改性生物沥青的制备方法和改性生物沥青及其用途与流程
本发明涉及改性生物沥青的制备方法和改性生物沥青及其用途。
背景技术:
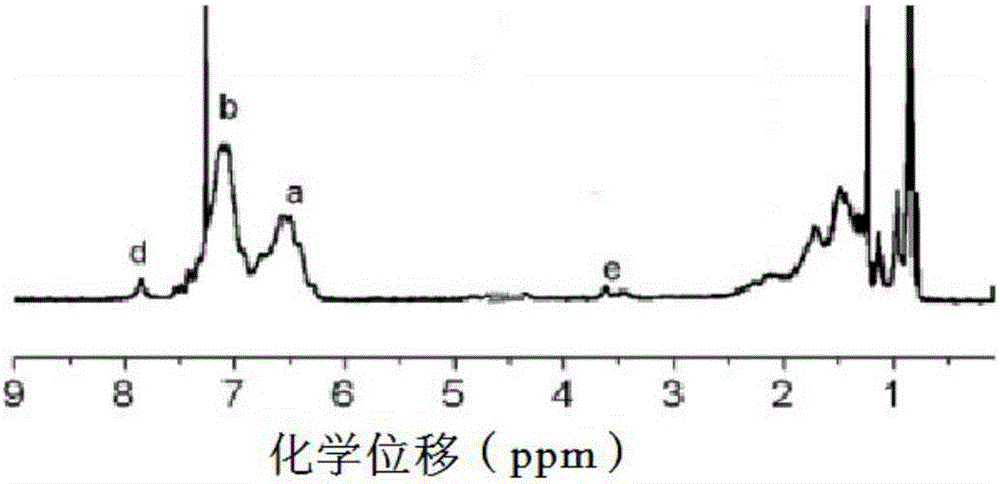
:近年来,随着我国经济的繁荣昌盛,我国公路特别是高速公路建设事业得到了快速发展。然而石油资源的日渐枯竭,道路建设的可持续发展面临着巨大的挑战。在此情况下,资源的再生利用成为交通行业重要的发展趋势。因此,研发新型的沥青材料来替代石油沥青已经成为国内外路面工程领域研究的热点,而生物沥青作为一种潜力巨大的新能源沥青材料,已被研究人员逐渐重视,对生物沥青的生产、性能改进及路面使用的研究迫在眉睫。生物沥青是由生物质热解油中的沥青组分(重质油)经过分离和深加工制备的新型沥青材料。与石油沥青相比生物沥青在生产成本、施工温度以及温室气体排放方面有较大优势,但在技术性能上也存在一些缺陷:(1)生物沥青低温下容易硬脆,延度值较小;(2)生物沥青在高温下易老化;(3)生物沥青成分复杂,稳定性差,长时间放置后,表现出非匀质特性。为克服生物沥青的缺陷,纵观生物沥青的改性方法,以往的生物沥青制备及改性方法是采用常压蒸馏、减压蒸馏、萃取、氧化工艺、催化加氢、酯化或者通过加醇类化学品(小分子改性剂),使生物沥青中的活性物质转变成非活性物质,从而提高生物沥青的稳定性。也有添加高分子改性剂(如废旧塑料和废旧胶粉),但其实质是简单的物理共混,并没有化学反应。技术实现要素:为了解决现有生物沥青存在的问题,本发明的目的是提供一种新的改性生物沥青的制备方法和由所述方法获得的改性生物沥青,所得改性生物沥青三大指标优良,具有较好的高温和低温性能。本发明涉及一种改性生物沥青的制备方法,其特征在于,所述方法包括如下步骤:1)采用可逆-休眠自由基聚合方法,以烯类单体制备高分子改性剂;2)将步骤1)中获得的高分子改性剂加入生物沥青中,在135℃下搅拌10-30min;搅拌均匀后加入基质沥青中,在165℃下搅拌10-30min,获得混合均匀的改性生物沥青;其中以改性生物沥青的总重量计,步骤2)中使用65~94%的基质沥青,5~25%的生物沥青和1~10%的高分子改性剂。在本发明的一个实施方案中,可逆-休眠自由基聚合包括退化链转移自由基聚合(DTRP)和可逆加成-断裂链转移聚合(RAFT)。在本发明的另一个实施方案中,可逆-休眠自由基聚合包括均相聚合和非均相聚合。在本发明的另一个实施方案中,均相聚合包括本体聚合、溶液聚合,非均相聚合包括悬浮聚合和乳液聚合。在本发明的另一个实施方案中,烯类单体包括苯乙烯类、丙烯酸酯类和丁二烯类单体。在本发明的另一个实施方案中,步骤1)中获得的高分子改性剂的数均分子量Mn在15000~100000的范围内。在本发明的另一个实施方案中,步骤1)中获得的高分子改性剂包括均聚物和嵌段共聚合物。在本发明的另一个实施方案中,步骤1)中的可逆-休眠自由基聚合在50~100℃下进行。本发明还涉及由上述制备方法获得的改性生物沥青。本发明还涉及上述改性生物沥青用于道路建设的用途。本发明制备的高分子改性剂用于改性生物沥青。基质沥青与生物沥青的数均分子量往往在几万以下,过高分子量的高分子改性剂往往在基质沥青和生物沥青中的相容性变差,因此,需要通过可控聚合制备得到分子量在合适的范围内(数均分子量在15000~100000的范围内)的高分子改性剂。本申请选择具有活性基团的苯乙烯类单体、丙烯酸酯类单体和丁二烯类单体进行聚合,得到具有活性基团的聚合物,该聚合物可与生物沥青中的活性基团产生反应,一方面提高了生物沥青的稳定性,另外该高分子改性剂本身具有的饱和主链与基质沥青相似,从而起到了偶联剂的作用,使得基质沥青与生物沥青的相容性得到了提高。在本发明的试验研究中将生物沥青与不同种类的高分子改性剂按不同的掺混比例进行混合,制得改性生物沥青。