一种含有邻苯二醌类官能团的聚乙二醇修饰剂及其制备方法和用途与流程
本发明属于生物医药领域,更具体地涉及生物偶联化学领域,特别是涉及一种含有邻苯二醌类官能团的聚乙二醇(PEG)修饰剂及其制备方法和用途。
背景技术:
随着生物技术的发展,越来越多的生物药物被用来治疗人类的疾病,如治疗性的多肽蛋白药物、蛋白亚单位疫苗、抗体或抗体片段等。然而,生物药物在使用过程中存在着许多问题,比如:稳定性差使得药物在制备、储存和使用过程中发生降解,易被蛋白酶水解导致药物活性降低,分子量小容易被肾小球过滤使得体内半衰期短,一些肿瘤治疗药物不具有靶向杀伤病变细胞的能力,对机体带来巨大副作用,外源性的蛋白治疗药物还存在着免疫原性过高,而另一方面,作为疫苗的抗原蛋白的免疫原性又太低等。
因此,治疗性的蛋白常常采用生物偶联的方法来改善其药效学性质,如用PEG修饰蛋白避免被蛋白酶水解而延长作用时间,或利用PEG作为两端偶联剂(linker)连接抗体和细胞毒性药物提高药物靶向性、降低在体内循环时的毒性,或抗原蛋白与载体蛋白偶联来提高其免疫原性。这一技术目前取得了较好的发展,已在长效药物、抗体药物偶联(ADC)以及结合疫苗中获得成功应用。在生物偶联药物中定点和修饰产物稳定性对保证安全给药、生产质控和稳定储存都至关重要。当前对于蛋白质类药物的定点选择有氨基、羧基、组氨酸咪唑基和半胱氨酸的巯基,半胱氨酸的巯基由于在蛋白质中存在较少,常被作为定点修饰位点。
当前巯基定点修饰剂有PEG-马来酰亚胺(maleimide)(PEG-Mal)、PEG-乙烯基砜(vinylsulfone)、PEG-SH(末端为游离巯基)等。PEG-乙烯基砜、PEG-SH由于修饰效率低而很少被使用,PEG-Mal是常用的巯基修饰剂。然而,PEG-Mal在应用过程中存在明显缺陷。一是马来酰亚胺官能团的水解,如果修饰反应时间过长,尤其是应用在两端同功能或异功能的linker时,马来酰亚胺的水解会导致修饰率过低。近年来发现马来酰亚胺使用过程中另一个严重的问题是PEG-mal修饰蛋白或药物会存在PEG脱落(dePEGylation)的现象。PEG链脱落后的药物便不再具备既定的要求,原本以为降低了细胞毒性的药物进行一定剂量的注射,会再次产生强的毒性。研究发现,PEG链的脱落源于马来酰亚胺与巯基形成的硫醚键的断裂(Aaron D.Baldwin,et al,.Bioconjugate chemistry 2011;22:1946-1953),断裂原因主要有:迈克尔加成的逆向反应和氧化条件下硫醚键断裂。已有研究通过对马来酰亚胺的结构进行改造,得到稳定性较高的化合物。但这些技术目前仍未被广泛应用,其原因主要是活化官能团的制备繁琐、不易获得。
一种更有效的、不易发生官能团水解且修饰产物更加稳定的巯基修饰剂是市场需要的,尤其要满足:巯基特异性定点、修饰活性高、修饰剂不水解和修饰产物不发生PEG脱落现象。近来有研究发现,邻苯二酚类化合物通过氧化成醌类结构可与亲核官能团巯基发生特异性反应,形成稳固的化学键,从而对含有巯基的界面实现化学修饰和功能改性(US 2008/0149566)。受此启发,有研究将具有两亲性特点的PEG与含有邻苯二酚官能团的多巴胺(DA)共价偶联,开发了PEG-DA,或分支型PEG-DA,实现对受伤组织的修复(Mehdizadeh M,et al,.Biomaterials.2012Nov;33(32):7972-83.)。有技术公开用PEG-DA尝试修饰脂肪酸酶,遗憾的是,这种修饰剂在酸性条件下不能进行修饰反应,且使用修饰剂与脂肪酸酶的比例达到30:1,修饰效率较低(Dongshuang Wang,et al,.Journal of Molecular Catalysis B:Enzymatic 2015;111:36–42)。
