即配的75%乙醇1ml,摇晃,洗涤RNA沉淀,7500rmp离心5分钟;
(5)RNA的再溶解轻轻倒掉上清,取沉淀置超净工作台,打开风机,吹干,加20-30ul的DEPC水溶解,在55-60℃下孵育10分钟;
(6)RNA的保存提取的RNA需保存于-80℃超低温冰箱中,或立即用于逆转录;
(7)RNA中痕量DNA的去除;
表2反应体系
置于37℃,1小时。将上述反应物加入DEPC水稀释至200μl,再用相同量Trizol去除蛋白质,重新加入异丙醇使RNA沉淀,然后再次用DEPC水溶解RNA;
(8)总RNA的鉴定取适量RNA,1.2%琼脂糖凝胶(含EB),100V,15分钟电泳。取RNA标本,按一定比例稀释后,测定A260和A280,计算RNA浓度和A260/A280的比值。
2.2逆转录
按vazyme(HiScriptTM Q RT Super Mix for qPCR)逆转录试剂盒说明书进行。基因组DNA去除
(1)在RNase free的离心管中配制如下混合液
表3混合液配置
用移液器轻轻吹打混匀。42℃2分钟。
(2)配制逆转录反应体系:
表4逆反应体系配置
用移液器轻轻吹打混匀。
(3)42℃水浴孵育15分钟,再85℃加热5分钟后终止反应
(4)cDNA产物可在-20℃储存或立即用于qPCR反应。
2.3Real-Time PCR
2.3.1引物设计
模板序列为NM_001311182.1,应用Primer5.0软件设计,本实验选择GAPDH为内参,引物由上海生工合成。具体引物序列如下:
表5引物序列
2.3.2反应体系:
表6q-PCR反应体系
2.3.3PCR反应程序
表7PCR反应程序
2.4统计学及数据分析方法
数据的统计学处理采用SPSS13.0软件包。Real-time PCR结果先分别获得目的基因和参照基因Ct,用设计为样本的目的基因的Ct值减去样本参照基因的Ct值,得到ΔCt(1);同样,用对照组目的基因的Ct减去对照组参照基因的Ct,得到ΔCt(2),然后用ΔCt(1)减去ΔCt(2),得到ΔΔCt,即ΔΔCt=ΔCt(1)-ΔCt(2)。用公式2-ΔΔCt计算样本组目的基因相对于对照组目的基因的量。采用t检验对每两组间ΔCt数据进行分析。
二、实验结果
我们对所有样本提取RNA进行总RNA浓度及纯度检测,微量紫外分光光度计测量显示发现所有样本无论RNA纯度RNA≥400ng/μl,吸光度A260/280≥1.80,均符合real time-PCR实验要求。
实时定量PCR扩增曲线拐点清楚,扩增曲线整体平行性好,表明各反应管的扩增效率相近,极限平而无上扬现在,曲线指数期斜率较大,说明扩增效率较高;样本扩增产物溶解曲线都是单峰,说明扩增产物只有一条,为特异性扩增;根据qRT-PCR的相对定量公式,比较KLK14基因在舌鳞癌组织和癌旁组织中的表达水平。结果显示:KLK14在舌鳞癌组织中的表达水平远远高于癌旁组织中的表达水平,前者是后者的约一百倍,以上结果验证了高通量转录组表达数据的整合分析KLK14在舌鳞癌组织中高表达的结果。
实施例4细胞系SCC15的培养
一、材料
SCC15是人口腔鳞状细胞癌细胞株,由首都医科大学口腔医学研究所馈赠。
二、细胞培养
细胞复苏:
(1)从-80℃冰箱取出细胞,迅速置于37℃温水中解冻;
(2)将冻存液吸出,置入5ml装有含血清的培养基的离心管中;
(3)将离心管置于4℃离心机中,900rpm,离心4分钟;
(4)倒掉上清,加入10ml培养基,混匀,吸出置于培养瓶中,置于37℃、5%CO2细胞培养箱中培养。
细胞传代:
(1)当细胞贴壁生长长至密度为80%左右时,弃原培养基,用PBS洗两次;
(2)倒掉PBS,加2ml胰酶至培养瓶中,置于培养箱消化3分钟,观察细胞完全脱落后,加入5ml含血清的细胞培养基,混匀,将悬液吸出置入15ml离心管中;
(3)将离心管置于4℃离心机中,1000rpm,离心7分钟;