并与基质沥青的性能对比,进行改性生物沥青的稠度(针入度试验)、温度稳定性(软化点试验)和塑性(延度试验)指标的研究,综合分析不同高分子改性剂和掺混比例对改性生物沥青性能的影响,最后通过采用合理的掺混比例和高分子改性剂来改善生物沥青的性能,以满足规范要求,同时也为进一步的应用奠定基础。附图说明图1显示了根据本申请的方法制备的高分子改性剂的典型GPC曲线图(Mn=30900,Mw/Mn=2.22);图2显示了根据实施例1a获得的聚合物1A的核磁氢谱图;图3显示了根据实施例1b获得的聚合物1B的核磁氢谱图;图4显示了根据实施例1c获得的聚合物1C的核磁氢谱图;图5显示了根据实施例2a获得的聚合物2A的核磁氢谱图;图6显示了根据实施例2b获得的聚合物2B的核磁氢谱图;图7显示了根据实施例2c获得的聚合物2C的核磁氢谱图;图8显示了根据实施例2d获得的聚合物2D的核磁氢谱图;图9显示了根据实施例3a获得的聚合物3A的核磁氢谱图;图10显示了根据实施例3b获得的聚合物3B的核磁氢谱图;图11显示了根据实施例4a获得的聚合物4A的核磁氢谱图;图12显示了根据实施例4b获得的聚合物4B的核磁氢谱图;图13显示了根据实施例5a获得的聚合物5A的核磁氢谱图;图14显示了根据实施例5b获得的聚合物5B的核磁氢谱图。具体实施方式以下通过具体实施例来进一步说明本发明。应该理解,本发明实施例所述方法仅用于说明本发明,而不限制本发明,在本发明的构思前提下对本发明制备方法和改性方法的简单改进都属于本发明要求保护的范围。实施例1-RAFT均相聚合实施例1a-单体苯乙烯(St)的RAFT均相聚合在单支口圆底反应瓶中按照配比([St]:[RAFT]:[引发剂]=450~5000:3~5:1)加入苯乙烯单体,作为RAFT试剂的2-乙氧羰基丙烷-2-二硫代苯甲酸酯(EPDTB),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,苯乙烯单体与RAFT试剂的摩尔浓度的比为(150~1000:3~5),RAFT试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚苯乙烯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物1A用于后续测试和试验。实施例1b-单体氯丁二烯(CP)的RAFT均相聚合在单支口圆底反应瓶中按照配比([CP]:[RAFT]:[引发剂]=450~5000:3~5:1)加入氯丁二烯单体,作为RAFT试剂的2-乙氧羰基丙烷-2-二硫代苯甲酸酯(EPDTB),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,氯丁二烯单体与RAFT试剂的摩尔浓度的比为(150~1000:3~5),RAFT试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚氯丁二烯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物1B用于后续测试和试验。实施例1c-单体甲基丙烯酸甲酯(MMA)的RAFT均相聚合在单支口圆底反应瓶中按照配比([MMA]:[RAFT]:[引发剂]=450~5000:3~5:1)加入甲基丙烯酸甲酯单体,作为RAFT试剂的2-氰基-2-丙基苯并二硫(CPDB),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,甲基丙烯酸甲酯单体与RAFT试剂的摩尔浓度的比为(150~1000:3~5),RAFT试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚甲基丙烯酸甲酯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物1C用于后续测试和试验。