本发明提供的含有邻苯二醌类官能团的聚乙二醇修饰剂能够实现在广泛的pH范围进行修饰,且实现巯基定点、修饰剂不易发生水解、修饰产物稳定的优点。到目前为止,还没有相关技术报道利用多巴醌的高反应活性和稳定的巯基连接进行修饰剂的开发和应用。
技术实现要素:
为解决上述问题,本发明的目的之一是提供一种基于邻苯二醌类官能团的具有巯基定点选择性的PEG修饰剂,包括一端封闭另一端为邻苯二醌衍生功能团的PEG修饰剂、两端均为邻苯二醌的同官能团的PEG修饰剂以及仅一端为邻苯二醌的两端异官能团的PEG修饰剂。
为了达到上述发明目的,本发明采用以下技术方案:
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为单端封闭的修饰剂,具有如下通式(I):
其中
X为单甲氧基(CH3O-)、葡萄糖或半乳糖残基中1种或2种以上的组合,是封闭末端;
Y为酰胺键(-CONH-)、氨基甲酸酯键(-O-CONH-)、酯键(-O-CO-)或醚键(-O-)中1种或2种以上的组合;
n为1~1000;
邻苯二醌的苯环上的取代基R’、R”、R”’选自以下基团之一:-H、-CH3、-CH3OH、-COOH、-S-CH3、-S-CH2CH2OH、-S-CH2COOH、-CH2CH3-OH和-S-CH2CH(NH2)COOH,其中至少一个取代基为-H;R’、R”、R”’可相同或不同。
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为两端同官能团修饰剂,具有如下通式(II):
其中
Y为酰胺键(-CONH-)、氨基甲酸酯(-O-CONH-)、酯键(-O-CO-)或醚键(-O-)中1种或2种以上的组合;
n为1~1000;
邻苯二醌的苯环上的取代基R’、R”、R”’或R1’、R1”、R1”’选自以下基团之一:-H、-CH3、-CH3OH、-COOH、-S-CH3、-S-CH2CH2OH、-S-CH2COOH、-CH2CH3-OH和-S-CH2CH(NH2)COOH,其中至少一个取代基为-H;R1’、R1”、R1”’可相同或不同。
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为两端异官能团修饰剂,具有如下通式((III):
其中
Y为酰胺键(-CONH-)、氨基甲酸酯(-O-CONH-)、酯键(-O-CO-)、醚键(-O-)中1种或2种以上的组合;
Z为氨基(-NH2)、羧基(-COOH)、琥珀酰亚胺酯基马来酰亚胺酯基琥珀酰亚胺碳酸酯基酰肼中1种或2种以上的组合;
n为1~1000;
邻苯二醌的苯环上的取代基R’、R”、R”’选自以下基团之一:-H、-CH3、-CH3OH、-COOH、-S-CH3、-S-CH2CH2OH、-S-CH2COOH、-CH2CH3-OH和-S-CH2CH(NH2)COOH,其中至少一个取代基为-H;R’、R”、R”’可相同或不同。
利用邻苯二醌作为活性官能团能够解决邻苯二酚不能在酸性条件下进行修饰反应的限制,提高修饰效率。利用邻苯二醌特异性与巯基反应的特点,可实现对蛋白质药物的特异性定点修饰;利用邻苯二醌与巯基反应的化学键稳定的特点,可获得稳定性极大提高的修饰产物。
本发明的目的之一还在于提供以上衍生物的制备方法,采用如下技术方案:
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为单端封闭的修饰剂的制备方法,包括如下步骤:
1)将一端为甲氧基、葡萄糖或半乳糖残基,另一端为游离羟基的聚乙二醇溶于溶剂中,加入催化剂和氧化剂将末端醇羟基氧化为羧基,经分离纯化后得到一端为甲氧基、葡萄糖或半乳糖残基,另一端为羧基的聚乙二醇修饰试剂;
2)将盐酸多巴胺溶于溶剂中,加入缚酸剂得到脱盐酸后的多巴胺;
3)将步骤1)获得的聚乙二醇溶于溶剂中,加入交联剂冰浴下反应一段时间后,升至室温,在惰性气体保护下,与步骤2)得到的脱盐酸后的多巴胺溶液混合,室温反应过夜,分离纯化后得到端基为含有邻苯二酚官能团的聚乙二醇修饰剂;
4)将步骤3)得到的聚乙二醇修饰剂氧化,分离纯化并干燥得到端基为含有邻苯二醌官能团的聚乙二醇修饰剂;
优选地,所述溶剂选自水、二氯甲烷、三氯甲烷、甲酸、乙酸、盐酸溶液、N,N’-二甲基甲酰胺、二甲基亚砜、甲醇、乙二醇、乙醇和乙腈中的至少一种。
优选地,所述分离或纯化采用溶剂沉淀法、重结晶法、色谱柱分离法、制备液相分离法、透析法中1种或2种以上的组合。
优选地,所述干燥采用冷冻干燥法、真空干燥法、自然干燥法中1种或2种以上的组合。
优选地,步骤1)中所述催化剂为2,2,6,6-四甲基哌啶-1-氧基和KBr。
优选地,步骤1)所述的氧化剂为NaCIO。
优选地,步骤2)中所述缚酸剂选自碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、乙酸钠、吡啶、三乙胺和二异丙基乙胺中的至少一种。
优选地,步骤3)中所述惰性气体为氮气、氦气、氖气、氩气中的至少一种。
优选地,所述交联剂为EDC、DCC、NHS、DIC、CMC中的至少一种。
优选地,步骤4)所述氧化剂选自NaIO4,高锰酸钾,30%H2O2,多酚氧化酶或弱碱性缓冲液中的至少一种。
优选地,步骤4)中所述氧化采用NaIO4或高锰酸钾的非均相氧化、30%H2O2氧化、多酚氧化酶的氧化或弱碱性pH水溶液内搅拌空气氧化。
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为两端同官能团修饰剂的制备方法,包括如下步骤:
1)将两端均为琥珀酰亚胺酯基的聚乙二醇溶于溶剂中;
2)将盐酸多巴胺溶于溶剂中,加入缚酸剂后得到脱盐酸后的多巴胺;
3)在惰性气体保护下,向步骤1)的聚乙二醇溶液加入步骤2)得到的脱盐酸后的多巴胺,避光过夜反应后,将反应混合物透析至去离子水中,并用盐酸酸化,继续透析至去离子水中并且冻干,得到两端端基为含有邻苯二酚官能团的聚乙二醇修饰剂;
4)将步骤3)得到的聚乙二醇修饰剂氧化,分离纯化并干燥得到两端端基均为含有邻苯二醌官能团的聚乙二醇修饰剂;
优选地,所述溶剂选自水、二氯甲烷、三氯甲烷、甲酸、乙酸、盐酸溶液、N,N’-二甲基甲酰胺、二甲基亚砜、甲醇、乙二醇、乙醇和乙腈中的至少一种。
优选地,所述分离或纯化采用溶剂沉淀法、重结晶法、色谱柱分离法、制备液相分离法、透析法中1种或2种以上的组合。
优选地,所述干燥采用冷冻干燥法、真空干燥法、自然干燥法中1种或2种以上的组合。
优选地,步骤2)中所述缚酸剂选自碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、乙酸钠、吡啶、三乙胺和二异丙基乙胺中的至少一种。
优选地,步骤3)中所述惰性气体为氮气、氦气、氖气、氩气中的至少一种。
优选地,步骤4)所述氧化的氧化剂选自NaIO4,高锰酸钾,30%H2O2,多酚氧化酶或弱碱性缓冲液中的至少一种;