(4)弃上清,加入10ml含血清培养基至离心管中,吹打混匀,吸出置于细胞培养瓶中,置于置于37℃、5%CO2细胞培养箱中培养。
细胞冻存:
(1)当细胞贴壁生长长至密度为80%左右时,弃原培养基,用PBS洗两次;
(2)弃PBS,取2ml胰酶加入培养瓶中,置于培养箱消化3分钟,观察细胞完全脱落后,加入5ml含血清的细胞培养基,混匀,将悬液吸出置入15ml离心管中;
(3)将离心管置于4℃离心机中,1000rpm,离心7分钟;
(4)弃上清,加入2ml冻存液于离心管中,吹打混匀,将细胞悬液移至于冻存管中,放入冻存盒,置于-80℃冰箱。
实施例5干扰实验抑制KLK14基因表达
一、材料
根据KLK14基因在GenBank(NCBI Reference Sequence:NM_001311182.1),shRNA慢病毒LV-KLK14-RNAi载体及包装系统均购自上海吉凯基因公司。
二、实验方法
1、SCC15细胞的培养
方法步骤同实施例4。
2、稳定干扰KLK14的细胞株建立及鉴定
重组慢病毒组和对照病毒组分别转染细胞SCC15.取对数生长期细胞SCC15按0.5×106/孔接种于6孔板中,生长到50%—60%融合后进行转染,吸去6孔板的培养液,按MOI值=(即感染复数MOI=感染病毒数/细胞数)向每孔加入稀释病毒液,72h后荧光显微镜观察细胞绿色荧光蛋白的表达情况,观察感染效率,如效率大于50%,则继续培养,如反之感染效率低于50%,重新进行感染。至感染后120h,收集细胞,进行后续试验。并提取细胞总RNA检测mRNA表达水平,判断转染水平。
3、细胞分组
将细胞分为三个组:SCC15未做任何处理的空白对照组,简称空白对照组;SCC15仅加入LV-NC组简称阴性对照组,即NC组;SCC-15加入LV-KLK14shRNA组称转染组,即RNAi组。
4、CCK-8法检测细胞增殖能力
(1)各组细胞成对数生长状态,用胰蛋白酶消化,完全培养基终止消化,离心(1000rpm,7分钟)后调整细胞密度,以100ul/孔种到96孔培养板中,细胞数量为2xl03个/孔,每组设6个平行孔,继续培养24h、48h、72h、96h、120h、144h、168h;
(2)将上述每个时间段的细胞取出,向每孔加入10ul的CCK-8溶液,以加入相应不含细胞的细胞培养液和CCK-8溶液为空白组;
(3)继续将96孔培养板放置细胞培养箱中培养2小时,用酶标仪检测吸光度,绘制细胞增殖曲线,实验重复3次。
5、统计学方法
对数据分析使用SPSS13.0统计学软件,结果采用均数±标准差表示,采用方差分析以及t检验,P值<0.05有统计学意义。
三、实验结果
1、转染慢病毒LV-KLK14shRNA质粒对人舌癌细胞株SCC15中KLK14mRNA和蛋白表达的影响
为了验证稳定细胞株的建立,使用real-time PCR技术检测KLK14mRNA表达水平。结果如图1所示。干扰组(RNAi组)细胞中KLK14mRNA表达水平明显减低,表达水平仅为空白对照组的45%左右,而空白对照组和转染空病毒载体细胞(NC)均未见明显变化,且两组之间的没有统计学差异(P>0.05)。
2、KLK14对细胞增殖的影响
采用CCK-8法检测KLK14对细胞增殖能力能力的影响,结果显示:在24小时后,与阴性对照组和空白对照组比较,干扰组(RNAi组)的活细胞数量明显减少,细胞增殖能力明显减弱,从生存曲线也可以看出在干扰组中曲线形状较平缓。将各组吸光值进行统计学分析,与阴性对照组和空白组比较,干扰组吸光度值在24h即显示出具有显著差异,并可持续维持(P<0.05),而阴性对照组和空白组比较比较,无明显统计学差异(P>0.05)。
本发明采用高通量测序筛选出舌鳞癌致病相关基因KLK14,结合分子细胞生物学实验验证,证实了KLK14与舌鳞癌病具有很好的相关性。本发明为舌鳞癌临床诊疗提供了新的靶标,具有很好的临床应用前景。
- 还没有人留言评论。精彩留言会获得点赞!