实施例2-DTRP均相聚合实施例2a-单体苯乙烯的DTRP均相聚合在单支口圆底反应瓶中按照预先设定的配比([St]:[CHI3]:[引发剂]=450~5000:3~5:1)加入苯乙烯单体,作为DTRP试剂的碘仿(CHI3),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,苯乙烯单体与DTRP试剂的摩尔浓度的比为(150~1000:3~5),DTRP试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚苯乙烯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物2A用于后续测试和试验。实施例2b-单体氯丁二烯的DTRP均相聚合在单支口圆底反应瓶中按照预先设定的配比([CP]:[CHI3]:[引发剂]=450~5000:3~5:1)加入氯丁二烯单体,作为DTRP试剂的碘仿(CHI3),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,氯丁二烯单体与DTRP试剂的摩尔浓度的比为(150~1000:3~5),DTRP试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚氯丁二烯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物2B用于后续测试和试验。实施例2c-单体丙烯酸正丁酯(BA)的DTRP均相聚合在单支口圆底反应瓶中按照预先设定的配比([BA]:[DTRP]:[引发剂]=450~5000:3~5:1)加入丙烯酸正丁酯单体,作为DTRP试剂的α-碘代异丁腈(CP-I),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,丙烯酸正丁酯单体与DTRP试剂的摩尔浓度的比为(150~1000:3~5),DTRP试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚丙烯酸正丁酯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物2C用于后续测试和试验。实施例2d-单体甲基丙烯酸甲酯的DTRP均相聚合在单支口圆底反应瓶中按照预先设定的配比([MMA]:[DTRP]:[引发剂]=450~5000:3~5:1)加入甲基丙烯酸甲酯单体,作为DTRP试剂的α-碘代异丁腈(CP-I),偶氮类引发剂(偶氮二异丁氰[AIBN]/偶氮二异庚氰[ABVN]),苯类溶剂(苯/甲苯/二甲苯),磁力搅拌溶解。在所述聚合中,甲基丙烯酸甲酯单体与DTRP试剂的摩尔浓度的比为(150~1000:3~5),DTRP试剂与引发剂的摩尔浓度的比为(3~5:1)。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,50~100℃油浴锅中恒温反应。反应至聚甲基丙烯酸甲酯的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物2D用于后续测试和试验。实施例3-RAFT/DTRP非均相聚合实施例3a-单体氯丁二烯的RAFT非均相聚合配制油相:900g氯丁二烯单体,加入34.2g歧化松香、2.97~5.85gRAFT试剂二苄基三硫代碳酸酯(DBTTC),充分溶解。