优选地,步骤4)中所述氧化采用NaIO4或高锰酸钾的非均相氧化、30%H2O2氧化、多酚氧化酶的氧化或弱碱性pH水溶液内搅拌空气氧化。
一种含有邻苯二醌类官能团的聚乙二醇修饰剂,为两端异官能团修饰剂的制备方法,包括如下步骤:
1)将一端为氨基(-NH2)、羧基(-COOH)、琥珀酰亚胺酯基、琥珀酰亚胺碳酸酯基、酰肼,另一端为马来酰亚胺酯基的聚乙二醇溶于溶剂中;
2)将盐酸多巴胺溶于溶剂中,加入缚酸剂后得到脱盐酸后的多巴胺;
3)在惰性气体保护下,向步骤1)的聚乙二醇溶液加入步骤2)得到的脱盐酸后的多巴胺,避光过夜反应后,分离纯化后得到端基为含有邻苯二酚官能团的聚乙二醇修饰剂;
4)将步骤3)得到的聚乙二醇修饰剂氧化,分离,纯化并干燥得到两端端基均为含有邻苯二醌官能团的聚乙二醇修饰剂;
优选地,所述溶剂选自水、二氯甲烷、三氯甲烷、甲酸、乙酸、盐酸溶液、N,N’-二甲基甲酰胺、二甲基亚砜、甲醇、乙二醇、乙醇和乙腈中的至少一种;
优选地,所述分离或纯化采用溶剂沉淀法、重结晶法、色谱柱分离法、制备液相分离法、透析法中1种或2种以上的组合;
优选地,所述干燥采用冷冻干燥法、真空干燥法、自然干燥法中1种或2种以上的组合。
优选地,步骤2)中所述缚酸剂选自碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、乙酸钠、吡啶、三乙胺和二异丙基乙胺中的至少一种。
优选地,步骤3)中所述惰性气体为氮气、氦气、氖气、氩气中的至少一种。
优选地,步骤4)所述氧化的氧化剂选自NaIO4,高锰酸钾,30%H2O2,多酚氧化酶或弱碱性缓冲液中的至少一种;
优选地,步骤4)中所述氧化采用NaIO4或高锰酸钾的非均相氧化、30%H2O2氧化、多酚氧化酶的氧化或弱碱性pH水溶液内搅拌空气氧化。
本发明的目的之一还在于提供本发明所述的含有邻苯二醌类官能团的聚乙二醇修饰剂的用途,包括但不限于修饰含有巯基的具有药理活性的药物,达到延长半衰期、降低免疫原性的目的,或作为linker两端偶联具有药理活性药物的目的。
作为优选,所述含有巯基的具有药理活性的药物为含有游离巯基且暴露于表面的或通过还原剂(TCEP、β-ME、DTT,β-MEA)还原得到游离巯基的蛋白质、多肽或化学小分子药物。
作为优选,修饰含有巯基的具有药理活性的药物的方法包括如下步骤:
1)将含有游离巯基的具有药理活性的药物,与本发明所述含有邻苯二醌官能团的修饰剂溶于缓冲溶液;
2)室温反应10min~24h后,加入终止反应物终止反应;检测并分离纯化得到目标修饰产物。
优选地,步骤1)中所述含巯基的具有药理活性的药物的浓度为0.5mg/ml~4mg/ml。
优选地,含巯基的具有药理活性的药物与所述修饰试剂的摩尔比为4:1~1:16。
优选地,所述缓冲溶液选为20mM tris-HCl、20mM NaAC-HAc、20mM PB、20mM Gly-NaOH中的1种或2种以上的混合。
优选地,所述缓冲溶液的pH为2~10。
优选地,步骤2)中室温反应的时间为0.5h~15h。
优选地,所述终止反应物为含有一个或多个游离巯基的化学小分子,优选分子量不超过1000Da,包括但不限于半胱氨酸、GSH、巯基乙醇或DTT中的1种或2种以上的混合。
优选地,所述半胱氨酸或GSH的添加量为0.01mg/ml~1mg/ml,巯基乙醇或DTT的添加量为0.1%~2%。
优选地,检测分析通过分离胶浓度8%~15%的SDS-PAGE、分离胶浓度8%~15%的Native-PAGE、RP-HPLC、HPSEC分析中的至少1种。
优选地,分离纯化通过盐析法、离子交换层析法、凝胶过滤层析法、亲和层析法、疏水层析法中的1种或2种以上的组合。
作为优选,作为linker两端偶联具有药理活性药物的方法包括如下步骤:
1)将所述含巯基的具有药理活性的药物分子中的一种与所述同端或异端基聚乙二醇修饰剂的一端共价偶联;终止修饰反应后,根据是否会影响步骤2)的偶联选择对步骤1)偶联产物分离与否;如影响,则分离;如不影响,则不分离;
2)将所述含巯基的具有药理活性的药物的另一种与步骤1)获得的产物共价偶联,检测分析并分离纯化得到目标修饰产物。
优选地,步骤1)中所述含巯基的具有药理活性的药物的浓度为0.5mg/ml~4mg/ml。
优选地,含巯基的具有药理活性的药物与所述修饰试剂的摩尔比为4:1~1:16。
优选地,所述缓冲溶液选为20mM tris-HCl、20mM NaAC-HAc、20mM PB、20mM Gly-NaOH中的1种或2种以上的混合。
优选地,所述缓冲溶液的pH为2~10。
优选地,步骤2)中室温反应的时间为0.5h~15h。
优选地,所述终止反应物为含有一个或多个游离巯基的化学小分子,优选分子量不超过1000Da,包括但不限于半胱氨酸、GSH、巯基乙醇或DTT中的1种或2种以上的混合。
优选地,所述半胱氨酸或GSH的添加量为0.01mg/ml~1mg/ml,巯基乙醇或DTT的添加量为0.1%~2%。
优选地,检测分析通过分离胶浓度8%~15%的SDS-PAGE、分离胶浓度8%~15%的Native-PAGE、RP-HPLC、HPSEC分析中的至少1种。
优选地,分离纯化通过盐析法、离子交换层析法、凝胶过滤层析法、亲和层析法、疏水层析法中的1种或2种以上的组合。
优选地,所述药物为临床上可接受的任一种剂型。
优选地,所述剂型为口服剂型或可注射剂型。
本发明具有以下特点:
1、本发明首次利用邻苯二醌作为活性反应分子与含有游离巯基的具有药理活性的药物分子或治疗性蛋白质、多肽发生反应,包括单方向的修饰及双方向的作为偶联linker,邻苯二醌结构反应活性高,条件温和,制备简单;
2、本发明提出的修饰剂在反应缓冲液中特异性针对巯基发生反应;
3、本发明提出的修饰修饰剂在修饰缓冲液中修饰效率高;
4、修饰剂在反应缓冲溶液中存放96h后,修饰能力没有发生显著降低,而与之对应的Mal修饰活性官能团的修饰能力下降比较明显;
5、本发明偶联的邻苯二醌与巯基形成的硫醚键在常规条件下是稳定存在的,不易发生断裂,即使在100℃热处理0.5h也未曾检测到出现断裂的现象;与之对应的mal则发生了明显的PEG脱落现象;
6、本发明的修饰剂的制备方法简单、操作方便,采用本领域常规的方法即可。
附图说明
图1是本发明实施例1中制备的单端封闭的修饰剂的结构表征,其中,(a)是H1NMR对修饰剂的结构表征,(b)是MALDI-TOF对修饰剂的结构表征;
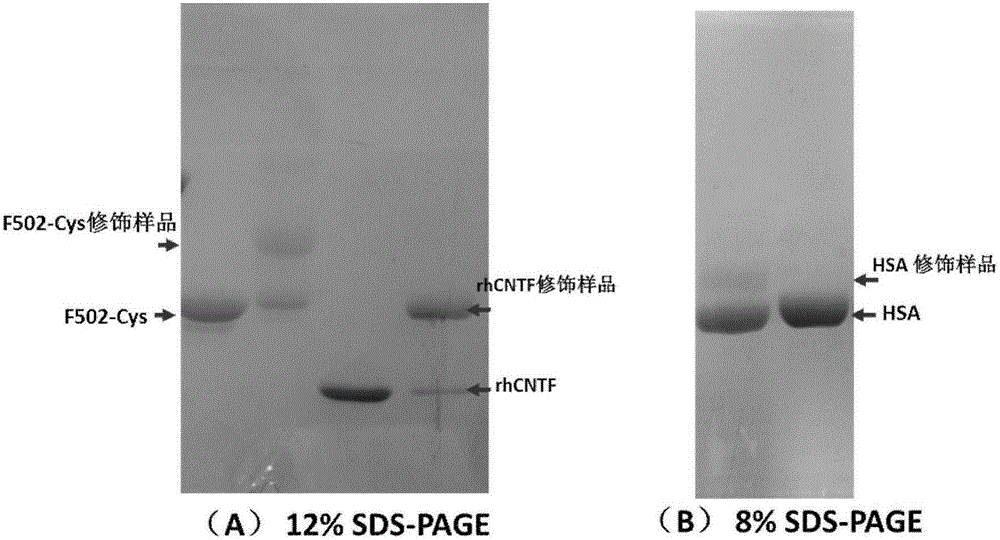
图2是本发明实施例4中修饰重组人睫状神经因子(含有一个半胱氨酸,rhCNTF)、鞭毛蛋白的突变蛋白(不含鞭毛蛋白的高可变区,且突变体含有一个半胱氨酸,F502-Cys)和人血清白蛋白(含有一个半胱氨酸,HSA)的SDS-PAGE图;
图3是本发明实施例5中定点修饰游离巯基的证明,修饰F502(鞭毛蛋白,不含有半胱氨酸)及F502突变蛋白(鞭毛蛋白突变,含有一个半胱氨酸)的SDS-PAGE图;
图4是本发明实施例6中mPEG-DAQ与商品化修饰剂mPEG-Mal、mPEG-VS修饰效率的比较;
图5是本发明实施例7中mPEG-DAQ修饰rhCNTF产物的纯化层析图,其中,(a)是离子交换层析图,(b)是凝胶过滤层析图;