配制水相:990g蒸馏水,加入6.57g氢氧化钠、1.80g十二烷基硫酸钠、3.60gLomarPW、1.80gNaCO3,充分溶解。乳液聚合过程:在低温乳液聚合反应体系中,加入配制好的水相,开动机械搅拌,加入配制好的油相,通氮气保护。首先在20℃下预乳化35min降温至10℃,然后加入氧化剂-K2S2O8水溶液,用蠕动泵加入配制好的还原剂Na2SO3和甲脒亚磺酸钠的水溶液,聚合温度精确控制在9℃,聚合过程中从反应容器底部放出一定量的聚合物乳液,称重法监测氯丁二烯单体转化率,聚合过程中及结束后的乳液在大量甲醇中凝胶沉淀,晾置干燥后,所得聚合物3A用于GPC、1HNMR核磁等性能测试及后续试验。实施例3b-单体氯丁二烯的DTRP非均相聚合配制油相:900g氯丁二烯单体,加入34.2g歧化松香、2.97~5.85gDTRP试剂(CHI3),充分溶解。配制水相:990g蒸馏水,加入6.57g氢氧化钠、1.80g十二烷基硫酸钠、3.60gLomarPW、1.80gNaCO3,充分溶解。乳液聚合过程:低温乳液聚合反应体系中,加入配制好的水相,开动机械搅拌,加入配制好的油相,通氮气保护。首先在20℃下预乳化35min降温至10℃,然后加入氧化剂-K2S2O8水溶液,用蠕动泵加入配制好的还原剂Na2SO3和甲脒亚磺酸钠的水溶液,聚合温度精确控制在9℃,聚合过程中从反应容器底部放出一定量的聚合物乳液,称重法监测氯丁二烯单体转化率,聚合过程中及结束后的乳液在大量甲醇中凝胶沉淀,晾置干燥后,所得聚合物3B用于GPC、1HNMR核磁等性能测试及后续试验。实施例4-通过RAFT聚合制备嵌段聚合物通过可逆加成-断裂链转移聚合制备嵌段聚合物类的高分子改性剂的思路为:首先按照实施例1中的方法制备大分子RAFT试剂,然后加入第二单体,进行聚合,制备得到嵌段聚合物类的高分子改性剂。实施例4a-通过RAFT聚合制备PSt-b-PCP嵌段聚合物首先制备大分子RAFT试剂PSt:在单支口圆底烧瓶中加入第一单体苯乙烯(20.8g,200mmol)、RAFT试剂EPDTB(1.072g,4mmol)、引发剂偶氮二异丁腈AIBN(0.168g,1mmol)、四氢呋喃THF(40g),在70℃油浴中反应20h。用冷冻甲醇沉淀洗涤三遍得到大分子RAFT试剂PSt(Mn=2900g·mol-1,Mw/Mn=1.23)。然后进行PSt为大分子RAFT试剂的氯丁二烯嵌段聚合。将PSt(2g)、第二单体氯丁二烯(20.21g,138mmol)、引发剂偶氮二异丁腈AIBN(0.0283g,0.17mmol)、溶剂苯(30.3g)进行磁力搅拌溶解。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,60℃油浴锅中恒温反应至嵌段聚合物的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物4A用于后续测试及试验。实施例4b-通过RAFT聚合制备PMMA-b-PCP嵌段聚合物同上,制备大分子RAFT试剂PMMA:单支口圆底烧瓶中加入第一单体甲基丙烯酸甲酯(6.20g,0.06mol)、RAFT试剂2-氰基-2-丙基苯并二硫(CPDB,0.465g,2.1mmol)、引发剂偶氮二异丁腈AIBN(0.16g,0.97mmol)、苯(16g),60℃油浴中反应8h。用冷冻甲醇沉淀洗涤三遍得到大分子RAFT试剂PMMA(Mn=1800g·mol-1,Mw/Mn=1.33)。然后进行PMMA为大分子RAFT试剂的氯丁二烯嵌段聚合。将PMMA(2g,1.11mmol)、第二单体氯丁二烯(25g,113mmol)、引发剂偶氮二异丁腈AIBN(0.0966g,0.588mmol)、溶剂苯(30g)进行磁力搅拌溶解。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,60℃油浴锅中恒温反应至嵌段聚合物的分子量达到15000~100000,加入冷冻的石油醚迅速停止反应。所得聚合物4B用于后续测试和试验。实施例5-通过DTRP聚合制备嵌段聚合物通过退化链转移自由基聚合制备嵌段聚合物高分子改性剂的思路为:首先按照实施例2中的方法制备大分子DTRP试剂,然后加入第二单体,进行聚合,制备得到嵌段聚合物类的高分子改性剂。实施例5a-通过DTRP聚合制备PCP-b-PMMA嵌段聚合物首先按照上面使用碘仿(CHI3)作为DTRP试剂的氯丁二烯溶液均相聚合制备大分子DTRP试剂PCP-I,氯丁二烯单体转化率较低时停止反应,将残留未反应的单体和溶剂旋蒸处理掉。然后加入第二单体甲基丙烯酸甲酯MMA、引发剂偶氮二异丁腈AIBN、溶剂甲苯(质量是甲基丙烯酸甲酯单体的1.5倍),配比为[MMA]:[PCP-I]:[AIBN]=115:1:0.29,磁力搅拌溶解。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,60℃油浴锅中恒温反应预定时间,加入冷冻的石油醚迅速停止反应。所得聚合物5A用于后续测试和试验。实施例5b-通过DTRP聚合制备PCP-b-PSt嵌段聚合物同上,首先制备得到大分子DTRP试剂PCP-I,然后加入第二单体苯乙烯、引发剂偶氮二异丁腈AIBN、溶剂甲苯(质量是甲基丙烯酸甲酯单体的1.5倍),配比为[St]:[PCP-I]:[AIBN]=62.5:1:0.16,磁力搅拌溶解。置于冷冻冰盐水中通氩气鼓泡10min除去体系中的氧气,密封,抽真空、通氩气置换5次。避光条件下,60℃油浴锅中恒温反应预定时间,加入冷冻的石油醚迅速停止反应。所得聚合物5B用于后续测试和试验。核磁氢谱图分析:图2中明显可见聚苯乙烯和RAFT端基的特征峰。化学位移在7.92ppm处的特征峰归属于RAFT试剂EPDTB苯环上的氢。化学位移在6.2-7.4ppm处的a、b峰归属于PSt苯环上次甲基上的氢。图3是聚合所得PCP的典型核磁氢谱图。图中明显可见聚氯丁二烯和RAFT端基的特征峰。化学位移在7.92ppm处的特征峰归属于RAFT试剂EPDTB苯环上的氢。4.15ppm处的d峰归属于–COO-CH2-CH3的亚甲基上的氢。1.2ppm处的b和c峰归属于–C(CH3)2-COO-CH2-CH3的甲基上的氢。化学位移在5.2-5.9ppm处的e峰归属于聚氯丁二烯主链上次甲基上的氢。图4可看出,化学位移在3.6ppm处的特征峰归属于PMMA酯甲基上的质子氢。b峰是-CH2C(CH3)(COOCH3)的甲基上的质子氢。7.92-8.01ppm(d)处的特征峰是RAFT试剂CPDB苯环的质子氢。图5中4.3-4.65ppm处的a、b峰分别是聚苯乙烯的α位和ω位碘原子封端的次甲基(-CHI2)和次甲基(-CHΦI)上的质子的特征峰;6.1-7.2ppm处的c峰是聚苯乙烯苯环上的次甲基上的质子的特征峰。图6碘仿为退化链转移剂制备得到的PCP核磁氢谱图。化学位移在5.1-5.9ppm处的特征峰归属于PCP主链次甲基上的氢。化学位移在4.5-4.8ppm处的特征峰归属于碘原子封端的亚甲基和次甲基上的氢。图7是CP-I为退化链转移剂制备得到的PBA核磁氢谱图。化学位移在3.9-4.1ppm处的e峰归属于PBA酯基邻位上亚甲基的氢。4.79ppm处的d峰归属于ω位碘原子封端的次甲基上的氢。图8中化学位移在1.20ppm处的a峰是聚合物主链α端基的两个甲基的特征峰,化学位移在2.80ppm处的b’峰属于聚合物ω末端与碘原子相连的亚甲基的特征峰,化学位移在1.80ppm处的b峰、0.80ppm处的双峰c以及3.55~3.63ppm处的d峰分别是聚合物主链亚甲基、侧甲基和酯甲基的特征峰。