图6是本发明实施例8的制备的修饰剂抗水解能力考察;
图7是本发明实施例9的考察修饰产物热稳定性的电泳图,其中,(a)是mono-mPEG-MAL-CNTF,(b)是mono-mPEG-DAQ-CNTF。
具体实施方式
为更好地说明本发明,便于理解本发明的技术方案,本发明的典型但非限制性的实施例如下:
实施例1单端单甲氧基封闭的mPEG5k-DAQ的制备过程。
称取mPEG-OH(5000Da)10克(2mM),溶于60-100ml的纯水中,向其内加入2,2,6,6-四甲基哌啶-1-氧基(TEMPO)2-4mg和KBr 48mg,冰水浴下搅拌溶解。用4N HCl调节次氯酸钠溶液(浓度为8%)的pH为10,于冰水浴下冷却;将冷却的NaClO溶液倒入前述的mPEG溶液中(6-8ml),反应搅拌进行。大约30秒后,体系的pH值开始下降,用冷的0.5N NaOH溶液调节,保持反应溶液的pH值为10,大约2-4h后体系的pH值不再改变;在室温下,继续搅拌6小时或过夜后,加入1ml乙醇湮灭反应;用4N HCl调节反应的pH值为2-3,用二氯甲烷萃取6次,合并萃取液并浓缩;冷乙醚沉淀,淋洗,真空干燥后得到末端为羧基的聚乙二醇修饰试剂(mPEG-COOH)。
称取567mg盐酸多巴胺(3mM)溶于3ml二氯甲烷溶剂中,同时加入催化剂量的三乙胺作为缚酸剂,反应15min~30min后,得到除盐酸的多巴胺。称取5.0g按照上述方法制备的mPEG-COOH(1mM),溶于27ml干燥的二氯甲烷中,加入138mg NHS(1.2mM)。在冰浴下搅拌1小时后,加入216.3mg DCC(1.05mM),密封反应系统,冰浴下反应2小时。升至室温,在惰性气体保护下,加入除盐酸多巴胺的二氯甲烷溶液,反应过夜。用布氏漏斗滤去反应中生成的DCU,将滤液浓缩至5ml左右,顷入冰冷的无水乙醚中沉淀。白色沉淀用热的异丙醇溶解-重结晶3次后,抽滤干燥,得活化的mPEG-DA 4.8g,充氮气、避光保存。
称取按照上述方法制备的20mg mPEG-DA于玻璃试管,溶于少量(约200~300ul)二氯甲烷中,加入适量NaIO4固体,充分搅拌震荡,反应30min。离心12000rpm,10min除掉NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得mPEG充分沉淀,快速离心,弃上清。加入少量二氯甲烷使得沉淀再次重悬,离心12000rpm,10min除掉残留NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得mPEG充分沉淀,快速离心,弃上清。在通风橱中充分挥发乙醚和二氯甲烷。此过程mPEG5k-DAQ收率约为50%~60%。
称取1mg制备的mPEG5k-DAQ溶于550ul CDCl3,进行600M H1NMR检测,检测结果如说明书附图1(a)所示,由结果可以分析邻苯二酚的酚羟基几乎转化为醌的结构。
称取1mg制备的mPEG-5k-DAQ溶于50ul乙腈中,用MALDI-TOF MASS分析其分子量,检测结果如说明书附图1(b)所示,由结果分析制备的mPEG-5k-DAQ分子量主要在5000Da~5500Da。
实施例2两端同官能团DAQ-PEG5k-DAQ的制备过程。
称取两端均为琥珀酰亚胺酯基的聚乙二醇(NHS-PEG5k-NHS)1克(0.2mM),溶于6-10ml的干燥、无水的二氯甲烷中,得溶液(A);称取盐酸多巴胺56.7mg(0.3mM)溶于二氯甲烷,在氮气保护下搅拌时加入催化量的三乙胺作为缚酸剂,反应15min,得溶液(B)。将溶液(A)和溶液(B)混合,避光、充氮气保护,过夜反应。
将反应混合物透析至去离子水中,浓缩至浓度达到15~20mg/ml,然后继续透析到水中,并用盐酸酸化至pH 3.5,继续透析到去离子水中去掉游离的盐酸,将其冻干,得到两端端基为含有邻苯二酚官能团的聚乙二醇修饰剂(DA-PEG5k-DA);
称取按照上述方法制备的20mg DA-PEG-DA于玻璃试管,溶于少量(约200~300ul)二氯甲烷中,加入适量NaIO4固体,充分搅拌震荡,反应30min。离心12000rpm,10min除掉NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得PEG充分沉淀,快速离心,弃上清。加入少量二氯甲烷使得沉淀再次重悬,离心12000rpm,10min除掉残留NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得PEG充分沉淀,快速离心,弃上清。在通风橱中充分挥发乙醚和二氯甲烷。此过程mPEG5k-DAQ收率约为50%~60%。
实施例3两端异官能团X-PEG5k-DAQ的制备过程。
称取一端为氨基(-NH2)、羧基(-COOH)、琥珀酰亚胺酯基、琥珀酰亚胺碳酸酯基、酰肼,另一端为琥珀酰亚胺酯基的聚乙二醇(X-PEG5k-NHS)1克(0.2mM),溶于6-10ml的干燥、无水的二氯甲烷中,得溶液(A);称取盐酸多巴胺56.7mg(0.3mM)溶于二氯甲烷,在氮气保护下搅拌时加入催化量的三乙胺作为缚酸剂,反应15min,得溶液(B)。将溶液(A)和溶液(B)混合,避光、充氮气保护,过夜反应。
将滤液浓缩至5ml左右,顷入冰冷的无水乙醚中沉淀。白色沉淀用热的异丙醇溶解-重结晶3次后,抽滤干燥,得活化的X-PEG-DA 4.8g,充氮气、避光保存。
称取按照上述方法制备的20mg X-PEG-DA于玻璃试管,溶于少量(约200~300ul)二氯甲烷中,加入适量NaIO4固体,充分搅拌震荡,反应30min。离心12000rpm,10min除掉NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得PEG充分沉淀,快速离心,弃上清。加入少量二氯甲烷使得沉淀再次重悬,离心12000rpm,10min除掉残留NaIO4固体。向滤液中加入足量的冷乙醚并震荡,使得PEG充分沉淀,快速离心,弃上清。在通风橱中充分挥发乙醚和二氯甲烷。此过程X-PEG-DA收率约为50%~60%。
实施例4单端单甲氧基封闭的mPEG5k-DAQ修饰剂修饰重组人睫状神经因子(含有一个半胱氨酸,以下简称rhCNTF)、鞭毛蛋白的突变蛋白(不含鞭毛蛋白的高可变区,且突变体含有一个半胱氨酸,以下简称F502-Cys)和人血清白蛋白(含有一个半胱氨酸,以下简称HSA)的应用。
将纯品的rhCNTF溶于20mM tris-HCl,pH7.4,浓度1mg/ml,按照摩尔比1:4的比例加入mPEG5k-DAQ,室温反应2h。用分离胶浓度12%的SDS-PAGE分析。由说明书附图2可以看出,rhCNTF已经被修饰剂成功修饰。
将纯品的F502-Cys溶于20mM tris-HCl,pH7.4,浓度1mg/ml,按照摩尔比1:4的比例加入mPEG5k-DAQ,室温反应2h。用分离胶浓度12%的SDS-PAGE分析。由说明书附图2可以看出,F502突变蛋白已经被修饰剂成功修饰。
将纯品的HSA溶于20mM tris-HCl,pH7.4,浓度1mg/ml,按照摩尔比1:4的比例加入mPEG5k-DAQ,室温反应2h。用分离胶浓度12%的SDS-PAGE分析。由说明书附图2可以看出,HSA已经被修饰剂成功修饰。