图9是DBTTC调控氯丁二烯RAFT聚合过程所得PCP的典型核磁氢谱图。化学位移在5.2-5.9ppm处的b峰归属于聚氯丁二烯主链上次甲基的氢。除去CDCl3在7.26ppm处残留的质子氢的峰外,化学位移在7.1-7.4ppm处的a峰归属于端基RAFT上苯环的氢。图10是碘仿为退化链转移剂乳液聚合制备得到的PCP核磁氢谱图。化学位移在5.1-5.9ppm处的特征峰归属于PCP主链次甲基上的氢。化学位移在4.5-4.8ppm处的特征峰归属于碘原子封端的亚甲基和次甲基上的氢。图11是EPDTB为RAFT试剂调控下制备得到的PSt-b-PCP两嵌段共聚物核磁氢谱图。核磁氢谱图的分析如下:化学位移在6.3-7.2pmm处和5.2-5.9ppm处的特征峰分别归属于聚苯乙烯和聚氯丁二烯主链上的次甲基质子氢。而且,也观察到了7.53-8.01ppm处的特征峰应该是RAFT端基苯环上的质子氢。这证明了PSt-b-PCP两嵌段共聚物的结构。结合此聚合反应过程的可控特征,确信成功制备得到了PSt-b-PCP两嵌段共聚物。图12是CPDB为RAFT试剂调控下制备得到的PMMA-b-PCP两嵌段共聚物核磁氢谱图。可看出,化学位移在3.6ppm处的特征峰归属于PMMA酯甲基上的质子氢。b峰是-CH2C(CH3)(COOCH3)的甲基上的质子氢。5.2-6.0ppm(e)处的特征峰是PCP主链上次甲基的质子氢。2.1-3.0ppm(f)处的特征峰是PCP主链上亚甲基的质子氢。7.92-8.01ppm(d)处的特征峰是RAFT试剂CPDB苯环的质子氢。这些表明氯丁二烯在以PMMA为大分子RAFT试剂时进行了聚合反应,说明成功制备得到了两嵌段聚合物PMMA-b-PCP。图13是碘仿为退化链转移剂调控下制备得到的PCP-b-PMMA两嵌段共聚物核磁氢谱图。分析如下:化学位移在5.1-5.9ppm处的a峰归属于聚氯丁二烯主链上次甲基的特征峰,化学位移在3.6ppm左右处的b峰是聚甲基丙烯酸甲酯酯甲基上质子氢的特征峰。化学位移在2.9ppm处的c峰是与链末端与I原子相邻的β末端亚甲基质子氢的特征峰。这些实验结果进一步证明成功制备得到了结构清晰的PCP-b-PMMA两嵌段聚合物。图14是碘仿为退化链转移剂调控下制备得到的PCP-b-PSt两嵌段共聚物核磁氢谱图。图中可看出,5.2-5.9ppm处的a峰是聚氯丁二烯主链上次甲基的特征峰,6.3-7.3ppm处的b峰是聚苯乙烯苯环上质子氢的特征峰。4.5-4.7ppm处的c峰是链上ω末端的次甲基(-CHΦI)质子氢的特征峰。这些实验结果进一步证明成功制备得到了结构清晰的PCP-b-PSt两嵌段聚合物。生物沥青改性试验:按照表1所示的配比,将上述实施例制备得到的高分子改性剂加入生物沥青中,在135℃下搅拌15min;搅拌均匀后加入90#基质沥青中,在165℃搅拌20min,即可制成混合均匀、优质的改性生物沥青。表1生物沥青改性试验配比高分子改性剂生物沥青基质沥青配比110g25g65g配比25g15g80g配比31g5g94g将所制备得到的改性沥青进行性能测试,测试方法采用本领域常规测试方法以及中华人民共和国行业标准JTGE20-2011,所得改性沥青试验见表2a-2c:表2a改性生物沥青三大指标测试结果(配比1)表2b改性生物沥青三大指标测试结果(配比2)表2c改性生物沥青三大指标测试结果(配比3)相比于例如李作恒的CN201410720791.5(题为“一种废旧高分子改性沥青组合物及其制备方法”,其中采用废旧高分子材料改性生物沥青),本发明得到的改性生物沥青稳定性更好,三大指标优良,具有较好的高温和低温性能。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1