实施例5单端单甲氧基封闭的mPEG5k-DAQ修饰剂修饰蛋白质的位点选择性确认。
分别将F502和F502-Cys溶于20mM bistris-HCl,pH5.0、20mM tris-HCl,pH7.0、20mM tris-HCl,pH8.5,浓度均为1mg/ml;按照摩尔比1:4的比例加入mPEG5k-DAQ,室温反应2h。用分离胶浓度12%的SDS-PAGE分析。由说明书附图3可以得出,未半胱氨酸定点突变的F502蛋白没有出现修饰条带,而与之对应的F502-Cys却存在相应修饰条带,证明mPEG-DAQ对F502的修饰出现在半胱氨酸位置,即巯基定点。
实施例6mPEG-DAQ与商品化修饰剂mPEG-Mal、mPEG-VS修饰效率的比较。
将纯品rhCNTF溶于20mM tris-HCl,pH7.4,浓度为1mg/ml,按照摩尔比
1:4和1:8的比例加入mPEG5k-DAQ、mPEG5k-Mal和mPEG20k-VS,室温反应2h和8h,用分离胶浓度12%的SDS-PAGE分析。由说明书附图4可以看出,在1:4和1:8的修饰比例下,mPEG-DAQ和mPEG-Mal的修饰率相近,而远远高于mPEG-VS。
实施例7单端单甲氧基封闭的mPEG5k-DAQ修饰剂修饰rhCNTF的层析纯化。
将实施例4制备的mPEG5k-DAQ-rhCNTF用蛋白质纯化系统AKTA进行纯化,采用的层析柱为Q FF(GE)和Superdex 75(GE)。QFF纯化时,层析bufferA选择50mM tris-HCl,pH7.4,bufferB选择50mM tris-HCl,1M NaCl,pH7.4;凝胶过滤层析条件为50mM tris-HCl,0.15M Na2SO4,pH7.4。层析结果图如说明书附图5(a)和(b)所示,由结果分析可以得知经过一步QFF层析纯化时主要除掉未反应的PEG,经过凝胶过滤层析时主要除掉原蛋白;且经过凝胶过滤后修饰样品的表观分子量符合理论值(考虑PEG较大的水力学半径)。
实施例8修饰剂抗水解能力的确认。
将实施例1制备的mPEG5k-DAQ与商品mPEG5k-Mal修饰剂分别溶于25mM tris-HCl,pH7.4,浓度为1mg/ml,分别置于4℃冰箱,放置0h、12h、24h、48h和96h,取样,与1mg/ml rhCNTF反应,rhCNTF与修饰剂摩尔比为1:4,通过12%SDS-PAGE。比较mPEG5k-DAQ与mPEG5k-Mal水解不同时间后的修饰剂的修饰能力变化情况。由说明书附图6可知,mPEG-Mal修饰剂随着水解时间的延长,修饰率会逐渐降低;而本发明公开的mPEG-DAQ修饰率却没有明显降低的现象。
实施例9单甲氧基封闭的mPEG5k-DAQ修饰产物的热稳定性的确认。
将实施例6纯化得到mono-mPEG-DAQ-rhCNTF用G25脱盐柱置换buffer至20mM PB,pH7.4缓冲体系中。同样的方法制备mono-mPEG-MAL-rhCNTF。各取两种修饰产物500ul与EP管中,在100℃加热0min、4min、8min、16min和32min。用12%SDS-PAGE考察PEG脱落情况,检测结果如图7所示。由结果分析可得,本发明公开的修饰试剂制备的修饰产物在经过加热处理后原蛋白的含量没有显著增加,而商品化的PEG-Mal修饰试剂修饰产物在经过加热处理后,会有原蛋白明显的量的增加。
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!